

Abstract
Dyskeratosis congenita (DC) is an inherited bone marrow failure syndrome that is characterized by lacey reticular hyperpigmentation of the skin, dystrophic nails, mucous membrane leukoplakia and pancytopenia. Diagnosis may be delayed until clinical signs are apparent. Severe pancytopenia frequently causes early mortality of DC patients, who have an increased risk of developing oropharyngeal squamous cell carcinoma. Several case reports have described oral changes in DC, which include oral leukoplakia, increased dental caries, hypodontia, thin enamel structure, aggressive periodontitis, intraoral brown pigmentation, tooth loss, taurodontism and blunted roots. We determined the prevalence of these previously reported findings in a cohort of 17 patients with DC and 23 family members. The most common oral changes in DC patients were oral leukoplakia (65% of the entire DC population), decreased root/crown ratio (75% with sufficient tooth development) and mild taurodontism (57% with sufficient tooth development). From the clinical perspective, a diagnosis of DC or other inherited bone marrow failure syndrome should be considered in young persons with oral leukoplakia, particularly those with no history of smoking. Multiple permanent teeth with decreased root/crown ratios further suggest DC.
Keywords: dyskeratosis congenita, leukoplakia, tooth development, DKC1, TERC, TERT
Introduction
Early in the 1900s, Zinsser, Cole and Engman described an inherited variant of ectodermal dysplasia that affected skin, nails and mucous membranes1. The syndrome eventually became known as dyskeratosis congenita (DC), and is classified as one of the Inherited Bone Marrow Failure Syndromes (IBMFS). Classic DC is characterized by dystrophic nails, reticular lacey hyperpigmentation of the skin, and mucous membrane leukoplakia2. Further studies have established DC as a multi-system disorder in which affected patients often develop single or pancytopenia, abnormalities of the eye, lacrimal duct stenosis, pulmonary fibrosis and malignancy, in addition to changes of skin and mucosa3. Although onset may occur in early childhood, diagnosis is often delayed until clinical signs are apparent. Early mortality of DC patients is frequently due to severe pancytopenia. More than 275 unique patients with DC have been reported from all major ethnic groups2.
The presumed underlying defect in DC is an inability to preserve telomere length secondary to mutations in the genes for RNA or proteins composing this ribonucleoprotein complex3. Chromosome ends are capped by telomeres, which are protective DNA-protein complexes. Telomeres shorten progressively with successive cell divisions in most cells, and cell division ceases when telomere length becomes critically short. Germ cells and selected stem cells must have a mechanism to preserve telomere length to continue cell division and provide new cells for many tissues3. In healthy cells, a ribonucleoprotein reverse transcriptase complex, telomerase, maintains telomere length by protecting telomeric end caps of chromosomes. To date, mutations of genes encoding the RNA template (TERC), telomerase catalytic reverse transcriptase (TERT) and dyskerin, a component of the telomerase complex, have been identified in various DC families4, 5. Clinically there appear to be three patterns of inheritance for DC: autosomal dominant (AD), autosomal recessive (AR) and X-linked recessive (XLR)2. Mutations of the gene DKC1 (located at Xq28), which encodes nucleolar protein dyskerin, are found in XLR DC, whereas mutations in TERC and TERT have been identified in some AD DC families. No mutations have been identified in more than half the known patients in large series, and thus additional DC genes await identification2.
Tissues with cell populations that must regenerate frequently are most affected in DC. The most serious complications are greatly increased risks of bone marrow failure, cancer at an early age, infection, and hemorrhage2. A literature review found 45 cases of early cancers reported out of an estimated 274 patients, and most cancers were squamous cell carcinoma (SCC)2. The oropharynx was the site of 17 of these reported SCC, assumed to arise from the chronic mucosal leukoplakia that develops at a very young age in many of these patients2. In addition to their increased risk of cancer, DC patients have a significant burden of disease, as they may become transfusion-dependent and require repeated hospitalizations for infections6. These increased susceptibilities to multiple complications necessitate early diagnosis to institute treatments such as hematopoietic stem cell transplant, androgens, or hematopoietic growth factors.
IBMFS are diagnosed by physical and laboratory findings, often in conjunction with evidence of familial segregation apparent from the family pedigree. Many times genetic testing will not identify a specific mutation2. Several case reports have described specific oral changes in association with DC. These include oral leukoplakia, increased dental caries, hypodontia, thin enamel structure, aggressive periodontitis, intraoral brown pigmentation, tooth loss, taurodontism and blunted roots7–10. To establish the phenotypes of various IBMFS, including DC, the Clinical Genetics Branch of the National Cancer Institute (NCI) is extensively evaluating patients and family members with these disorders. A thorough oral examination is part of the evaluation. This report presents the oral and dental findings of 17 patients with DC and 23 family members.
Methods and Materials
Study design
This was a cross-sectional study of patients with an IBMFS and their family members. All consented to a protocol approved by the NCI Institutional Review Board. This study includes evaluations conducted between January 2003 and March 2006. The majority of patients were evaluated only once.
Subjects
Seventeen patients with DC and 23 family members, representing twelve different families, were evaluated. In nine families, the members affected with DC were examined. Three families were evaluated after the affected member had died. No patients had known consangineous parentage. Fourteen healthy IBMFS family members (< 31 years of age) and one additional healthy male served as an age and sex appropriate control group for studies of dental indices (Decayed, Missing and Filled permanent Surfaces or DMFS) and root development. These controls were relatives of patients with Fanconi Anemia, Diamond-Blackfan Anemia and Shwachman-Diamond Syndrome.
Examination
A standardized, detailed health history, including smoking history, was collected at the time of the physical examination. Concurrent laboratory studies were performed at the National Institutes of Health Clinical Center. Every patient had a comprehensive oral examination, including radiographs in 16/17. Panographic radiographs were evaluated from twelve DC patients, a panel of 6 intraoral radiographs (4 periapical and 2 bite-wing exposures) from four DC patients, while one 3 year old patient was too young to obtain diagnostic radiographs. Clinical and radiographic evidence of caries and existing restorations were recorded. The location and type of soft tissue lesions were noted and photographed. The World Health Organization (WHO) definition of oral leukoplakia (“a white patch or plaque that cannot be characterised, clinically or pathologically as any other disease”) was used to classify white lesions11. Root lengths and crown lengths of all permanent first and second molars and premolars with radiographically closed apices were calculated from panographic radiographs12. Evidence of past or present aggressive periodontitis (>50% generalized alveolar bone loss) was determined radiographically for 12 patients. Taurodontism was calculated from radiographs using the method described by Shifman and colleagues13 that involves determining the distance between the lowest point of the roof of the pulp chamber to the apex of the longest root, and distance between the baseline connecting the two CEJ and the highest point in the floor of the pulp chamber. Three degrees of taurodontism can be calculated from this index (hypotaurodontism or mild, mesotaurodontism or moderate and hypertaurodontism or severe).
Mutational analyses
Mutations in DKC1, TERC, and TERT were identified by bi-directional sequencing of PCR-amplified fragments (GeneDx, Inc., Gaithersburg, MD).
Statistical methods
DMFS scores and the percentage of posterior teeth with decreased root/crown ratios of the control and DC groups were compared using Student’s t-test. Frequencies of leukoplakia, brown pigmentation, decreased root/crown ratios, taurodontism, thin enamel, hypodontia, and aggressive periodontitis in the control and DC groups were compared with Fisher’s exact test. The p <0.05 value was used for significance.
Results
Patient and family characteristics
The 17 patients with DC represented nine different families. Mutational analyses established the distinct mutation in nine patients, of whom five had mutant DKC1, three had mutant TERC and one had mutant TERT (Table 1). The other eight did not have mutations in those genes, and were diagnosed by clinical findings and pedigree. Thirteen patients were male and four were female, ranging in age from 3 to 46 years (mean ± 1 standard deviation (SD) = 18.1 ± 10.5 years, median 15 years), with 16 of 17 patients under the age of 30 years. Fifteen of the 17 had anemia, neutropenia, and/or thrombocytopenia when evaluated at the clinic (Table 1). Three patients required regular transfusions of red blood cells and platelets. Three were treated with androgens (only one was responding), one was receiving both granulocyte colony stimulating factor and erythropoietin, and one was receiving granulocyte colony stimulating factor.
Table 1.
Features of DC patients*
| Family Number | Case Number | Age on exam, yrs | Sex | WBC** (K/μL) | ANC** (K/μL) | Hb** (g/dL) | MCV** | Platelet Count (K/μL) | Inheritance Pattern | Mutant gene | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 46 | F | 5.150 | 2.735 | 12.8 | 115 | 28 | AD** | TERC** | Halotestin |
| 1 | 2 | 15 | M | 4.440 | 2.202 | 14.7 | 101 | 99 | AD | TERC | |
| 2 | 3 | 25 | M | 3.550 | 1.945 | 10.7 | 113 | 77 | AD | TERT** | |
| 3 | 4 | 26 | F | 2.460 | 1.707 | 8.1 | 110 | 64 | AD | unknown | Oxymetholone |
| 3 | 5 | 25 | M | 3.460 | 1.817 | 14.6 | 96 | 148 | AD | unknown | |
| 3 | 6 | 13 | M | 3.290 | 1.415 | 13.9 | 89 | 188 | AD | unknown | |
| 3 | 7 | 29 | M | 4.340 | 2.209 | 15.1 | 96 | 142 | AD | unknown | |
| 3 | 8 | 22 | M | 3.790 | 1.766 | 14.5 | 99 | 117 | AD | unknown | |
| 3 | 9 | 21 | M | 3.130 | 1.681 | 12.1 | 109 | 24 | AD | unknown | |
| 4 | 10 | 10 | M | 3.570 | 1.571 | 9.6 | 106 | 25 | XLR** | DKC1** | |
| 5 | 11 | 22 | M | 2.740 | 1.403 | 10.0 | 118 | 24 | XLR | DKC1 | |
| 6 | 12 | 9 | M | 2.400 | 0.71 | 10.7 | 103 | 14 | XLR | DKC1 | G-CSF**, Aranesp, RBC** and platelet transfusions |
| 6 | 13 | 3 | M | 2.160 | 0.616 | 9.5 | 98 | 16 | XLR | DKC1 | |
| 6 | 14 | 13 | M | 3.600 | 1.897 | 12.9 | 97 | 45 | XLR | DKC1 | |
| 7 | 15 | 15 | F | 2.180 | 0.392 | 7.9 | 114 | 17 | AD | TERC | |
| 8 | 16 | 9 | F | 2.620 | 1.022 | 9.5 | 87 | 12 | unknown | unknown | G-CSF, RBC and platelet transfusions |
| 9 | 17 | 6 | M | 3.380 | 1.213 | 9.1 | 112 | 110 | unknown | unknown | Oxymetholone, prednisone, RBC and platelet transfusions |
Shaded values are outside the reference range.
WBC = white blood cells, ANC = absolute neutrophil count, Hb = hemoglobin, MCV = mean corpuscular volume, AD = autosomal dominant, TERC = gene encoding the RNA component of telomerase2, 4, 5, TERT = gene encoding human telomerase reverse transcriptase3, XLR = X-linked recessive, DKC1 = gene encoding the dyskerin protein3, G-CSF = granulocyte colony stimulating factor, RBC = red blood cells
A total of 23 clinically unaffected family members were evaluated, including six individuals from three families in which the proband was deceased. The family group consisted of 14 females and 9 males, ranging in age from 7 years to 63 years (mean ± 1 SD = 37.9 years ± 14.8 years; median 36 years, Table 2). The sex ratio in the family members was not significantly different than in the patients, but the family members were significantly older (p <0.001). Four females were heterozygous for mutant DKC1, and six (three males and three females) did not have the mutated gene in families with known mutations. Carrier status of the other 13 family members was unknown, or presumed normal from the pedigree.
Table 2.
Features of DC family members
| Family Number | Age on exam (yrs) | Sex | Inheritance | Carrier Status |
|---|---|---|---|---|
| 3 | 30 | Female | AD | Unknown |
| 3 | 33 | Male | AD | Unknown |
| 3 | 28 | Male | AD | Unknown |
| 3 | 51 | Female | AD | Presumed normal |
| 4 | 13 | Male | XLR | Unknown |
| 4 | 36 | Male | XLR | Unknown |
| 4 | 41 | Female | XLR | Unknown |
| 5 | 34 | Female | XLR | Normal |
| 5 | 25 | Female | XLR | Normal |
| 5 | 50 | Male | XLR | Normal |
| 5 | 54 | Female | XLR | Heterozygous for mutant DKC1 |
| 6 | 36 | Male | XLR | Normal |
| 6 | 36 | Female | XLR | Heterozygous for mutant DKC1 |
| 7 | 48 | Female | AD | Normal |
| 8 | 48 | Male | AD | Unknown |
| 8 | 42 | Female | AD | Unknown |
| 8 | 7 | Female | AD | Unknown |
| 10 | 29 | Female | XLR | Heterozygous for mutant DKC1 |
| 10 | 61 | Male | XLR | Normal |
| 10 | 58 | Female | XLR | Heterozygous for mutant DKC1 |
| 11 | 63 | Female | Unknown | Unknown |
| 12 | 24 | Female | Unknown | Unknown |
| 12 | 24 | Male | Unknown | Unknown |
DC, dyskeratosis congenital; AD, autosomal dominant
Soft tissue lesions
Intraoral soft tissue changes were found in 12 of 17 DC patients (Table 3). Findings were classified by appearance and by location into the following categories: leukoplakia (excluding prominent linea alba), erythema, brown pigmentation, and papillary atrophy on the tongue. Oral leukoplakia, present in 11 DC cases, was the most frequently found oral change in this group, with the most common sites being the tongue (n = 9, Table 3, Figure 1), buccal mucosa (n=5), palate (n=2) and gingiva (n=1). The leukoplakia of the tongue varied in presentation. It was found in isolated patches (Figure 1A), as a fine reticular pattern (1B) or as a plaque-like leukoplakia distributed across the entire tongue dorsum (Figure 1C and 1D). In three younger patients (ages 9, 10 and 13 years), leukoplakia accompanied papillary atrophy of the tongue (Figure 1A, 1B). We could not find an age-related pattern or genotype/phenotype correlation in the varied clinical patterns of leukoplakia. Patients with either TERC or DKC1 mutations presented with leukoplakia on the tongue and/or buccal mucosa. The one patient with a known mutation of TERT did not have leukoplakia. Only three of the eleven patients with leukoplakia had a history of any tobacco use, defined as >100 cigarettes smoked in a lifetime or use of any other tobacco product.
Table 3.
Abnormal soft tissue findings
| Case number | Age on exam, yrs | Findings | Tongue | Buccal mucosa | Gingiva | Palate | Floor of mouth | Smoking history* | Mutant Gene |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 15 | Brown pigmentation | XX | no | TERC | ||||
| 4 | 26 | Leukoplakia | XX | yes | unknown | ||||
| Erythema | XX | ||||||||
| 7 | 29 | Plaque-like leukoplakia | XX | no | unknown | ||||
| Erythema | XX | ||||||||
| 8 | 22 | Leukoplakia | XX | XX | yes | unknown | |||
| 9 | 21 | Plaque-like leukoplakia | XX | XX | XX | no | unknown | ||
| 10 | 10 | Leukoplakia | XX | XX | no | DKC1 | |||
| Erythema with mild papillary atrophy | XX | XX | |||||||
| 11 | 22 | Plaque-like leukoplakia | XX | XX | XX | yes | DKC1 | ||
| Erythema | XX | XX | XX | ||||||
| 12 | 9 | Reticular leukoplakia | XX | no | DKC1 | ||||
| Erythema | XX | ||||||||
| 13 | 3 | Plaque-like leukoplakia | XX | no | DKC1 | ||||
| Erythema | XX | ||||||||
| 14 | 13 | Leukoplakia | XX | no | DKC1 | ||||
| Erythema with papillary atrophy | XX | XX | |||||||
| 15 | 15 | Plaque-like leukoplakia | XX | XX | no | TERC | |||
| Brown pigmentation | XX | XX | |||||||
| 16 | 9 | Leukoplakia | XX | no | Unknown | ||||
| Erythema with papillary atrophy | XX |
Smoking = >100 cigarettes in life or use of any other type of tobacco product
Figure 1. Varied presentation of leukoplakia in patients with dyskeratosis congenita.
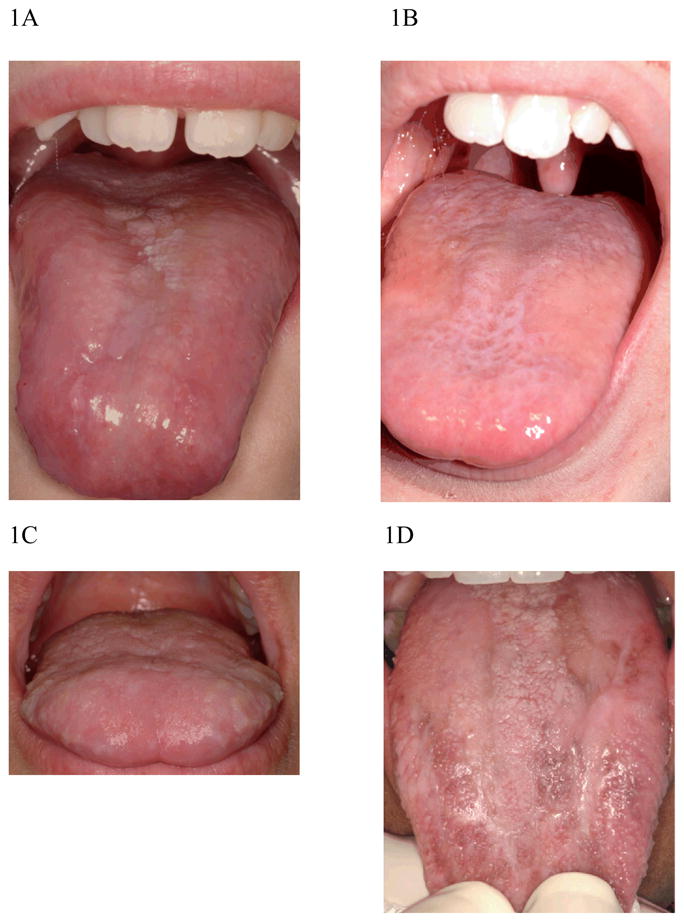
1A: Erythema, leukoplakia and papillary atrophy of the tongue in a 9 year old female, mutant gene unknown.
1B: Mild Papillary atrophy and leukoplakia of the tongue in a reticular pattern in a 10 year old male with mutant DKC1.
1C. Plaque-like leukoplakia on the dorsum of the tongue in a 21 yr old male, mutant gene unknown.
1D. Plaque-like leukoplakia with brown pigmentation of tongue in a patient with mutantb TERC.
Erythema, the second most common soft tissue finding in DC patients, was found in eight patients with or without thrombocytopenia. These erythematous areas on either the buccal mucosa or tongue appeared to be sites that had lost the superficial epithelium without disruption of the underlying connective tissue (Figure 1A, Figure 2). The erythematous patches on the buccal mucosa were at the level of the occlusal plane (Figure 2), suggesting the surface tissue sloughed after contact with the teeth. These areas were only present in patients who also had oral leukoplakia. Five patients had petechiae of the buccal mucosa, and one patient with significant thrombocytopenia had a hematoma on the lip.
Figure 2.
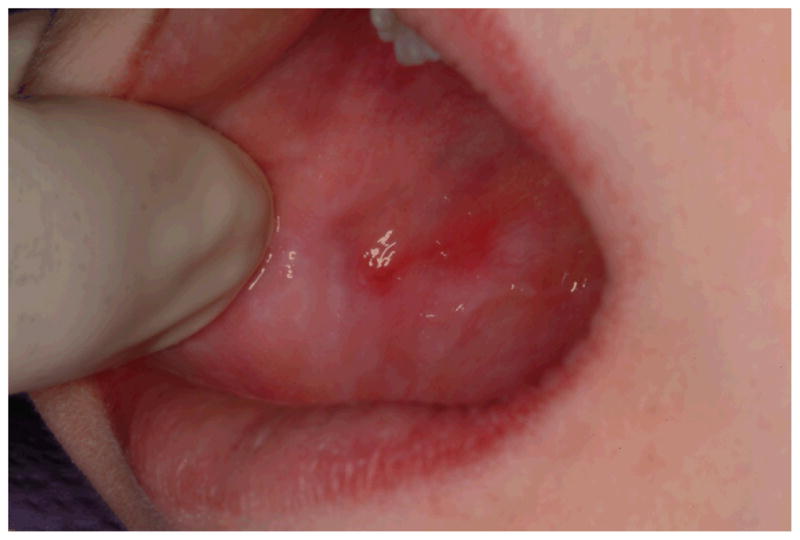
Erythema and diffuse leukoplakia of the buccal mucosa in a 10 year old male with mutant DKC1.
The last distinct soft tissue change found in the DC patient group was brown pigmentation of the tongue (Figure 1D), which was present in only two patients. These individuals, an African-American female and a Caucasian male, both had mutations in TERC.
Biopsies of the oral lesions were not obtained from patients enrolled in this study as most had very low platelet counts and were followed by other practitioners in their home communities.
No notable soft tissue changes were identified in family members.
Periodontal changes
No patient had radiographic evidence of past or present aggressive periodontitis.
Tooth changes
Since previous studies reported increased caries in DC, DMFS scores of 15 DC patients between the ages of 6 and 29 years (mean ± 1 SD = 17.2 years ± 7.3 years; median 15 years) were compared with sex and aged matched IBMFS family controls (age range 7 to 31 years, mean ± 1 SD = 16.1 years ± 7.4 years; median 14 years). There was no significant difference (p >0.05) in the number of decayed surfaces, missing teeth, filled surfaces or overall DMFS scores (data not shown). There was no evidence of thin enamel in the 16 DC patients with radiographs. Hypodontia (consisting only of congenitally missing premolars) was noted in two patients with DC and one control.
The difference between the proportion of teeth with decreased root/crown ratios was compared for the DC group and the family member control group and found to be significantly different (p <0.007, Fisher’s exact test). This finding was the most pronounced hard tissue change in DC (Tables 4 and 5) and was visible on panographic radiographs from several patients (Figures 3 and 4). This decreased ratio was secondary to decreased root length, as the heights of the crowns were not significantly different between the two groups (data not shown). The decreased root/crown ratio was found in at least four posterior teeth in 6 of 8 DC patients with sufficient radiographs and tooth development for evaluation (at least 13 years of age), and appeared to be developing in three children ages 9 or 10 years old (data not shown). Only one DC family member had multiple teeth with decreased root/crown ratios. This female who had one copy of mutant DKC1 (and three male children with DC) had eight teeth with decreased ratios. Shortened incisor roots were visible in a few panographic radiographs from DC patients, but these teeth were excluded from analysis as anterior teeth are often indistinct in this type of radiographic image. Since this was a cross-sectional rather than longitudinal study, it was not possible to determine whether the rate of root growth was also retarded in DC. Shortened roots were observed in individuals with mutations of the TERT, DKC1 or TERC genes. Taurodontism of the mildest Shifman class was noted in four of seven patients (Figures 3 and 4, Table 5) and none of the controls. Statistical analyses (two-sided Fisher’s exact test) confirmed that oral leukoplakia (p <0.001), decreased root/crown ratios of the posterior teeth (p = 0.007) and taurodontism (p = 0.015) occurred more frequently in the DC group (Table 5).
Table 4.
Root/crown ratio of posterior teeth, DC patients and IBMFS family controls
| Controls | DC patients | |||
|---|---|---|---|---|
| Sex | Age | Number of teeth below published norms* | Age | Number of teeth below published norms* |
| Female | 41 | 1/16 | 46 | 2/15 |
| Male | 14 | 1/16 | 15 | 0/16 |
| Male | 31 | 1/15 | 25 | 9/16 |
| Male | 29 | 2/16 | 29 | 6/9 |
| Male | 21 | 0/14 | 22 | 5/16 |
| Male | 16 | 0/16 | 19 | 9/16 |
| Male | 24 | 0/16 | 22 | 16/16 |
| Male | 13 | 2/16 | 13 | 4/4 ** |
DC, dyskeratosis congenita
Abnormal = Less than normal mean – 2 s.d. The difference between the proportion of teeth with decreased root/crown ratios in the DC group and control group is significant, P <0.003 (unpaired t test). Not all posterior teeth could be evaluated.
Other posterior teeth had orthodontic brackets.
Table 5.
Frequency of oral hard and soft tissue abnormalities in DC patients and family members
| Finding | Controls | DC Patients | P value* | ||
|---|---|---|---|---|---|
| Yes | No | Yes | No | ||
| Leukoplakia | 1 | 22 | 11 | 6 | P < 0.001 |
| Erythema | 2 | 21 | 8 | 9 | P < 0.01 |
| Brown pigmentation | 0 | 23 | 2 | 15 | NS |
| Papillary atrophy | 0 | 23 | 3 | 14 | NS |
| Decreased root/crown | |||||
| ratio in at least 4 | 0 | 8 | 6 | 2 | P = 0.007 |
| posterior teeth** | |||||
| Taurodontism** | 0 | 10 | 4 | 3 | P = 0.015 |
| Thin enamel** | 0 | 16 | 0 | 16 | NS |
| Hypodontia | 1 | 15 | 2 | 14 | NS |
| Aggressive periodontitis | 0 | 23 | 0 | 16 | NS |
DC, dyskeratosis congenital; NS, not significant
The frequencies of the findings in the DC group and the family member control group were compared using Fisher’s exact test. DC family members were used as controls for all comparisons except decreased root/crown ratio. Eight age and sex matched healthy IBMFS family members were used as controls for this comparison.
Not all DC patients had evaluable radiographs, or were too young to determine if root formation was abnormal or taurodontism was present.
Figure 3.

Panorex radiograph from a 13-year-old male with mutant DKC1 showing decreased root/crown ratios in the posterior teeth and taurodontism
Figure 4.
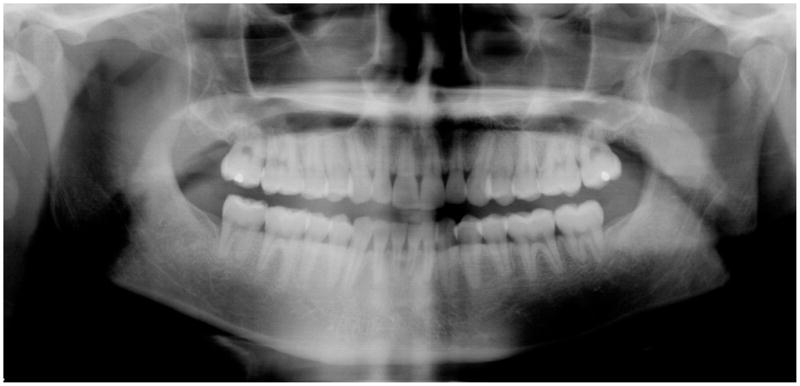
Radiograph from a 25-yr-old with mutant TERT and generalized decreased root/crown ratios in the posterior teeth.
Discussion
Controlled studies of the associated oral and dental findings in DC have not been reported previously. Although the prevalence of oral leukoplakia has been reported in larger cohorts, other previously reported dental findings were identified from case reports of one or two patients. In this study of 17 individuals with DC, the most commonly found oral changes were oral leukoplakia (65% of the entire population), decreased root/crown ratio (75% of patients with sufficient tooth development to permit evaluation) and mild taurodontism (57% with radiographs and sufficient tooth development to permit evaluation). Other previously reported oral features of this disease, increased dental caries, hypodontia, thin enamel and aggressive periodontitis, were not detected in this DC population.
The most common change of the oral mucosal tissues in DC was leukoplakia, one of the three features originally described in this disease. It was not present in every patient, and the prevalence in our cohort (65%) is similar to the 78% prevalence reported from a registry of 118 male patients with presumed XLR DC14. The clinical presentation of oral leukoplakia in these patients was very heterogeneous, which is consistent with the clinical variability reported for the skin changes associated with DC14. Other authors have proposed that the heterogeneity of the clinical DC presentation suggests modification of the phenotype by other genetic factors and/or the environment.
Very little is known about the histopathological features of oral leukoplakia in DC. A longitudinal study of one patient characterized histological features associated with progression from hyperkeratosis to dysplasia of a lesion on the tongue15. The dysplastic tissue from the ventral tongue exhibited abnormal keratin expression, consisting of co-expression of keratins K16, K10 and K13 15. p53 expression was not found in the first biopsy, but was present in all subsequent specimens. Unfortunately, no biopsies of leukoplakic tissues were available to permit similar studies from our patients.
In addition to leukoplakia, the tongues in some patients demonstrated atrophy of the papilla. The mechanism underlying this atrophy may be provided by studies with a mouse model (mTR+/− mice on the CAST/EiJ) of autosomal dominant DC16. Interbreeding of these mice that are heterozygous for deficiency of the RNA subunit of telomerase creates progressive telomere shortening. Histopathological examination of the intestines of later generation mice found crypt depletion and villi atrophy, suggesting normal telomere length is needed for maintenance of these tissues16. It is possible there is a similar requirement to maintain adequate telomere length to sustain healthy tongue epithelium, which is in constant need of replenishment from basal cells17.
Intraoral brown pigmentation, found in two patients with TERC mutations, may reflect an imbalance in telomerase activity. Recent work has demonstrated chromosomal imbalances involving TERC and TERT in various types of malignant melanoma18.
The finding of shortened roots in a high percentage of DC patients suggests normal telomere length or adequate telomerase activity is needed for complete root development. Two other reports further support this hypothesis. A recent study found cementoblast progenitor cells, believed to be responsible for root development, were immortalized by expression of human telomerase reverse transcriptase (TERT) and the gene for polycomb group protein, Bmi-119. Transduced single cell clones subsequently expressed mRNA for bone sialoprotein, osteocalcin, osteopontin, and type I collagen when implanted in immunodeficient mice. In another report, stem cells, which require functional telomerase, were identified in the apical root during certain stages of root formation of the rat tooth20. Alternatively, it is possible that the proteins encoded by the mutated genes in DC have functions in root and oral epithelial development by mechanisms not yet defined.
Root development of teeth may also provide information about the time of disease progression in patients with DC. Root development begins after the crown of the tooth is formed. The earliest root development of the permanent teeth occurs at about age 3 years, beginning with the central incisors21–23. The second permanent molar roots begin to form between age 7 and 8 years. Insults to the developing teeth, such as radiation, chemotherapy and/or hematopoietic stem cell transplant during that time can disturb root development22. Children surviving cancer may present later in life with clinically normal crowns and abnormally short roots of certain teeth22, marking the time of their cancer therapy. It is likely that a portion of dental follicular cells responsible for root development in DC cease functioning as telomeres shorten through repeated divisions. If all of the roots of the permanent teeth in a DC patient are short, telomerase activity or telomere length may have been inadequate since early childhood. If the only teeth with short roots are the second permanent molars, then telomerase activity may have been adequate until the early teenage years.
Taurodont teeth have enlarged pulp chambers and short roots13. Since root length is reduced in the DC patients, it was not surprising that taurodontism was found in four of seven evaluated patients. In addition, pulp chambers normally decrease in size with age because of secondary dentin deposition24. This process is believed to be mediated by stem cells in dental pulps that express dentin sialoprotein (DSP)25. It is possible that pulp stem cells that produce secondary dentin also are dysfunctional in DC. However, this presumed dysfunction did not appear to be profound, as the taurodontism in our patients was of the mildest form using the classification system of Shifman13.
Previously reported oral findings (aggressive periodontal disease, increased dental caries, thin enamel and hypodontia) associated with DC were not part of the oral phenotype found in this cohort. We were surprised that none of our patients had evidence of past or current aggressive periodontitis, and feel this may reflect the better dental hygiene of these DC patients or their absence of profound neutropenia. Most had absolute neutrophil counts (ANC) >1000 cells/μL (Table 1). One of the patients with an ANC <1000 cells/μL had radiographic suggestions of alveolar bone loss in the posterior teeth, but she was also undergoing full orthodontic therapy. Therefore, alveolar bone height assessments using radiographs were considered unreliable. Dental radiographs were not obtainable on the 3 year old with decreased ANC. Other previously reported findings of increased caries, hypodontia and thin enamel7, 9, 26 were not confirmed in this study. Given the heterogeneous nature of this patient group, these findings might be found in a larger study of patients and appropriate controls. In particular, studies of dental caries must be of significant size to control for the multiple other factors associated with caries in children 27, 28 to determine whether the frequency of caries is increased in DC.
In summary, the oral phenotype of DC is characterized by leukoplakia, decreased root/crown ratios and mild taurodontism. From the clinical perspective, a diagnosis of DC or another inherited bone marrow failure syndrome should be considered in any young person with oral leukoplakia, particularly those with no history of tobacco use. Continued studies with this cohort should define the oral phenotype more completely and determine what factors are most associated with the development of oral squamous cell carcinomas in these patients.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research and the National Cancer Institute.
References
- 1.Zinsser F. Atrophia cutis reticularis cum pigmentatione, dystrophia et leukoplakia oris. Ikonogr Dermatol. 1910;5:219–23. doi: 10.1007/BF00476707. [DOI] [PubMed] [Google Scholar]
- 2.Alter BP. Inherited Bone Marrow Failure Syndromes. In: Nathan D, Orkin SH, Ginsburg D, Look AT, editors. Hematology of Infancy and Childhood. 6. Saunders; Philadelphia: 2003. pp. 281–365. [Google Scholar]
- 3.Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol. 2006;43:157–66. doi: 10.1053/j.seminhematol.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34:257–63. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36:447–9. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 6.Steele JM, Sung L, Klaassen R, Fernandez CV, Yanofsky R, Wu J, et al. Disease progression in recently diagnosed patients with inherited marrow failure syndromes: A Canadian inherited marrow failure registry (CIMFR) report. Pediatr Blood Cancer. 2006;47:918–25. doi: 10.1002/pbc.20876. [DOI] [PubMed] [Google Scholar]
- 7.Wald C, Diner H. Dyskeratosis congenita with associated periodontal disease. Oral Surg Oral Med Oral Pathol. 1974;37:736–44. doi: 10.1016/0030-4220(74)90139-x. [DOI] [PubMed] [Google Scholar]
- 8.Loh HS, Koh ML, Giam YC. Dyskeratosis congenita in two male cousins. Br J Oral Maxillofac Surg. 1987;25:492–9. doi: 10.1016/0266-4356(87)90142-2. [DOI] [PubMed] [Google Scholar]
- 9.Yavuzyilmaz E, Yamalik N, Yetgin S, Kansu O. Oral-dental findings in dyskeratosis congenita. J Oral Pathol Med. 1992;21:280–4. doi: 10.1111/j.1600-0714.1992.tb01011.x. [DOI] [PubMed] [Google Scholar]
- 10.Handley TP, Ogden GR. Dyskeratosis congenita: oral hyperkeratosis in association with lichenoid reaction. J Oral Pathol Med. 2006;35:508–12. doi: 10.1111/j.1600-0714.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- 11.WHO collaborating center for oral precancerous lesions. Definition of leukoplakia and related lesions. An aid to studies on oral precancer. Oral Surg Oral Med Oral Pathol. 1978;4:518–539. [PubMed] [Google Scholar]
- 12.Holtta P, Nystrom M, Evalahti M, Alaluusua S. Root-crown ratios of permanent teeth in a healthy Finnish population assessed from panoramic radiographs. Eur J Orthod. 2004;26:491–7. doi: 10.1093/ejo/26.5.491. [DOI] [PubMed] [Google Scholar]
- 13.Tsesis I, Shifman A, Kaufman AY. Taurodontism: an endodontic challenge. Report of a case. J Endod. 2003;29:353–5. doi: 10.1097/00004770-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110:768–79. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 15.Ogden GR, Lane DP, Chisholm DM. p53 expression in dyskeratosis congenita: a marker for oral premalignancy? J Clin Pathol. 1993;46:169–70. doi: 10.1136/jcp.46.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao LY, Armanios M, Strong MA, Karim B, Feldser DM, Huso D, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123:1121–31. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 17.Potten CS, Booth D, Cragg NJ, Tudor GL, O’Shea JA, Appleton D, et al. Cell kinetic studies in the murine ventral tongue epithelium: thymidine metabolism studies and circadian rhythm determination. Cell Prolif. 2002;35(Suppl 1):1–15. doi: 10.1046/j.1365-2184.35.s1.1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirker C, Holzmann K, Spiegl-Kreinecker S, Elbling L, Thallinger C, Pehamberger H, et al. Chromosomal imbalances in primary and metastatic melanomas: over-representation of essential telomerase genes. Melanoma Res. 2003;13:483–92. doi: 10.1097/00008390-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Saito M, Handa K, Kiyono T, Hattori S, Yokoi T, Tsubakimoto T, et al. Immortalization of cementoblast progenitor cells with Bmi-1 and TERT. J Bone Miner Res. 2005;20:50–7. doi: 10.1359/JBMR.041006. [DOI] [PubMed] [Google Scholar]
- 20.Hosoya A, Nakamura H, Ninomiya T, Yoshiba K, Yoshiba N, Nakaya H, et al. Immunohistochemical Localization of Alpha-Smooth Muscle Actin During Rat Molar Tooth Development. J Histochem Cytochem. 2006;54:1371–8. doi: 10.1369/jhc.6A6980.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas HF. Root formation. Int J Dev Biol. 1995;39:231–7. [PubMed] [Google Scholar]
- 22.Holtta P, Hovi L, Saarinen-Pihkala UM, Peltola J, Alaluusua S. Disturbed root development of permanent teeth after pediatric stem cell transplantation. Dental root development after SCT. Cancer. 2005;103:1484–93. doi: 10.1002/cncr.20967. [DOI] [PubMed] [Google Scholar]
- 23.Ten Cate AR. The role of epithelium in the development, structure and function of the tissues of tooth support. Oral Dis. 1996;2:55–62. doi: 10.1111/j.1601-0825.1996.tb00204.x. [DOI] [PubMed] [Google Scholar]
- 24.Paewinsky E, Pfeiffer H, Brinkmann B. Quantification of secondary dentine formation from orthopantomograms--a contribution to forensic age estimation methods in adults. Int J Legal Med. 2005;119:27–30. doi: 10.1007/s00414-004-0492-x. [DOI] [PubMed] [Google Scholar]
- 25.Batouli S, Miura M, Brahim J, Tsutsui TW, Fisher LW, Gronthos S, et al. Comparison of stem-cell-mediated osteogenesis and dentinogenesis. J Dent Res. 2003;82:976–81. doi: 10.1177/154405910308201208. [DOI] [PubMed] [Google Scholar]
- 26.Brown CJ. Dyskeratosis congenita: report of a case. Int J Paediatr Dent. 2000;10:328–34. doi: 10.1046/j.1365-263x.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- 27.Psoter WJ, Pendrys DG, Morse DE, Zhang H, Mayne ST. Associations of ethnicity/race and socioeconomic status with early childhood caries patterns. J Public Health Dent. 2006;66:23–9. doi: 10.1111/j.1752-7325.2006.tb02547.x. [DOI] [PubMed] [Google Scholar]
- 28.Ramos-Gomez FJ, Weintraub JA, Gansky SA, Hoover CI, Featherstone JD. Bacterial, behavioral and environmental factors associated with early childhood caries. J Clin Pediatr Dent. 2002;26:165–73. doi: 10.17796/jcpd.26.2.t6601j3618675326. [DOI] [PubMed] [Google Scholar]