

Summary
Severe congenital neutropenia (SCN) is a heterogeneous bone marrow failure syndrome predisposing to myelodysplastic syndrome and acute myeloid leukaemia (MDS/AML). We studied 82 North American and Australian SCN patients enrolled in the Severe Chronic Neutropenia International Registry who were on long-term treatment with granulocyte colony-stimulating factor and for whom the neutrophil elastase (ELA2) gene was sequenced. There was no significant difference in the risk of MDS/AML in patients with mutant versus wild-type ELA2: the respective cumulative incidences at 15 years were 36% and 25% (P = 0·96). Patients with either mutant or wild-type ELA2 should be followed closely for leukaemic transformation.
Keywords: severe congenital neutropenia, neutrophil elastase ELA2, acute myeloid leukaemia, myelodysplastic syndromes, granulocyte colony-stimulating factor
Severe congenital neutropenia (SCN) is an inherited bone marrow failure syndrome that is usually diagnosed in very young children on the basis of absolute neutrophil count (ANC) values persistently below 0·50 × 109/l, with maturation arrest of neutrophil precursors in the bone marrow (Ancliff, 2003). Maintenance therapy with granulocyte colony-stimulating factor (G-CSF) is standard of care to prevent the occurrence of life-threatening bacterial sepsis (Dale et al, 1993). SCN is considered to be a pre-leukaemic disorder, and the cumulative incidence of myelodysplastic syndrome and acute myeloid leukaemia (MDS/AML) is extraordinarily high (Rosenberg et al, 2006).
Severe congenital neutropenia is genetically heterogeneous. A majority of cases are attributed to autosomal dominant mutations (inherited or sporadic) in the gene encoding neutrophil elastase (ELA2) (Dale et al, 2000). Other rare causative genes have recently been found, including mutations in HAX1 (Klein et al, 2007), but the prevalence of mutations in these genes in the general SCN population has yet to be determined (Boxer & Newburger, 2007).
Following the identification of disease-associated genes, an emerging clinical concern is to identify genotype-phenotype associations that might inform therapeutic decisions. The largest published studies addressing this question have presented clinical data for a total of 23 (Ancliff et al, 2003) and 54 (Bellanne-Chantelot et al, 2004) SCN patients stratified according to the presence or absence of any mutation in ELA2. This report describes associations of ELA2 mutation status with responsiveness to G-CSF therapy and risk of MDS/AML in a larger cohort of 82 North American and Australian patients on long-term G-CSF, prospectively followed in the Severe Chronic Neutropenia International Registry (SCNIR) (Dale et al, 2003).
Patients, materials and methods
The Severe Chronic Neutropenia International Registry
We studied patients enrolled in the SCNIR who had been diagnosed with SCN by their treating haematologist and who had blood or fibroblast samples stored in the University of Washington repository serving SCNIR patients from North America or Australia. Each patient specifically had a diagnosis of SCN; those with a diagnosis of cyclic neutropenia, Shwachman-Diamond Syndrome, or other related disorder were excluded. The 82 SCN patients included here (from 104 with any available samples) were all those with sufficient DNA for ELA2 sequencing, irrespective of baseline ANC or clinical response to G-CSF. ELA2 mutation analysis was conducted using standard polymerase chain reaction methods, Big Dye terminator technology, and an ABI analyzer (Applied Biosystems, Foster City, CA, USA). Validated data on G-CSF dose, and results of complete blood cell counts, were extracted from the database created for a prior study (Rosenberg et al, 2006).
Follow-up for occurrence of MDS/AML, bone marrow transplant (BMT), or death from bacterial sepsis or other causes was current up to October 12, 2006 for all patients. This study was conducted in accordance with the Declaration of Helsinki, under the auspices of the Human Subjects Committee of the University of Washington and other participating institutions. Patients provided informed consent for genetic studies. All statistical analyses were performed on anonymized samples.
Statistical methods
We used a generalized linear model for dose–response analysis. For analysis of leukaemia risk, BMT or death from sepsis or other causes were considered to be censoring events. We tested for differences in rates using an exact binomial test, and differences in cumulative incidence using Cox models and likelihood ratio tests. All statistical tests were two-sided. P < 0·05 was significant.
Results
The 82 patients were all treated with G-CSF. Treatment was initiated between 1987 and 2000 at a median age of 24 months. Subsequently, the patients contributed 904 person-years of follow-up, during which time 20 transformed to MDS/AML. Median follow-up was 10 years, ranging from 0·7 to 18·4 years.
Fifty-two patients had ELA2 mutations (63%), and 30 patients had wild-type ELA2 (37%). A total of seven, five, four and three patients carried the Pro110Leu, Ser97Leu, Gly185Arg, Val72Met missense mutations respectively. The 33 remaining patients harboured 27 other mutations.
Patients with mutant ELA2 had 2·8-fold [95% confidence interval (CI): 1·4–5·5-fold] higher mean ANC than patients with wild-type ELA2, after controlling for G-CSF dose. In patients with mutant ELA2, the mean ANC on treatment was close to or above the therapeutic target of 1·5 × 109 cells/l at every dose level (Fig 1). In contrast, patients with wild-type ELA2 reached the therapeutic target only if they responded to comparatively low doses of G-CSF.
Fig 1.
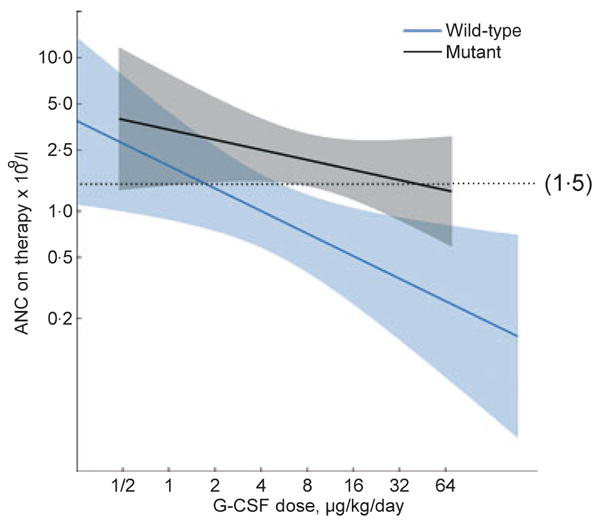
Granulocyte colony-stimulating factor (G-CSF) dose and absolute neutrophil count (ANC) response in patients with SCN, by ELA2 mutation status. Average ANC on therapy during the 6–18 month follow-up period on G-CSF, for patients with wild-type (blue) and mutant (black) ELA2, according to G-CSF dose (μg/kg/d) at 6 months. Dose was analysed on the log2 scale and ANC on the log10 scale. Solid lines show mean values from a generalized linear model that allows for a different linear relationship in patients with mutant versus wild-type ELA2. Shaded areas correspond to 95% point-wise confidence limits. Reference line at 1·5 × 109 cells/l shows the therapeutic target ANC. Median G-CSF dose was 5·2 μg/kg/d in patients with wild-type ELA2 and 9·9 μg/kg/d in patients with mutant ELA2.
Among 52 patients with mutant ELA2, 14 (27%) eventually developed MDS/AML, compared with six (20%) of 30 patients with wild-type ELA2; the crude proportions transforming to MDS/AML were not significantly different (P = 0·67). Controlling for years-on-G-CSF, there was no significant difference in the cumulative incidence of MDS/AML in patients with mutant versus wild-type ELA2 (P = 0·96): the cumulative incidences at 15 years were 36% (mutant) and 25% (wild-type), with broad and overlapping confidence intervals (Fig 2). In all patients combined, the cumulative incidence was 34% (95% CI: 19–46%) at 15 years.
Fig 2.
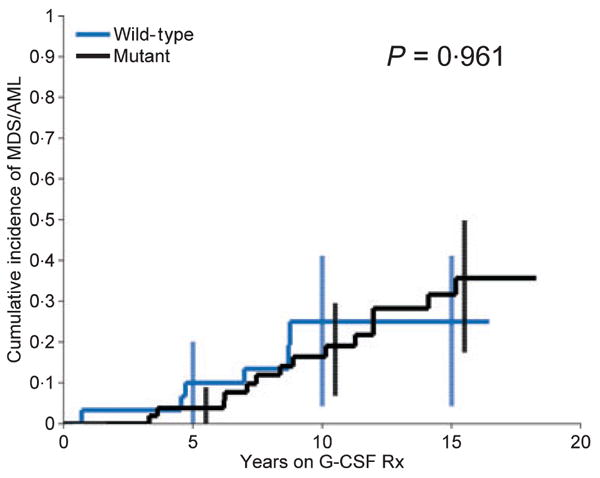
Cumulative incidence of MDS/AML in patients with SCN, by ELA2 mutation status. Cumulative incidence, by years on G-CSF therapy, and 95% confidence intervals at selected years (error bars), in patients with mutant (black) and wild-type (blue) ELA2. The Kaplan–Meier actuarial method was used to estimate the cumulative incidence curves.
We considered the effects of specific ELA2 mutations among the 52 patients with any mutation. Two preliminary findings emerged in exploratory analysis. First, the four patients with Gly185Arg had a particularly severe course. On treatment, ANC values were low despite high doses of G-CSF, and two patients maintained on comparatively high doses of 48 and 21 μg/kg/d G-CSF developed MDS/AML at 10·1 and 15·1 years. Second, no MDS/AML events were observed in seven patients with the Pro110Leu missense mutation or in five patients with the Ser97Leu missense mutation. The crude rate of MDS/AML was significantly lower in these 12 patients than in the 40 with any other ELA2 mutation (0 events in 153 person-years versus 14 events in 468 person-years, respectively, P = 0·04).
Discussion
Our study is the largest to date of ELA2 mutations and clinical outcomes in SCN. It is well known that almost all SCN patients respond to G-CSF, but there is considerable heterogeneity in the dose of G-CSF required to obtain a haematological response. In this study, we observed that patients with mutant ELA2 usually responded with higher ANC values at lower doses of G-CSF than patients with wild-type ELA2. As an exception to this general trend, we observed a particularly severe course in the four patients in our study with the ELA2 mutation Gly185Arg, consistent with the report from the French Neutropenia Registry that also had four SCN patients with this mutation (Bellanne-Chantelot et al, 2004).
Notwithstanding these differences in response to therapy, an important finding was that patients with or without mutations in ELA2 were at extraordinary risk of MDS/AML. Indeed, there was no significant difference in risk between the two groups. In all patients combined, the cumulative incidence of MDS/AML was 34% after 15 years on G-CSF.
We do not believe that selection or referral bias had a major impact on our analysis. Considering patients who enroled during the same calendar periods, we found no differences in the rates of loss-to-follow-up according to ELA2 mutation status (data not shown). Furthermore, the incidence of MDS/AML was similar in the patients included here, compared to other SCN patients in the SCNIR who were not included. Together, these observations suggest that our analyses of MDS/AML are statistically valid.
In contrast to our results, the French Neutropenia Registry reported a significantly reduced leukaemia risk in patients with wild-type ELA2, based on their observation that no patient with wild-type transformed (Bellanne-Chantelot et al, 2004). Our study was larger, with 20 MDS/AML events versus three MDS/AML events, and one acute lymphoblastic leukaemia event in the French study. We attribute the divergent results to statistical heterogeneity, and possibly, genetic heterogeneity between ELA2 wild-type patients in the respective studies. Our results suggest that it is the marrow disorder that leads to leukaemia, not simply the presence of an ELA2 mutation, and leads to the prediction that patients with wild-type ELA2 who develop leukaemia may ultimately be found to harbour mutations in a number of other causative genes.
Although the average incidence of MDS/AML in our cohort was similar in patients with and without ELA2 mutations, exploratory analysis supports the concept that susceptibility to MDS/AML varies by mutation. The subgroup of SCN patients with the Gly185Arg missense mutation may be at increased risk, given our findings and the leukaemias in this group reported by the French Neutropenia Registry (Bellanne-Chantelot et al, 2004). In addition, in our data, SCN patients with the Pro110Leu or Ser97Leu missense mutations were at significantly decreased risk. Interestingly, these specific ELA2 mutations are among the limited number associated with both SCN and cyclic neutropenia (Horwitz et al, 2007).
From the clinical perspective, SCN patients with a wide diversity of ELA2 mutations, and SCN patients without ELA2 mutations, have been observed to transform to leukaemia. Therefore, the average risk is extraordinarily high. Until more refined predictions are available, all SCN patients should be considered at high risk. Our data also suggest that patients with wild-type ELA2 who do not respond to standard doses of G-CSF are comparatively unlikely to respond to high doses. These patients should be considered for early haematopoietic stem cell transplantation, because they may be at high risk of mortality from sepsis. It is now essential to develop new practice guidelines for SCN (Touw & Bontenbal, 2007). These guidelines will need to carefully weigh the potential risks and benefits of G-CSF therapy versus transplantation, and specify the appropriate role and timing of genetic and molecular tests in counselling and decision-making.
Acknowledgments
We thank the patients and doctors for participating in the registry, and the data collection centre in Seattle, Washington for maintaining the North American and Australian registry. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Division of Cancer Epidemiology and Genetics; NIH Grant 1R24AI049392; NIH Grant R01DK54369, and a gift from the Amgen Foundation, Thousand Oaks, CA.
Footnotes
Contribution: P.S.R., D.C.L., B.P.A., and D.C.D designed the study; P.S.R. analyzed the data and wrote the paper; S.S., E.R., and A.A.A. conducted ELA2 sequencing; A.A.B. set the database; M.A.B., Y.D., G.K., L.A.B., P.E.N. and D.C.D. were involved in patient care; all participated in the writing of the paper.
Conflict of interest: D.C.D. and Y.D. receive research support from Amgen, Inc.; D.C.D. is a consultant and speaker for Amgen, Inc.; L.A.B. has family with stock options in Amgen, Inc.; authors are members of the SCNIR Medical Advisory Board (except S.S., E.R. and A.A.A).
References
- Ancliff PJ. Congenital neutropenia. Blood Reviews. 2003;17:209–216. doi: 10.1016/s0268-960x(03)00019-5. [DOI] [PubMed] [Google Scholar]
- Ancliff PJ, Gale RE, Liesner R, Hann I, Linch DC. Long-term follow-up of granulocyte colony-stimulating factor receptor mutations in patients with severe congenital neutropenia: implications for leukaemogenesis and therapy. British Journal of Haematology. 2003;120:685–690. doi: 10.1046/j.1365-2141.2003.04160.x. [DOI] [PubMed] [Google Scholar]
- Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, Vaury C, Barkaoui M, Fenneteau O, Maier-Redelsperger M, Chomienne C, Donadieu J. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103:4119–4125. doi: 10.1182/blood-2003-10-3518. [DOI] [PubMed] [Google Scholar]
- Boxer LA, Newburger PE. A molecular classification of congenital neutropenia syndromes. Pediatric Blood & Cancer. 2007;49:609–614. doi: 10.1002/pbc.21282. [DOI] [PubMed] [Google Scholar]
- Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, Wang W, Jakubowski A, Winton E, Lalezari P. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496–2502. [PMC free article] [PubMed] [Google Scholar]
- Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K, Benson KF, Horwitz M. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–2322. [PubMed] [Google Scholar]
- Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannourakis G, Kinsey SE, Davis R, Scarlata D, Schwinzer B, Zeidler C, Welte K. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. American Journal of Hematology. 2003;72:82–93. doi: 10.1002/ajh.10255. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817–1824. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nature Genetics. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S, Schwinzer B, Zeidler C, Welte K, Dale DC. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touw IP, Bontenbal M. Granulocyte colony-stimulating factor: key (f)actor or innocent bystander in the development of secondary myeloid malignancy? Journal of the National Cancer Institute. 2007;99:183–186. doi: 10.1093/jnci/djk057. [DOI] [PubMed] [Google Scholar]