

Abstract
Cardiovascular disease is a leading cause of mortality among patients with diabetes, and heart failure exists even in the absence of coronary disease. Myocardial metabolism is altered in the diabetic heart as a result of changes in substrate availability secondary to insulin resistance. The nuclear receptor peroxisome proliferator activated receptor-alpha (PPARα) and PPAR-gamma coactivator-1alpha (PGC-1α) play important roles in transcriptional regulation of myocardial metabolism and contribute significantly to the changes that occur in the diabetic heart. This review summarizes the role of PPARα and PGC-1α in myocardial metabolism in the normal heart and in the diabetic heart.
Keywords: Cardiovascular disease, Diabetes, Myocardial metabolism, Proliferator activated, receptor-alpha, PPAR-gamma coactivator-1alpha
Overview of Myocardial Energy Metabolism
To meet the energy demands of diverse physiologic and pathologic conditions, the mammalian heart has a continuous and high requirement for adenosine triphosphate (ATP) production. The myocardium uses a variety of fuels including glucose, lactate, and fatty acids to produce ATP through high-level mitochondrial oxidative metabolism. The ATP produced in the mitochondrion is transported to the cytoplasm for important cellular work, including myocyte contraction.
The healthy adult heart relies predominantly on fatty acids but can rapidly switch substrate preference depending on the developmental or physiologic state (e.g., exercise) [39]. This metabolic flexibility is believed to be important for normal cardiac function.
Several pathologic circumstances are associated with derangements in myocardial fuel utilization. Pathologic cardiac hypertrophy and congestive heart failure caused by pressure overload result in the myocardium switching to glucose as a predominant substrate. This change in metabolic programming often is referred to as a “fetal” shift because the myocardium of the developing embryo relies mostly on glycolysis and lactate metabolism for its ATP production [39]. In contrast, because the heart requires insulin for glucose uptake, the diabetic heart relies almost exclusively on fatty acids as an energy substrate [46, 53, 55].
There is ongoing debate as to whether these derangements in fuel metabolism contribute directly to pathologic cardiac remodeling. The shift in metabolism may be adaptive initially, but the consequences may be maladaptive. Our knowledge concerning inborn errors of metabolism, such as genetic defects in fatty acid oxidation (FAO) enzymes, suggests that conditions in which the heart is forced to use a single substrate (e.g., glucose) frequently result in cardiomyopathy.
Emerging evidence demonstrates that the capacity for mitochondrial oxidative metabolism is mediated at least in part at the level of gene transcription [15]. This gene regulatory control allows for reprogramming of enzyme expression in a dynamic manner to respond to various physiologic or pathologic conditions. Peroxisome proliferator activated receptor alpha (PPARα) and its cardiac-enriched coactivator, PPAR gamma coactivator-1alpha (PGC-1α), play important roles in the transcriptional regulation of myocardial metabolism. This review focuses on PPARα and PGC-1α signaling in both the normal myocardium and the diabetic heart.
PPARα, a Ligand-Activated Transcriptional Regulator of Fatty Acid Metabolism
As one of three PPAR isoforms, PPARα is a member of the nuclear hormone receptor superfamily. The PPARs are ligand activated transcriptional regulators. The endogenous ligands for PPARs have not been established with complete certainty, but it appears that long-chain fatty acids and/or their metabolites are the most likely source of the endogenous ligand [10].
At binding of their ligand, PPARs form heterodimers with 9-cis retinoid X receptors (RXRs) and bind to DNA response elements in target gene promoter regions (Fig. 1). In the adult cardiomyocyte, PPARα is expressed at high levels and plays an important role in transcriptional regulation of fatty acid metabolism [3, 24]. Additionally, PPARα has served as an important pharmacologic target due to its large hydrophobic ligand-binding site, which can be activated by a variety of compounds, including lipid-lowering fibrates [14, 27].
Fig. 1.
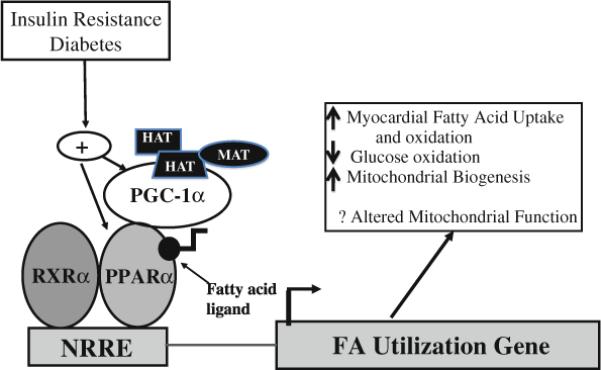
Both peroxisome proliferator activated receptor-alpha (PPARα) and PPAR-gamma coactivator-1alpha (PGC-1α) are activated by insulin resistance and diabetes. Forming a heterodimer with RXRα, PPARα binds to a DNA response element, namely, the nuclear receptor response element (NRRE). At binding of the NRRE within the promoter of PPAR target genes, gene transcription is activated. The transcriptional activity of PPARα is influenced by binding of endogenous ligands and by the coactivator PGC-1α. Recruiting additional coactivators with histone acetyltransferase (HAT) activity, PGC-1α interacts with other regulators such as ménage-a-trois 1 (MAT) to promote chromatin remodeling and to facilitate gene transcription
In the heart, PPARα activation induces expression of genes encoding nearly every step in the fatty acid utilization pathway including transport proteins to facilitate fatty acid uptake, acyl-coA synthetases for esterification of fatty acids to coenzyme A, fatty acid-binding proteins that facilitate delivery of fatty acids to different cellular compartments, mitochondrial carnitine system proteins that catalyze fatty acid transfer into the mitochondrion, every enzyme in the fatty acid β-oxidation pathway, and other accessory components of fatty acid metabolism such as uncoupling proteins.
Whereas PPARα ligand administration to rodents has clear effects on the induction of PPARα target gene expression in the liver, its effect in the myocardium appears to be minimal [13]. As a result of the hepaticspecific effect of PPARα ligand administration, most of the knowledge regarding PPARα's effect on cardiac metabolism has been generated by gain-of-function and loss-of-function models in mice. Mice with generalized deletion of PPARα (PPARα-null mice) demonstrate decreased FAO rates [9, 16, 32, 58] and concomintant increased glucose oxidation rates [9, 40]. The PPARα-null mice also have mild cardiac fibrosis with aging and an inability to compensate for an increased cardiac workload [9, 40, 58].
In contrast, mice with transgenic overexpression in the heart exclusively (myosin heavy chain (MHC)-PPARα mice) demonstrate upregulation of genes involved in fatty acid uptake and oxidation, with concomitant decreased expression of genes involved in glucose uptake and utilization [21, 41]. The MHC-PPARα mice exhibit an accumulation of myocardial triglyceride that is exacerbated by fasting or a high-fat diet [21, 22]. Relying heavily on FAO for ATP production, the MHC-PPARα animals experience left ventricular hypertrophy and cardiac dysfunction that is worsened with a high fat diet [21, 22]. Taken together, the opposing phenotypes demonstrated by deletion and over-expression of PPARα suggest an important role for PPARα in regulating cardiac energy metabolism.
PGC-1, a Master Metabolic Regulator
In addition to ligand-mediated activation, PPARs also are activated by transcriptional coactivators and corepressors. Transcriptional coactivators interact indirectly with the PPARs and other nuclear receptors to establish a platform for recruitment of other proteins important to chromatin remodeling and recruitment of the RNA polymerase II complex.
One of the most well-studied PPARα coactivators is PGC-1 [20, 25]. The PGC-1 coactivators (PGC-1α, PGC-1β, and PGC-related coactivator [PRC]) have been well described as important regulators of mitochondrial metabolism (see multiple recent reviews) [20, 29]. These coactivators have an impact on cellular biologic responses, enabling the cell to meet changing energy demands associated with various physiologic stimuli. These responses include augmenting mitochondrial biogenesis, respiratory rates, and uptake and metabolism of substrate.
Highly versatile, the PGC-1 coactivators have the ability to interact with multiple different transcription factors, many of which have an important role in cellular metabolism. In addition to serving an important role in transcriptional regulation, the expression of the PGC-1 coactivators is highly regulated by various hormones and signal transduction pathways [52].
The first member of the PGC-1 family to be identified was PGC-1α. It was discovered as a result of its interactions with the nuclear receptor, PPAR-gamma, in brown adipose tissue, a mitochondria-rich tissue specialized for thermogenesis. Both PGC-1β and PRC were subsequently identified but have been much less intensively studied. Expression of PGC-1α, induced in the heart shortly after birth, also is increased in association with physiologic and pathologic stimuli such as exercise and starvation [20, 25]. Findings have shown that PGC-1α plays a role in recruiting other coactivator proteins with histone acetyltransferase activity such as SRC1 and CBP/p300 to assist in chromatin remodeling (Fig. 1) [43, 57]. Recently, it has been demonstrated that PGC-1α also docks with a protein called ménage-a-trois 1 (MAT1), which phosphorylates RNA polymerase II to modulate its activity [49]. In addition, PGC-1α has its own RNA processing domain that also may contribute to its transcriptional regulatory activity [37].
In the heart, PGC-1α is known to interact with several different transcription factors including estrogen-related receptors, nuclear respiratory factors, and the PPAR family. The interaction between PGC-1α and PPARα in the heart plays an important role in regulating expression of enzymes involved in FAO and uptake [56] and may also be involved in regulating mitochondrial biogenesis [17]. Much of our understanding regarding the role of PGC-1α in the heart comes from genetically engineered mouse models.
Mice with constitutive overexpression of PGC-1α in the heart have robust proliferation of mitochondria, experience cardiomyopathy, and succumb to early mortality [31]. To study the pathophysiology of PGC-1α overexpression in the heart further, a tetracycline-inducible PGC-1α overexpression model was generated [48]. In this model, neonatal overexpression of PGC-1α resulted in significant mitochondrial proliferation. In contrast, overexpression in adult mice resulted in a modest mitochondrial proliferation, but the mitochondrial architecture was abnormal, and the mice had severe cardiac dysfunction. The cardiomyopathy in this model was reversible by discontinuing PGC-1α overexpression [48].
Two separate lines of PGC-1α-deficient mice have been described, both of which support an important role for PGC-1α in cardiac metabolism and mitochondrial function [2, 33]. Both murine models demonstrate impaired mitochondrial respiratory function and decreased expression of genes involved in multiple mitochondrial metabolic pathways. In addition, PGC-1α deficiency leads to cardiac dysfunction in the setting of pathologic stimuli such as pressure overload [1, 2, 33]. Taken together, the gain- and loss-of-function models demonstrate a clear role for PGC-1α in regulating mitochondrial number and metabolism.
Altered Cardiac Metabolism in Diabetes
The past decade has witnessed an emerging epidemic of obesity fueling an overall rise of almost 50% in diagnosed cases of type 2 diabetes in adults [35]. Alarmingly, the incidence of type 2 diabetes also has increased in the pediatric population in association with the epidemic of childhood obesity [36, 42]. Cardiovascular disease is the leading cause of death in the diabetic population. Although coronary artery disease and hypertension contribute to the incidence of cardiovascular disease, cardiomyopathy is common in diabetics independent of these risk factors [19, 28, 44, 45, 47]. Furthermore, more recent echocardiographic studies with obese adolescents have demonstrated that cardiovascular abnormalities may begin at an early age [34].
The pathogenesis of diabetic cardiomyopathy remains unclear. However, emerging data suggest that cardiac dysfunction is linked to alterations in myocardial lipid and energy metabolism [46, 53]. In models of both type 1 and type 2 diabetes, the energy substrate flexibility becomes constrained, and the diabetic heart begins to rely almost exclusively on FAO for ATP production [4, 23, 53].
In human patients, findings have shown that fatty acid metabolism is increased in type 1 diabetics [26]. It also has been noted that diabetics have triglyceride accumulation in the myocardium [38, 50, 54]. Interestingly, several investigators have documented increased PPARα and PGC-1α expression in murine insulin-resistant and diabetic hearts [5, 8, 17]. The role of these transcriptional regulators in altering the cardiac metabolism in the setting of diabetes is an area of active investigation.
PPARα and PGC-1α in the Diabetic Heart
Various murine models have helped to increase our understanding of the role played by PPARα and PGC-1α in the diabetic heart. Studies have shown that PPARα-deficient animals with insulin resistance experience blunted activation of FAO gene expression, suggesting that PPARα is necessary for this metabolic change [5, 17]. Furthermore, mice that overexpress PPARα exclusively in the heart (MHC-PPARα mice) have a cardiac phenotype similar to that of the diabetic heart, with increased fatty acid uptake and oxidation, decreased glucose uptake, and myocardial triglyceride accumulation [21].
In addition, the MHC-PPARα mice experience cardiac hypertrophy and contractile dysfunction that we believe is related to the excess fatty acid utilization and triglyceride accumulation [21, 22]. This accumulation of triglycerides is likely toxic to the myocardium and has been linked with insulin resistance and cardiac dysfunction [11, 12, 22, 59, 61]. Evidence suggests that PPARα may drive this lipotoxic response. The MHC-PPARα mice experience cardiomyopathy only after being challenged with a high-fat diet. The contractile dysfunction is associated with marked lipid accumulation but is reversible with removal of the high-fat diet [22]. In addition, deficiency of CD36 or lipoprotein lipase (LpL) (two important regulators of fatty acid uptake) in the context of PPARα overexpression rescues the lipid accumulation and cardiomyopathy in high-fat-fed MHC-PPARα mice [18, 60]. These data thus suggest that PPARα plays an important role in lipotoxicity and cardiomyopathic remodeling in the diabetic heart.
In addition to the lipotoxic effects associated with changes in fatty acid uptake, the diabetic heart may suffer from mitochondrial derangements, including changes in mitochondrial ultrastructure and function [6–8, 17, 51]. Alteration of mitochondrial gene expression has been noted, although reports are conflicting, with some evidence of increased expression and other evidence of decreased expression [17, 51].
Our data suggest that PGC-1α and mitochondrial oxidative phosphorylation gene expression may change with the progression from insulin resistance to full-blown diabetes (unpublished data). We previously demonstrated a mitochondrial biogenesis response and an increase in gene expression for enzymes involved in mitochondrial oxidative phosphorylation in the myocardium of insulin-resistant mice [17]. This effect was blunted in the setting of PPARα deficiency and recapitulated in MHC-PPARα animals, suggesting that PPARα is involved in the mitochondrial changes in the insulin-resistant heart [17].
Furthermore, this mitochondrial biogenesis phenomenon was associated with upregulation of PGC-1α gene expression in wild-type animals and a lack of change in PGC-1α in the setting of PPARα deficiency [17]. Thus, our data suggest that both PPARα and PGC-1α may regulate the mitochondrial biogenesis response. Indeed, we have demonstrated that PPARα is capable of activating the PGC-1α promoter in skeletal muscle cells (unpublished data), suggesting the presence of an autoregulatory loop between the transcriptional regulator and its coactivator.
More recently, we have noted that the mitochondrial changes found in MHC-PPARα hearts are associated with downregulation of PGC-1α and that mitochondrial architecture and PGC-1α expression levels are rescued in the setting of lipoprotein lipase deficiency [18]. Importantly, the rescue of PGC-1α and mitochondrial morphology was associated with improvement in cardiac function as detected by echocardiography. These data, suggest that lipid accumulation in the myocardium in the setting of PPARα overexpression may have detrimental effects on PGC-1α expression and mitochondrial function and that this may in turn contribute to contractile dysfunction. The specific role of PGC-1α in the insulin-resistant heart is an ongoing area of investigation. In addition, the role of the other PGC-1 isoforms, such as PGC-1β, which has overlapping roles with PGC-1α [30], remains to be investigated.
Conclusion
Diabetes is being diagnosed at alarming rates and at younger ages. Cardiovascular disease, the leading cause of mortality, is in large part related to metabolic abnormalities in the diabetic cardiomyocyte. The diabetic heart has limited substrate availability and thus depends on fatty acids for its major source of fuel. The shift in metabolic fuel source is driven, at least in part, by PPARα and its coactivator PGC-1α. Although the metabolic changes driven by PPARα and PGC-1α may be adaptive, they likely have maladaptive consequences, contributing to diabetic cardiomyopathy.
Acknowledgments
Jennifer G. Duncan, a Faculty Scholar of the Children's Discovery Institute at Washington University School of Medicine, is supported by an NHLBI K08 award (HL084093).
References
- 1.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPARγ coactivator 1α. Proc Natl Acad Sci USA. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-α during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–1730. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 5.Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, Coleman T, Mecham RP, Kelly DP, Semenkovich CF. Dexamethasone induction of hypertension and diabetes is PPAR-α dependent in LDL receptor-null mice. Nat Med. 2003;9:1069–1075. doi: 10.1038/nm898. [DOI] [PubMed] [Google Scholar]
- 6.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology. 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 7.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Jeong Yun U, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 9.Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, Severson DL, Kelly DP, Lopaschuk GD. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–4103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 10.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of FATP1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 13.Cook WS, Yeldandi AV, Rao MS, Hashimoto T, Reddy JK. Less extrahepatic induction of fatty acid beta-oxidation enzymes by PPAR alpha. Biochem Biophys Res Commun. 2000;278:250–257. doi: 10.1006/bbrc.2000.3739. [DOI] [PubMed] [Google Scholar]
- 14.Dashti N, Ontko JA. Alterations in rat serum lipids and apolipoproteins following clofibrate treatment. Atherosclerosis. 1983;49:255–266. doi: 10.1016/0021-9150(83)90137-5. [DOI] [PubMed] [Google Scholar]
- 15.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 16.Djouadi F, Bastin J, Kelly DP, Merlet-Benichou C. Transcriptional regulation by glucocorticoids of mitochondrial oxidative enzyme genes in the developing rat kidney. Biochem J. 1996;315:555–562. doi: 10.1042/bj3150555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-α/PGC-1α gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ, Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426–435. doi: 10.1161/CIRCULATIONAHA.109.888735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fein FS, Sonnenblick EH. Diabetic cardiomyopathy. Prog Cardiovasc Dis. 1985;4:255–270. doi: 10.1016/0033-0620(85)90009-x. [DOI] [PubMed] [Google Scholar]
- 20.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARαa overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamble J, Lopaschuk GD. Glycolysis and glucose oxidation during reperfusion of ischemic hearts from diabetic rats. Biochim Biophys Acta. 1994;1225:191–199. doi: 10.1016/0925-4439(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 24.Gilde AJ, van der Lee KAJM, Willemsen PHM, Chinetti G, van der Leij FR, van der Vusse GJ, Staels B, van Bilsen M. PPARα and PPARβ/δ, but not PPARγ, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- 25.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 26.Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, Gropler RJ. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47:598–604. doi: 10.1016/j.jacc.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 27.Ide T, Oku H, Sugano M. Reciprocal responses to clofibrate in ketogenesis and triglyceride and cholesterol secretion in isolated rat liver. Metabolism. 1982;31:1065–1072. doi: 10.1016/0026-0495(82)90153-6. [DOI] [PubMed] [Google Scholar]
- 28.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham Study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 29.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 30.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-1beta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros D, Kelly DP. PPARγ coactivator-1 (PGC-1) promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1α-deficient mice exhibit multisystem energy metabolic derangements: muscle dysfunction, abnormal weight control, and hepatic steatosis. PLoS Biol. 2005;3:672–687. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorch SM, Sharkey A. Myocardial velocity, strain, and strain rate abnormalities in healthy obese children. J Cardiometab Syndr. 2007;2:30–34. doi: 10.1111/j.1559-4564.2007.06001.x. [DOI] [PubMed] [Google Scholar]
- 35.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 36.Molnar D. The prevalence of the metabolic syndrome and type 2 diabetes mellitus in children and adolescents. Int J Obes Relat Metab Disord. 2004;28:70–74. doi: 10.1038/sj.ijo.0802811. [DOI] [PubMed] [Google Scholar]
- 37.Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell. 2000;6:307–316. doi: 10.1016/s1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 38.Murthy VK, Shipp JC. Accumulation of myocardial triacylglycerols in ketotic diabetes. Diabetes. 1977;26:222–229. doi: 10.2337/diab.26.3.222. [DOI] [PubMed] [Google Scholar]
- 39.Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 40.Panagia M, Gibbons GF, Radda GK, Clarke K. PPAR-α activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H2677–H2683. doi: 10.1152/ajpheart.00200.2004. [DOI] [PubMed] [Google Scholar]
- 41.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, Lee MK, Danton C, Deshmukh S, Cline GW, Wu JJ, Bennett AM, Rothermel B, Kalinowski A, Russell KS, Kim YB, Kelly DP, Kim JK. Cardiac-specific overexpression of peroxi-some proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes. 2005;54:2514–2524. doi: 10.2337/diabetes.54.9.2514. [DOI] [PubMed] [Google Scholar]
- 42.Pinhas-Hamiel O, Zeitler P. The global spread of type 2 diabetes mellitus in children and adolescents. J Pediatr. 2005;146:693–700. doi: 10.1016/j.jpeds.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 43.Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B, Spiegelman BM. Activation of PPARγ coactivator-1 through transcription factor docking. Science. 1999;286:1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- 44.Regan TJ. Congestive heart failure in the diabetic. Ann Rev Med. 1983;34:161–168. doi: 10.1146/annurev.me.34.020183.001113. [DOI] [PubMed] [Google Scholar]
- 45.Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmed MR, Haider B. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest. 1977;60:885–899. doi: 10.1172/JCI108843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodrigues B, Cam MC, McNeill JH. Myocardial substrate metabolism: implications for diabetic cardiomyopathy. J Mol Cell Cardiol. 1995;27:169–179. doi: 10.1016/s0022-2828(08)80016-8. [DOI] [PubMed] [Google Scholar]
- 47.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 48.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 49.Sano M, Izumi Y, Helenius K, Asakura M, Rossi DJ, Xie M, Taffet G, Hu L, Pautler RG, Wilson CR, Boudina S, Abel ED, Taegtmeyer H, Scaglia F, Graham BH, Kralli A, Shimizu N, Tanaka H, MÑkelÑ TP, Schneider MD. MÇnage-Ö-Trois 1 is critical for the transcriptional function of PPARγ coactivator 1. Cell Metab. 2007;5:129–142. doi: 10.1016/j.cmet.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 51.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM, Jr, Klein JB, Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type I diabetes. Am J Physiol Endocrinol Metab. 2004;287:E896–E905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 52.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 53.Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res. 1997;34:25–33. doi: 10.1016/s0008-6363(97)00047-3. [DOI] [PubMed] [Google Scholar]
- 54.Szczepaniak LS, Victor RG, Orci L, Unger RH. Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res. 2007;101:759–767. doi: 10.1161/CIRCRESAHA.107.160457. [DOI] [PubMed] [Google Scholar]
- 55.van Bilsen M, Smeets PJH, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burnout syndrome? Cardiovasc Res. 2004;61:218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 56.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallberg AE, Yamamura S, Malik S, Spiegelman BM, Roeder RG. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1α. Mol Cell. 2003;12:1137–1149. doi: 10.1016/s1097-2765(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor α associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293–22299. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 59.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 61.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human disease. Proc Natl Acad Sci USA. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]