

Abstract
Despite extensive research to develop an effective neuroprotective strategy for the treatment of ischemic stroke, therapeutic options remain limited. Although caspase-dependent death is thought to play a prominent role in neuronal injury, direct evidence of active initiator caspases in stroke and the functional relevance of this activity have not previously been shown. Using an unbiased caspase-trapping technique in vivo, we isolated active caspase-9 from ischemic rat brain within 1 h of reperfusion. Pathogenic relevance of active caspase-9 was shown by intranasal delivery of a novel cell membrane-penetrating highly specific inhibitor for active caspase-9 at 4 h postreperfusion (hpr). Caspase-9 inhibition provided neurofunctional protection and established caspase-6 as its downstream target. The temporal and spatial pattern of expression demonstrates that neuronal caspase-9 activity induces caspase-6 activation, mediating axonal loss by 12 hpr followed by neuronal death within 24 hpr. Collectively, these results support selective inhibition of these specific caspases as an effective therapeutic strategy for stroke.
Introduction
Stroke is the third-leading cause of death and the leading cause of disability in the industrialized world. Despite decades of research, the pivotal inciting molecular events of ischemia remain elusive, and successful translational strategies are lacking. In ischemic stroke (85% of strokes) thrombosis or embolism occludes a major cerebral artery. The injured territory is comprised of the ischemic core and surrounding penumbra. In the ischemic core perfusion is decreased below the threshold for viability, and cells are electrically silent and irreversibly injured. Injury to the core occurs primarily via necrosis; however, there is evidence that apoptosis also occurs (Yuan, 2009). Cell death in the penumbra is caused mainly by apoptosis (Ribe et al., 2008). With timely reperfusion, either spontaneous or therapeutic, this territory may be salvaged. Restoration of blood flow can also induce “reperfusion injury,” which exacerbates inflammation, excitotoxicity, and apoptotic cell injury (Ribe et al., 2008). In humans, apoptotic markers, including cleaved caspases, can be observed in the peri-infarct region from 1 to 26 d after a stroke (Mitsios et al., 2007; Broughton et al., 2009). Although these data implicate caspases, they do not pinpoint the caspase pathways that lead to dysfunction and neuronal death. To allow unequivocal identification of active initiator caspase postischemia we adapted an unbiased caspase trapping assay for use in vivo. Both caspase-8 and caspase-9 were active by 1 h postreperfusion (hpr), whereas caspase-1 and caspase-2 were not.
To determine whether caspase-9 activation was critical for neurodegeneration, we inhibited caspase-9 activity using the X-linked inhibitor of apoptosis (XIAP)-baculoviral IAP repeat 3 (BIR3) domain (XBIR3). We linked XBIR3, a highly specific inhibitor of caspase-9 (Eckelman et al., 2006), to Penetratin1 (Pen1), a cell transduction peptide. We demonstrate that intranasal delivery of Pen1-XBIR3 before occlusion or at 4 hpr provides postischemic functional neuroprotection.
Axon degeneration occurs during stroke with diffusion tensor imaging revealing extensive loss of axonal tracts in the stroke penumbra (Lie et al., 2004; Thomalla et al., 2004). We have identified a new role for caspase-6 in axonal degeneration in stroke. Caspase-6 has been selectively implicated in axonal degeneration during neuronal apoptosis. In AD, neuropil contains caspase-6-cleaved tau and tubulin, implicating caspase-6 in axonal degeneration in AD (Guo et al., 2004; Klaiman et al., 2008). Caspase-6 mediates neuronal death and axonal degeneration induced by activation of the p75 neurotrophin receptor (Troy et al., 2002; Park et al., 2010) or death receptor 6 (Nikolaev et al., 2009). These studies implicate caspase-6 in selective axonal degeneration during neuronal apoptosis.
The present study demonstrates that active caspase-9 leads to activation of caspase-6, axonal degeneration, and subsequent neuronal death. Active caspase-6 is temporally generated in axons and cell bodies after ischemia in rats and mice. Genetic knockout of caspase-6 in mice provides functional and histologic neuroprotection after middle cerebral artery occlusion (MCAo). The time course of caspase-6 activation corresponds with axonal degeneration observed in human stroke as well as other rodent models. Most importantly, the delayed time course of axonal caspase-6 activation, occurring between 12 and 24 hpr, makes it an attractive molecular target for neuroprotection.
Materials and Methods
Antibodies.
For immunohistochemistry, anti-Tuj1 (Abcam ab7751), anti-neurofilament-L (Cell Signaling Technology 2835), anti-GFAP (Thermo Scientific PA1–10004), anti-full-length and -cleaved caspase-9 (Abcam ab28131; also used for Western blotting), anti-cleaved caspase-6 (Cell Signaling Technology 9761), anti-cleaved caspase-3 (Cell Signaling Technology 9661), and anti-cleaved caspase-7 (MBL BV-3147-3) were used. For Western blotting, THE anti-His (GenScript A00186), anti-caspase-6 (BD 556581), anti-ERK1, anti-Tau V-20 (sc-1996 and sc-93, respectively; Santa Cruz Biotechnology), and anti-Lamin A/C (MBL International JM-3267-100) were used.
Transient middle cerebral artery occlusion: rodent stroke models and neurofunctional assays.
All rodent procedures were approved by the Columbia University Institutional Animal Care and Use Committee. Caspase-6 null (C6−/−) mice (Jackson Laboratories) (Zheng et al., 2000; Watanabe et al., 2008) on a C57BL/6 background were bred with wild-type C57BL/6 mice to generate C6+/− heterozygotes. Heterozygotes were then bred to generate C6−/− and wild-type littermates for studies. Male C6−/− and wild-type littermate mice (23–30 g; aged 2–3 months) as well as adult Wistar male rats (250–300 g; aged 12 weeks; Taconic Laboratories) were subjected to transient MCAo occlusion (tMCAo) as published previously (Connolly et al., 1996; Komotar et al., 2007). Rats received 2 h of occlusion, whereas mice received 1 h. Postprocedure rodents were placed in a 37°C postoperation incubator and maintained at normothermia for 1 h. Brains were harvested and processed for Western blotting or immunohistochemistry as described below. For rat neurofunctional analysis, a 24 point neurological functional exam was developed by adaptation of previous studies (Wahl et al., 1992; Garcia et al., 1995; Petullo et al., 1999, Reglodi et al., 2003). Groups were analyzed by ANOVA. For mouse neurofunctional analysis, a 28 point neurological functional exam was performed as described previously (Clark et al., 1997), with group comparisons made with Student's t test.
Convection-enhanced delivery.
Adult male Wistar rats (250–300 g) were anesthetized using isoflurane (2%) delivered via an anesthesia mask for stereotactic instruments (Stoelting) and positioned in a stereotactic frame. Convection-enhanced delivery (CED) was performed as described previously (Bruce et al., 2000) with stereotactic coordinates of 1 mm anterior, 3 mm lateral, and 5 mm depth. Infusion of the therapeutic was then instituted at a rate of 0.5 μl/min. After infusion, the cannula was removed at a rate of 1 mm/min, the burrhole was sealed with bonewax, and the skin incision was closed with skin adhesive. Postprocedure rats were placed in a 37°C postoperation incubator and maintained at normothermia for 1 h.
Pen1-XBIR3.
XBIR3 was purified as described previously (Sun et al., 2000). Pen1 (Q-Biogene) was mixed at an equimolar ratio with purified XBIR3 and incubated overnight at 37°C to generate disulfide-linked Pen1-XBIR3. Linkage was assessed by 20% SDS-PAGE and Western blotting with anti-His antibody. For CED, 30 μl of Pen1-XBIR3 (36.8 μm) was infused immediately before induction of ischemia. Animals were housed at room temperature (RT) and then killed, and their brains were processed for immunohistochemistry (see below) or protein isolation (brain tissue dissection followed by snap-freezing in liquid nitrogen). An equivalent volume of saline was infused as a negative control.
In vivo caspase activity assay.
Biotin-Val-Ala-Asp(OMe)-fluoromethylketone (bVAD; MP Biomedicals) was used as an in vivo molecular trap for active caspases. bVAD (200 nmol) was diluted in 30 μl of sterile saline and infused by CED before stroke or 3 hpr in rats. Brain tissue was harvested after treatment with bVAD and tMCAo and flash-frozen in liquid nitrogen. Tissue was lysed by pestle disruption in cold CHAPS buffer containing protease inhibitors (Roche). For bVAD-caspase complex precipitation, protein lysates were precleared by rocking with Sepharose beads (GE Healthcare) for 1 h at 4°C. Precleared lysate was centrifuged, and the supernatant was transferred to 30 μl of streptavidin-agarose beads (Sigma) and rocked gently overnight at 4°C. Beads were washed/centrifuged (300 μl washes, 5000 rpm for 5 min) 15 times with CHAPS buffer. After the final wash/pelleting, caspase-bVAD complexes were boiled off of streptavidin beads into 1× SDS sample buffer without reducing agent. Beads were pelleted at 14,000 rpm for 10 min, and the supernatant was transferred to a fresh tube and resolved by SDS-PAGE. Saline was used in vehicle animals to act as a precipitation control for bVAD.
Intranasal delivery of Pen1-XBIR3.
While the rats were under isoflurane anesthesia and lying on their backs, Pen1-XBIR3 (36.8 μm) was delivered by administering 6 μl drops to alternating nares every 2 min for 20 min (60 μl total delivered) (Thorne et al., 2004). Intranasal treatment was done either before stroke or 4 hpr. Saline was used as a negative control. For nonstroked controls, animals were given 60 μl of vehicle or Pen1-XBIR3 and anesthetized for the same period of time as stroked animals (∼1.25 h). Brains were harvested for immunohistochemistry or Western blotting.
Immunohistochemistry, cell process quantification, and statistical analysis.
Rats and mice were killed with 0.3 ml of ketamine (10 mg/ml) and xylazine (0.5 mg/ml) and perfused followed by fixation with 4% paraformaldehyde. Sections were blocked for 1 h with 10% normal goat serum/1% BSA with 0.1% Triton X-100, incubated with primary antibody overnight at 4°C, washed with PBS-Triton X-100 (0.1%), and incubated with the species-appropriate Alexa Fluor-conjugated secondary antibody (Invitrogen) for 2 h at RT. Slides were also stained with Hoechst 33342 for 15 min at RT (1 μg/ml; Invitrogen) to visualize nuclei or NeuroTrace fluorescent Nissl stain (1:300; Invitrogen) for 30 min to visualize neuronal nuclei. Human samples were additionally treated with Sudan Black (1% in 70% EtOH) for 5 min at RT and washed with three changes of PBS (3 min each). For detection of fluorescent staining, sections were imaged with an upright Nikon fluorescent microscope using a SPOT digital camera (Morell Instruments) and a PerkinElmer Spinning Disc Confocal Imaging System.
Quantification of neurons and axons was accomplished using the Cell Counter plug-in for ImageJ (National Institutes of Health). For quantification in the rat brain, 20× magnification images were acquired from the dorsal motor cortex and the S1 somatosensory cortex forelimb region; both regions are contained within the infarct penumbra. Single blind counts of processes or neurons were made in both regions of interest and then pooled for each individual animal. Three animals were used per cohort. For mouse brains, 20× magnification images were taken in the S1 somatosensory cortex forelimb region, and similar counts were made as described below. Counts were made for neurofilament-light chain (NF-L)-positive processes and NeuN-positive cell bodies. Comparisons between groups used the Student's t test or ANOVA.
Human samples were also analyzed with DAB staining. Samples were incubated with 0.3% H2O2 for 30 min, followed by blocking with 10% normal goat serum/1% BSA in PBS, and primary antibody incubation diluted in blocking buffer overnight at 4°C. After washing with PBS, slides were incubated with a species-appropriate biotin-conjugated secondary antibody (Vector Laboratories) for 30 min at RT. Samples were then incubated with ABC reagent (Vector Laboratories) for 30 min and DAB stain for 10 min. Samples were counterstained with hematoxylin and subsequently dehydrated with ethanol and cleared with two washes of xylene.
Calculation of infarct volume.
Twenty four hours after MCA occlusion, rats and mice were killed with 0.3 ml of ketamine (10 mg/ml) and xylazine (0.5 mg/ml). Sections were prepared for immunohistochemistry (see above) and stained using an H&E kit from American MasterTech (KTHNEPT). Infarcted brain was visualized as an area of hematoxylin-negative (pink) tissue in a surrounding background of viable (blue and pink) tissue. Serial sections were photographed and projected on tracing paper at a uniform magnification. Direct and indirect infarct volumes were calculated from serial sections. Direct stroke volume was expressed as the area of the infarct divided by the area of the ipsilateral hemisphere. Indirect stroke volume was expressed as the area of the infarct divided by the area of the contralateral hemisphere.
Rat hippocampal cultures.
Hippocampal neurons from E18 rat embryos were dissected, dispersed in a defined serum-free media, and plated on poly-d-lysine-coated (0.1 mg/ml) tissue culture wells. The neurons were maintained in a serum-free environment with Eagle's MEM and Ham's F12 (Invitrogen) containing glucose (6 mg/ml), insulin (25 μg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5 U/ml), and streptomycin (0.5 μg/ml). Glial cells made up <2% of the culture. All cells were cultured for 8–10 d before treatment.
Neuronal survival assay.
4-Hydroxynonenal (HNE; 3 μm; Cayman Chemicals) was added to cultures in triplicate with and without Pen1-XBIR3 (80 nm) as described previously (Rabacchi et al., 2004). After 1 d of treatment cell number was quantified as described previously (Rabacchi et al., 2004). Briefly, the cells were lysed in counting buffer, and intact nuclei were counted using a hemocytometer. Nuclei of the healthy cells appeared bright and had a clearly defined nuclear membrane, whereas nuclei of dead cells disintegrated or appeared irregularly shaped. Cell counts were performed in triplicate wells and averaged. Survival was relative to control wells.
Results
Caspase-9, an initiator caspase, is active early in stroke
The activity of individual caspases has not been specifically assayed in the setting of stroke. It is not known when caspase activity is initiated during a stroke, and defining the time course of caspase activation is important from a mechanistic and therapeutic perspective. The participation of individual caspases is also not understood, but it is generally accepted that initiator caspases are active first, leading to effector caspase activation, and ultimately to cell death. With 11 individual caspases present in the human or rodent brain, specific measures are required to understand the caspase pathways initiated by stroke. At present, the caspase activity probe bVAD is the best way to determine whether caspases are active after a death stimulus. bVAD is an irreversible pan-caspase inhibitor that has been used in vitro to identify caspase activation after various death stimuli (Denault and Salvesen, 2003; Tu et al., 2006; Tizon et al., 2010). This method was recently adapted for use in cultured primary neurons (Tizon et al., 2010). We now apply it for use in vivo in the CNS. bVAD binds irreversibly to all caspases that are active. In other words, if a caspase is active and its active site is available, bVAD will bind to it. Once bVAD is bound it also inhibits that caspase, blocking any downstream events. Because bVAD is biotinylated, it can be isolated on streptavidin agarose along with any active caspase that is bound to it. Administration of bVAD before induction of death will trap active initiator caspases; activation of effector caspases will be inhibited.
To determine which initiator caspases were activated early in stroke, rats were injected with 200 nmol of bVAD via CED to the rat striatum immediately before MCAO followed by reperfusion with death at 1 hpr. The injected region was dissected, and bVAD-caspase complexes were isolated on streptavidin-agarose beads and analyzed by Western blotting. bVAD captured caspase-9 (Fig. 1A) and caspase-8 (data not shown), showing activation of these initiator caspases is an early event in stroke. Caspase-1 and caspase-2 were not trapped by bVAD (data not shown).
Figure 1.
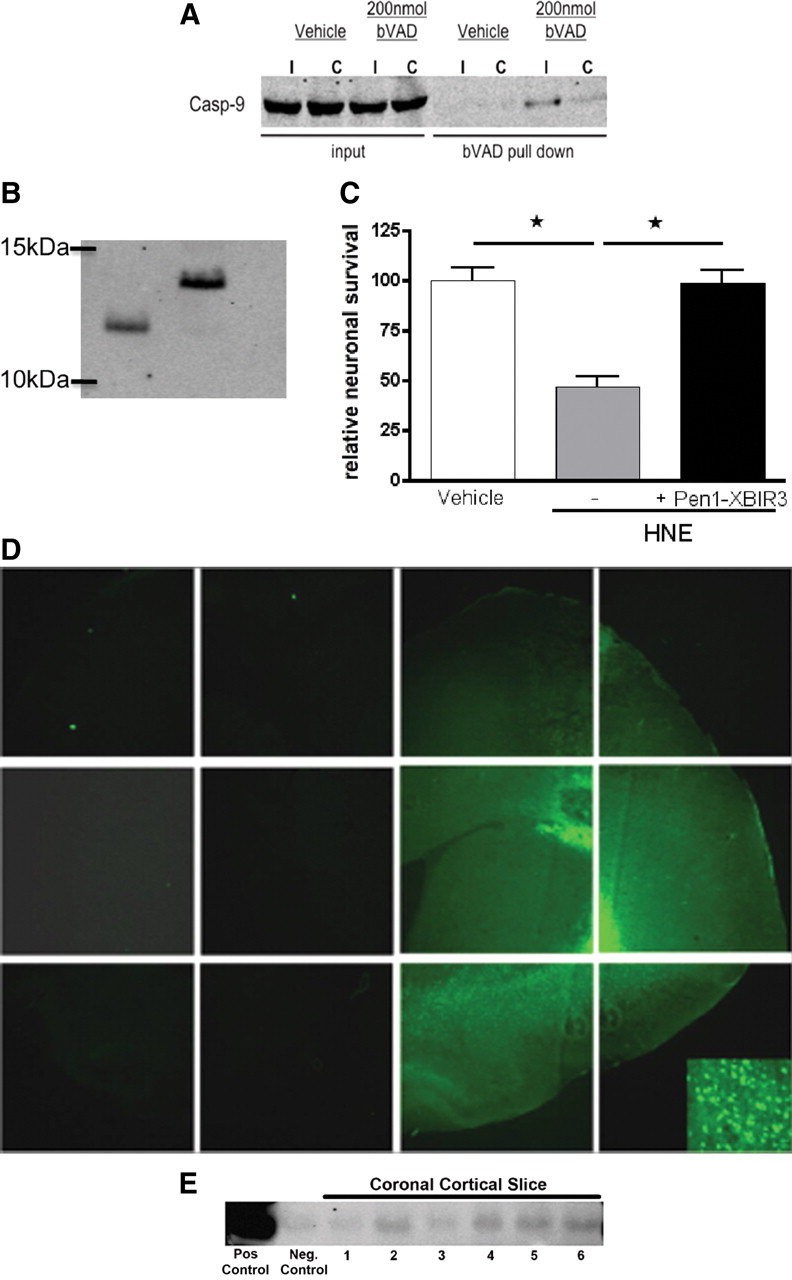
Caspase-9 is active early in stroke and causes neurodegeneration. A, Active caspase-9 is induced by tMCAo. bVAD was infused using CED into the predicted stroke area of rats before tMCAo. Animals were harvested at 1 hpr, and bVAD-caspase complexes were isolated and analyzed by Western blotting. I, ipsilateral (stroked) hemisphere; C, contralateral (nonstroked) hemisphere. B, Pen1-XBIR3 linkage. Purified XBIR3 (left lane) was linked to Pen1 (right lane), a cell transducing peptide. Equimolar amounts of Pen1 and XBIR3 were incubated overnight at 37°C. Linkage was mediated by formation of disulfide bonds. Western blot with anti-His is shown; recombinant BIR3 peptide has a His tag (see Materials and Methods). C, HNE-induced hippocampal neurodegeneration in vitro is blocked by Pen1-linked XBIR3. Primary hippocampal neurons were treated with 3 μm HNE with and without 80 nm Pen1-XBIR3. Relative neuronal survival was quantified after 1 d treatment (n = 3; p < 0.001, ANOVA). D, Delivery of Pen1-fluorescent peptide to CNS by CED. Pen1 was linked to a FITC-labeled peptide, which was delivered to the striatum using CED. Diffuse spreading is observed throughout the ipsilateral hemisphere in the striatum and surrounding cortical matter (4× magnification). Inset at higher magnification (20×) shows cellular uptake of FITC. Epifluorescence microscopy was used. E, Intranasal application delivers Pen1-XBIR3 throughout the rat CNS. The rat was killed 1 h after intranasal delivery of Pen1-XBIR3 (60 μl). The brain was sliced into six 2 mm coronal sections from anterior (olfactory bulbs) to posterior (occipital pole). Slices were solubilized and protein analyzed by SDS-PAGE and Western blotting with anti-HIS. Lane 1, positive control, Pen1-XBIR3. Lane 2, negative control, protein from noninjected animal. Lanes 3-8, coronal sections anterior (1) to posterior (6).
Intranasal delivery of Pen1-XBIR3 provides therapeutic protection from stroke
To determine the functional relevance of caspase-9 activity in stroke pathogenesis, we inhibited caspase-9 activity both before and after ischemia. Currently available small-molecule inhibitors are not sufficiently specific to dissect the contribution of individual caspases (McStay et al., 2008). Mammals express a family of cell death inhibiting proteins known as IAPs. IAPs contain BIR domains, which perform specific functions. One member of this family, XIAP, is a potent specific inhibitor of active caspase-9, caspase-3, and caspase-7. IAPs contain BIR domains, and, for XIAP, caspase inhibition specificity depends on the BIR domains. The BIR3 domain is a specific inhibitor of active caspase-9, and the BIR2 domain inhibits active caspase-3 and caspase-7 (Eckelman et al., 2006).
Therapeutic access of compounds to damaged neurons in the brain requires overcoming several obstacles including the blood–brain barrier (BBB) and the plasma membrane. To provide intracellular delivery, XIAP-BIR3 was disulfide-linked to Pen1, a cell-permeating peptide (Davidson et al., 2004) (Fig. 1B). Upon entry into the cell the disulfide linkage is broken by the reducing environment of the cytoplasm. This releases the peptide cargo and allows it to act at its target. Functional efficacy of this construct was confirmed using primary hippocampal neuron cultures that were subjected to HNE-mediated death, which depends on caspase-9 (Rabacchi et al., 2004). Treatment of HNE-treated cultures with Pen1-XBIR3 abrogated death compared with vehicle treatment alone (Fig. 1C). To ensure that a Pen1 peptide could be delivered to the brain, Pen1 was linked to a FITC-labeled control peptide and delivered directly to the striatum using CED. Brains were harvested 1 h after delivery (Fig. 1D). The FITC peptide entered cells and was distributed throughout the ipsilateral hemisphere.
CED administration provided proof-of-principle that Pen1 peptide can be delivered into the cells in the CNS, but we also used a method with greater therapeutic potential: intranasal delivery. Intranasal delivery of neurotrophins and other compounds has been demonstrated to provide access to the CNS to prevent neurodegeneration in a number of models (Dhuria et al., 2010) including stroke (Liu et al., 2001, 2004). This delivery method takes advantage of the olfactory and trigeminal pathways to bypass the BBB. Until now, proteins and compounds delivered via this method to rodent CNS have targeted extracellular targets, such as cell surface receptors. Because we were targeting caspases, which are intracellular proteins, we needed to deliver the cargo across the plasma membrane. As shown above, Pen1 provides the necessary intracellular uptake of linked peptides. Pen1-XBIR3 was delivered intranasally to rats, brains were sliced coronally, and the presence of XBIR3 in the brain was determined by Western blotting (Fig. 1E). Pen1-XBIR3 was delivered to all slices of the brain, similar to the delivery pattern for IGF (Thorne et al., 2004).
The effects of Pen1-XBIR3 on ischemia-induced changes in neurons were assessed by immunostaining for NeuN. At 24 hpr Pen1-XBIR3 provided significant protection against neuron loss (Fig. 2A). Fluorescent nissl (NeuroTrace) staining of neuron nuclei yielded similar results (data not shown). Direct stroke volume (infarct area/ipsilateral hemisphere area) at 24 hpr, measured by H&E staining, was also reduced in the Pen1-XBIR3 cohort (vehicle: 55.08 ± 3.546 vs Pen1-XBIR3: 38.56 ± 4.749). Direct stroke volume automatically corrects the size of the infarct for its level of edema. Indirect stroke volume, on the other hand, does not account for the contribution of edema to infarct size. For vehicle-treated animals, indirect stroke volume was 65.95 ± 4.933, meaning edema accounted for approximately an 11% expansion in the size of the infarct. For the Pen1-XBIR3 cohort, stroke volume was essentially identical between direct and indirect measurements (38.56 ± 4.749 vs 39.24 ± 6.276). This finding may indicate that Pen1-XBIR3 reduces edema during MCAo, in addition to decreasing stroke volume. These results show that caspase-9 mediates neuronal death, and that intranasally delivered Pen1-XBIR3 is neuroprotective against stroke.
Figure 2.
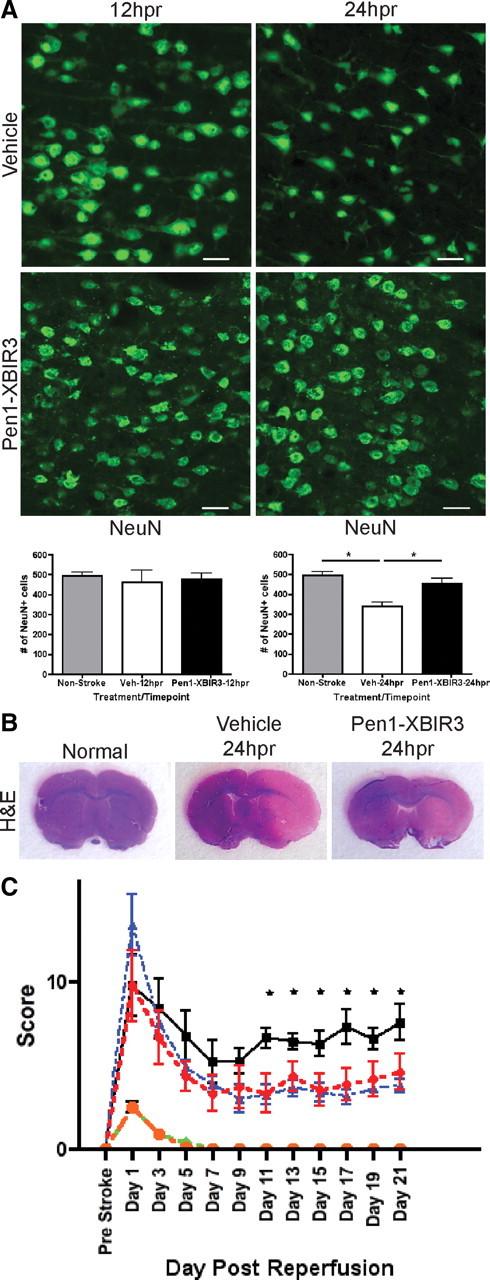
Pen1-XBIR3 is neuroprotective against cerebral ischemia. A, Intranasal Pen1-XBIR3 protects neurons. Vehicle or Pen1-XBIR3 was delivered, and rats were harvested at 12 hpr (left) and 24 hpr (right). Sections were immunostained for NeuN (green), and NeuN-positive cells were quantified. Epifluorescence microscopy was used. Scale bars, 50 μm. Graphs are means (with SEM) of axon counts and neuron counts at 12 hpr (left) and 24 hpr (right). Nonstroked, 494.7 ± 18.52, n = 3; vehicle-12hpr, 463.7 ± 57.53, n = 3; Pen1-XBIR3-12 hpr, 477.3 ± 28.95, n = 3; vehicle-24 hpr, 338.0 ± 22.91, n = 3; Pen1-XBIR3-24 hpr, 453.7 ± 25.44, n = 3 (ANOVA, *p < 0.05). B, Intranasal Pen1-XBIR3 reduces ischemic infarct volume. Vehicle or Pen1-XBIR3 was delivered as in A, and rats were harvested at 24 hpr. Sections were stained with H&E. Direct stroke volume (infarct area/ipsilateral hemisphere area) was quantified. Vehicle-24 hpr, 55.08 ± 3.546, n = 3; Pen1-XBIR3–24 hpr, 38.56 ± 4.749, n = 3 (ANOVA, p < 0.05). C, Intranasal Pen1-XBIR3 provides long-term protection from stroke. Two hour tMCAo was performed on rats given either prophylactic (prestroke) intranasal vehicle (black squares) or prophylactic Pen1-XBIR3 (blue triangles) or therapeutic (poststroke) Pen1-XBIR3 (red circles). Rats were monitored for 21 d. Means (with SEM) of neurofunctional score are shown. Control vehicle (green diamond) and Pen-XBIR3 (orange circle) animals displayed no loss of neurofunction. ANOVA is shown. Asterisks indicate stroked prophylactic vehicle versus prophylactic or therapeutic Pen1-XBIR3 (n = 8 per group, p < 0.05).
The efficacy of Pen1-XBIR3 to prevent sensory-motor disability caused by stroke was tested by administering either a prophylatic (preocclusion) or therapeutic (4 hpr) intranasal bolus of vehicle or Pen1-XBIR3. Rats were assayed with a 24 point neurofunctional scale beginning 1 d postischemia with testing every other day for 3 weeks after the ischemic event. Animals treated with Pen1-XBIR3, either prophylatically or at 4 hpr, exhibited significantly less stroke-related disability than their vehicle-treated counterparts (Fig. 2C). Therapeutic protection by Pen1-XBIR3 indicates that caspase-9 activation is persistent at least until 4 hpr during stroke and that this pathway is critical for the acute neurodegeneration elicited by stroke.
Caspase-6 is active in neuronal processes and soma after stroke
After identifying which initiator caspases were active early in the stroke, we analyzed which effector caspases were activated. The trap method used for initiators blocks activation of effectors. Unlike initiators, which can be activated by dimerization without cleavage (Pop and Salvesen, 2009), cleavage of effectors is indicative of activation. We therefore performed immunohistochemical analysis of cleaved effector caspases. Cleaved caspase-6 (cl-C6) and cleaved caspase-3 (data not shown) were detected. Cleaved caspase-7 was not observed. Although there is evidence that caspase-3 plays a role in stroke (Le et al., 2002), caspase-6 has not previously been reported to have a role in stroke.
Rats were subjected to tMCAo and brains were imaged for cl-C6 at increasing times postreperfusion. The penumbral region in the forebrain, specifically cortical layers I-IV in the granular insular, somatosensory, and dorsal motor cortices (Fig. 3A), revealed a temporal increase in staining for cl-C6 (Fig. 3B). No cl-C6 was detected in control nonischemic animals. By 4 hpr there was minimal staining in the penumbra, but by 12 hpr there was abundant cl-C6 staining in processes and cell bodies (Fig. 3B). There was progressive activation of C6 in the nuclei by 24 hpr, which continued through 3 d postreperfusion (dpr). At 7 dpr cl-C6 was seen only in nuclei. In wild-type mice subjected to tMCAo, the pattern of staining was similar, with cell body and process staining detected at 24 hpr (data not shown) and 3 dpr (Fig. 3C). This time course corresponds neurologically to both the progression of the infarct, with expansion of the infarct over the first 3 d, and axon degeneration. Costaining with NeuN showed cl-C6 was located in neurons (Fig. 3D), whereas there was no colocalization with GFAP, a marker for astrocytes (data not shown).
Figure 3.
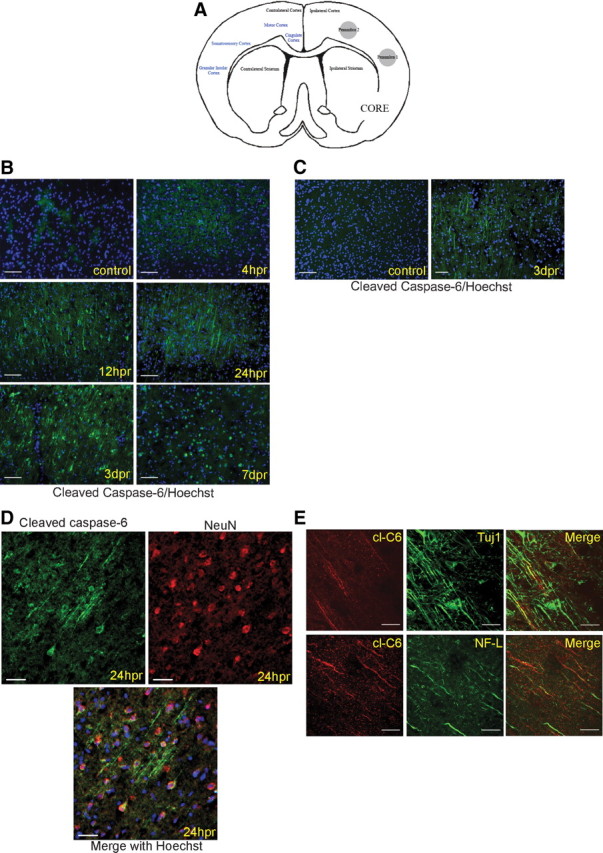
tMCAo induces activation of caspase-6 in neuronal processes and soma. A, Schematic of core and penumbra region in frontocorticostriatal region. B–E, Images were taken from cortical layers III-IV of the cingulate, primary motor, primary + secondary somatosensory, and granular insular cortices in the penumbra. The MCA to the ipsilateral hemisphere has been transiently occluded, whereas the contralateral hemisphere has not been manipulated. B, Caspase-6 is activated in cell bodies and processes in stroke penumbra in rat tMCAo. Rats were subjected to 2 h tMCAo followed by reperfusion for the indicated duration. Rodent tissue was immunostained for cl-C6 (green), and nuclei were stained with Hoechst (blue). cl-C6 appears in cell bodies and processes at 12 hpr. Cell body and process staining is observed through 3 dpr. By 7 dpr, nuclei and cell structures that resemble apoptotic bodies are positive for cl-C6. Scale bars, 50 μm. C, Caspase-6 is activated in cell bodies and processes in stroke penumbra in mouse tMCAo. Mice were subjected to 1 h tMCAo followed by reperfusion for the indicated duration. Tissue was immunostained for cl-C6 (green), and nuclei were stained with Hoechst (blue). cl-C6 appears in processes. Epifluorescence microscopy was used. Scale bars, 50 μm. D, cl-C6 is neuron specific. Cortical penumbra sections from stroked rats (24 hpr) were immunostained for cl-C6 (green), NeuN (red), a neuronal marker, and Hoechst (blue). Top left, cl-C6; top right, NeuN; bottom, the merge of cl-c6, NeuN, and Hoechst. cl-C6 does not colocalize with the astrocyte marker GFAP (data not shown). Epifluorescence microscopy was used. Scale bars, 50 μm. E, cl-C6 is present in axons. Top, cortical penumbra sections from stroked rats (12 hpr) were immunostained for cl-C6 (red) and Tuj1 (green), an axonal marker. Left, cl-C6; center,Tuj1; right, a merge of both. Single processes that contain cl-C6 are apparent. Regions of axons with nonfragmented Tuj1 staining do not have cl-C6 staining. In contrast, regions with cl-C6 exhibited fragmented Tuj1 staining. Bottom, brain sections were immunostained for cl-C6 (red) and NF-L (green), another axon marker. Left, cl-C6; center, NF-L; right, a merge of both. The staining pattern is similar to cl-C6 and Tuj1: regions of axons with nonfragmented NF-L staining do not have cl-C6 staining. In contrast, regions with cl-C6 exhibited fragmented NF-L staining. Confocal microscopy was used. Scale bars, 50 μm.
To identify whether cl-C6 was present in axons, sections were costained for cl-C6 and Tuj1 or NF-L (axon markers). At 24 hpr, Tuj1 and cl-C6 were found in single neuron processes (Fig. 3E). Immunostaining of these processes was not continuous, and gaps in the processes were positive for cl-C6. Interestingly, previous work in AD suggests caspase-6 cleaves tubulin and tau (Guo et al., 2004; Klaiman et al., 2008), which may disrupt axon stability. cl-C6 is also found in single processes containing NF-L (Fig. 3E), with similar cl-C6-filled gaps in the process staining.
Caspase-9 activates caspase-6 in processes and soma of neurons
Although caspase-9 can be isolated with bVAD early in the stroke (1 hpr) (Fig. 1A), it is still active at 4 hpr (Fig. 4A). If all of the caspase-9 was activated by 1 hpr, bVAD administered at 3 hpr would not capture any caspase-9. Therefore, the pool of caspase-9 molecules in the infarct area is not activated all at one time, but is activated gradually during the stroke. Concurrently, after stroke there is a detectable increase in caspase-9 immunofluorescence, and caspase-9 and cl-C6 were colocalized in processes and soma along with cl-C6, shown at 4 and 24 hpr (Fig. 4B). In healthy rodent brain caspase-9 and cl-C6 are not detectable with immunostaining (Fig. 4C). The colocalization of caspase-9 and cl-C6 supports a mechanism for caspase-9 activating caspase-6.
Figure 4.

Caspase-9 and active caspase-6 colocalize in the same cells and processes during stroke. A, Caspase-9 is active at 4 hpr. bVAD or vehicle was infused using CED into the predicted stroke area of rats at 3 hpr. Animals were harvested immediately after the 1 h infusion, and bVAD-caspase complexes were isolated and analyzed by Western blotting. B, Caspase-9 and cl-C6 are induced in the same cells after tMCAo. Rats were subjected to 2 h tMCAo followed by 4 or 24 hpr. Caspase-9 and cl-C6 immunostaining reveals cells colabeled with caspase-9 and cl-C6 at both time points. Caspase-9 is visible in the processes along with cl-C6. Nonstroked rodent brain does not display caspase-9 or cl-C6 staining (C). Confocal microscopy was used. Scale bars, 25 μm. Cortical penumbra staining is shown. C, Intraparenchymal CED of Pen1-XBIR3 blocks tMCAo induction of cl-C6 in neuronal soma and processes. Pen1-XBIR3 or vehicle was infused with CED into the predicted stroke area of rats before tMCAo. Rats were harvested at 24 hpr for immunohistochemistry for cl-C6 (green), caspase-9 (red), and Hoechst (blue). Top, A nonstroked animal. Middle, Vehicle. Bottom, A Pen1-XBIR3-treated animal. The caspase-9-specific inhibitor, Pen1-XBIR3, blocks the induction of cl-C6 observed at 24 hpr. Epifluorescence microscopy was used. Scale bars, 50 μm. D, Intranasal delivery of Pen1-XBIR3 decreases cleaved caspase-6 expression in processes at 24 hpr. Vehicle or Pen1-XBIR3 was delivered intranasally before tMCAo and rats were harvested at 12 hpr (left) and 24 hpr (right). Sections were immunostained for cl-C6 (red), and cl-C6-positive processes were quantified. Epifluorescence microscopy was used. Scale bars, 50 μm. Vehicle-12 hpr: 144.0 ± 28.50, n = 3 versus Pen1-XBIR3–12 hpr: 102.7 ± 23.15, n = 3; p = 0.3233. Vehicle-24 hpr: 109.7 ± 12.73, n = 3 versus Pen1-XBIR3–24 hpr: 57.33 ± 10.04, n = 3 (t test, p < 0.05). E, Intranasal Pen1-XBIR3 blocks the reduction in NF-L-positive processes induced by tMCAo in rats. An intranasal bolus of vehicle or Pen1-XBIR3 was prophylactically delivered, and rats were harvested at 12 hpr (left) and 24 hpr (right). Sections were immunostained for NF-L (green), and NF-L-positive axons were quantified at 12 and 24 hpr. Epifluorescence microscopy was used. Scale bars, 50 μm. Vehicle-12 hpr: 118.7 ± 14.88, n = 3; Pen1-XBIR3–12 hpr: 179.7 ± 14.89, n = 3; Vehicle-24 hpr: 138.0 ± 9.074, n = 3; Pen1-XBIR3–24 hpr: 213.7 ± 11.84, n = 3 (t test, p < 0.05).
To determine unequivocally whether caspase-9 was activating caspase-6 in vivo, we tested whether inhibition of caspase-9 would block caspase-6 activation. Pen1-XBIR3 was delivered to the striatum 1 h before tMCAo using CED. Animals were harvested at 24 hpr, and brain sections were immunostained for caspase-9 and cl-C6. Pen1-XBIR3 completely inhibited the appearance of cl-C6 in cell bodies and axons (Fig. 4C). Thus, caspase-9 activity is necessary for the activation of caspase-6 in neuronal soma and processes after a transient ischemic event in rats.
The preceding findings demonstrate that intraparenchymal delivery of Pen1-XBIR3 prevents activation of caspase-6. To determine whether intranasal delivery of Pen1-XBIR3 also altered caspase-6 activity resulting from stroke, animals were treated with Pen1-XBIR3 1 h before tMCAo and harvested at the indicated times of reperfusion. Brains were analyzed for expression of activated caspase-6 at 12 and 24 hpr. Intranasal Pen1-XBIR3 provided significant reduction of caspase-6 activation. At 12 hpr, there was a trend toward reduction of caspase-6 activation in processes (Fig. 4D) (vehicle: 144.0 ± 28.50 vs Pen1-XBIR3: 102.7 ± 23.15). By 24 hpr, the reduction was significant (Fig. 4D) (vehicle: 109.7 ± 12.73 vs Pen1-XBIR3: 57.33 ± 10.04) compared with rats treated with saline. Therefore, caspase-9 inhibition using this delivery technique reduced caspase-6 activation. Interestingly, cl-C6 levels at 24 hpr inversely correlated with the NF-L density reduction (Fig. 4E) (vehicle-24 hpr: 138.0 ± 9.074 vs Pen1-XBIR3–24 hpr: 213.7 ± 11.84).
Cleaved caspase-3 was also observed in nuclei after 4 hpr. At 24 hpr, Pen1-XBIR3 did not alter the presence of cleaved caspase-3 (vehicle: 89.33 ± 2.333 vs Pen1-XBIR3: 81.67 ± 7.055).
Genetic knockout of caspase-6 is neuroprotective
Next, we examined whether caspase-6 inhibition is sufficient for neuroprotection against stroke using caspase-6−/− null mice (Watanabe et al., 2008). We subjected wild-type and caspase-6−/− mice to tMCAo, and caspase-6−/− mice (Fig. 5A) showed significantly better neurological function at 24 hpr compared with wild-type mice based on a 28 point exam (Clark et al., 1997) (wild type: 19.21 ± 1.931, n = 14 vs caspase-6−/−: 12.64 ± 1.525, n = 14). H&E staining showed a significant difference in infarct volume at 24 hpr (wild type: 61.56 ± 2.853, n = 3; caspase-6−/−: 45.37 ± 1.482, n = 4), commensurate with the significant difference in neurofunction.
Figure 5.

Caspase-6 knock-out mice demonstrate retention of processes and neurons and improved neurological function after tMCAo. A, Characterization of caspase-6−/− mice. Western blot analysis of caspase-6 expression in wild-type and caspase-6−/− mouse spleen is shown. ERK (extracellular signal-regulated kinase) expression is used as a loading control. B, Caspase-6 knockout preserves neurons. Wild-type and caspase-6−/− mice were subjected to 1 h tMCAo and killed at 24 hpr. NeuN staining of cortical penumbra brain tissue reveals a significant decrease in the number of neurons in stroked wild-type mice (148.0 ± 20.22, n = 3) compared with noninfarcted wild-type mice (282.7 ± 32.97, n = 3; p = 0.0253). Caspase-6−/− mice subjected to tMCAo retained more neurons than stroked wild-type mice (225.0 ± 8.114, n = 4 vs 148.0 ± 20.22, n = 3; p = 0.0108). Nonstroked wild-type and nonstroked caspase-6−/− mice did not exhibit a difference in the numbers of neurons (282.7 ± 32.97, n = 3 vs 296.3 ± 9.207, n = 3) (t test, *p < 0.05). Epifluorescence microscopy was used. Scale bars, 50 μm. Nissl staining yielded similar results (data not shown). C, Caspase-6 knockout preserves neuronal processes. Brain sections from wild-type and caspase-6−/− mice subjected to 1 h tMCAo and 24 hpr were immunostained for NF-L. Stroked wild-type mice had fewer NF-L-positive processes compared with stroked caspase-6−/− mice (47.67 ± 7.219, n = 3 vs 70.00 ± 4.916, n = 4; p = 0.0447) (t test, *p < 0.05). NF-L-positive processes were also shorter and more fragmented in wild-type mice compared with caspase-6−/−. Epifluorescence microscopy was used. Scale bars, 50 μm. D, Caspase-6 knockout prevents reduction in tau. Brain lysates from wild-type and caspase-6−/− mice subjected to 1 h tMCAo and 24 hpr were isolated and analyzed by Western blot. Tau expression was analyzed with anti-Tau (V-20), which recognizes the C-terminal end of Tau, the putative location of a caspase-6 cleavage site (Guo et al., 2004; Horowitz et al., 2004). Stroked caspase-6−/− mice (density: 1.154 ± 0.08655, n = 2) contained more tau than stroked wild-type mice (density: 0.7216 ± 0.03246, n = 2). ERK was used for loading control and normalization (n = 2).
To determine whether the improvement in neurofunction was related to the preservation of neurons and axons, quantification of neurons and axons was performed. Wild-type mice subjected to 1 h tMCAo followed by 24 hpr showed a 47% decrease in neuronal number compared with nonstroked wild-type mice; this decrease was partially rescued in caspase-6−/− mice (Fig. 5B). Fluorescent Nissl (NeuroTrace) staining yielded similar results (data not shown). Additionally, wild-type mice subjected to tMCAo had fewer NF-L-positive processes compared with caspase-6−/− mice (Fig. 5C) (47.67 ± 7.219, n = 3 vs 70.00 ± 4.916, n = 4). Processes from wild-type mice were shorter and exhibited more fragmented NF-L staining, suggestive of axon fragmentation and degeneration. Tau is a putative axonal substrate for caspase-6 with potential cleavage sites in N- and C-terminal regions of tau (Guo et al., 2004; Horowitz et al., 2004). Analysis with an antibody specific to the C-terminal region of tau revealed that caspase-6−/− brain retained more intact tau than wild-type brain at 24 hpr (Fig. 5D) (densitometry: 0.7216 ± 0.03246 vs 1.154 ± 0.08655, n = 2, mean ± SD). This suggests that caspase-6 reduces tau levels during stroke. This loss of tau may lead to microtubule instability and loss of process integrity.
Caspase-6 is activated in human stroke
Postmortem tissue from brains of patients who had died after ischemic stroke was immunostained for cl-C6. DAB imaging (Fig. 6 A) showed staining of cell bodies and processes in the infarcted tissue; NF-L staining of adjacent sections showed a decrease in process density. Using immunofluorescence, we found that cl-C6 colocalized with a marker for axons (Fig. 6B). cl-C6 was found in a process in the ischemic tissue, and the pattern of colocalization with Tuj1 was very similar to that observed in the rodent models of ischemia.
Figure 6.
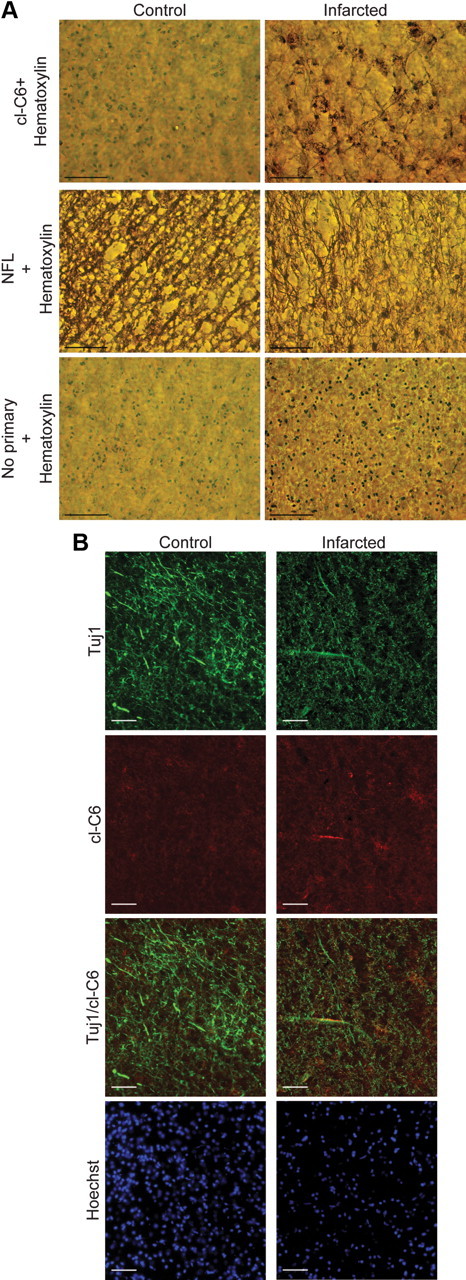
Active caspase-6 observed in human ischemia. Postmortem brain tissue from a patient who had suffered an infarct and from an age-matched control is shown. A, Immunohistological analysis (DAB processing) for cl-C6. DAB processing for cl-C6 showed cell body and process staining. Sections stained without primary antibody show no cell body or process staining. cl-C6 process staining resembles NF-L process staining. Sections from age-matched control brain show no cl-C6 staining. Scale bars, 100 μm. B, Immunofluorescent staining for cl-C6 and Tuj1. The infarct area shows the presence of cl-C6 in a process, Tuj1 appears in the same process. The control brain has no evidence of cl-C6. Epifluorescence microscopy was used. Scale bars, 50 μm.
Discussion
We have used an unbiased approach to identify caspases that are critical for neurodegeneration caused by cerebral ischemia. Our data show that caspase-6 and caspase-9 are regulators of neurodegeneration in cerebral ischemia. We have used active caspase trapping and identified caspase-9 as an early-stage mediator of cell death in stroke that leads to caspase-6 activation. We show that caspase-6 is activated in axons, supporting its role in axon degeneration during neuronal death (Nikolaev et al., 2009). Inhibiting these caspases, either through peptide inhibition of caspase-9 or genetic knockout of caspase-6, reduced stroke injury and improved sensorimotor abilities, providing sustained functional neuroprotection out to 3 weeks postischemia.
It is common practice to use short peptide caspase substrates, such as Asp-Glu-Val-Asp (DEVD) for caspase-3 and Ile-Glu-Thr-Asp (IETD) for caspase-8, for assaying caspase activity; however, these peptides are highly promiscuous (McStay et al., 2008) and can generate misleading data. bVAD, an irreversible pan-caspase inhibitor, provides a reliable measurement of caspase activity through biochemical pulldown of active caspases and has been shown to isolate caspases 1, 2, 8, or 9, depending on the death stimulus used. Originally used to assay caspase activity in cell lines (Tu et al., 2006), and, more recently, in primary neuron cultures (Tizon et al., 2010), we have adapted this procedure for in vivo use in the CNS. In the present study, we demonstrate that caspase-9 is active early in the progression of the infarct (1 hpr) by isolating active caspase-9 complexes with bVAD. This is the first demonstration of a catalytically active initiator caspase in the brain during stroke.
The small peptide caspase inhibitors, such as zVAD (benzyloxycarbonyl-Val-Ala-Asp) and DEVD, have been shown to confer neuroprotection in rodent models of stroke (Endres et al., 1998). These inhibitors, as mentioned above, are not specific for individual caspases (McStay et al., 2008) and thus do not specify which caspases are involved. Given the nonapoptotic functions of caspases in the nervous system, including synaptic plasticity and dendrite pruning (Huesmann and Clayton, 2006, Feinstein-Rotkopf and Arama, 2009), nonspecifically inhibiting the entire caspase family might prove detrimental in the CNS.
Because caspase-9 activity is stimulated early in stroke and elevated caspase-9 is observed in neurons with cl-C6 and caspase-9 cleaves caspase-6 in vitro (data not shown), we hypothesized that caspase-9 leads to caspase-6 activation during stroke. The BIR3 domain from XIAP (XBIR3) is a highly specific inhibitor of caspase-9. We linked XBIR3 by a reducible disulfide bond to Pen1, a transduction peptide that efficiently delivers cargo across the plasma membrane (Davidson et al., 2004; Fan et al., 2006; Guegan et al., 2006), to inhibit caspase-9 activity in cells. The use of a transient disulfide linkage between Pen1 and the cargo peptide ensures that the cargo peptide can assume its native functional conformation once it is transported into the cell. This method contrasts with cell-permeant peptide fusion domains, such as TAT (transactivator of transcription), which can lead to nonfunctional misfolded cargo protein (G. Salvesen, personal communication). Prior studies showed that intraperitoneal delivery of a similar caspase-9 inhibitor, a fusion protein of PTD (protein transduction domain)-XBIR3-RING (really interesting new gene), reduces infarct volume after tMCAo (Fan et al., 2006; Guegan et al., 2006), but did not explore the downstream mechanism. In our study, we used two different strategies to deliver this inhibitor to the brain. CED provides direct delivery to the region of the infarct; CED of this inhibitor before stroke abrogated the activation of caspase-6 in neuronal soma and processes. Therefore, caspase-9 activity regulates caspase-6 activity in stroke.
From a therapeutic perspective, intranasal delivery is a very attractive treatment strategy for CNS disorders because it provides direct, noninvasive access to the brain via the olfactory pathway. Intranasal delivery combined with the cell-permeant peptide Pen1 provides intracellular delivery to the CNS.
In the present study, intranasal delivery of Pen1-XBIR3 inhibited caspase-6 activation, reduced axon degeneration, and was neuroprotective. Neuroprotection was substantial even 3 weeks after the initial insult. Our data reveal that caspase-6 activation corresponds to axon degeneration in stroke and provide insight into how this process occurs in ischemia. Because caspase-6 activation is relatively delayed after ischemic onset, efficacious inhibition of caspase-6 in stroke may provide substantial postischemic functional neuroprotection and a valuable therapeutic strategy for cerebral ischemia.
Previous investigations have used genetic strategies of caspase inhibition to delineate the postischemic function of both initiator and effector caspase activation. Of the initiator caspases, caspase-1 null mice and mice expressing a caspase-1 dominant negative transgene are partially protected from tMCAo (Friedlander et al., 1997; Schielke et al., 1998). Caspase-2 null mice do not exhibit neuroprotection (Bergeron et al., 1998). Deletion of caspase-8 or caspase-9 is embryonic lethal, thus mice lacking these caspases have not been studied in tMCAo. Of the three effector caspases (3, 6, and 7) only caspase-3 has been studied extensively in stroke (Endres et al., 1998; Benchoua et al., 2001). Most compelling are the data that show that caspase-3 null mice are protected from tMCAo, showing a critical role for this caspase (Le et al., 2002). However, neuroprotection after caspase-3 gene deletion is not complete and other effector caspases may also have independent functions in stroke. Moreover, a recent study revealed that caspase-3 is induced in nonapoptotic cells during stroke (Wagner et al., 2011).
We describe here that genetic ablation of caspase-6 provides neuroprotection at the structural and functional levels against stroke. Functions for caspase-6 in neurons include processing huntingtin, which is associated with neurodegeneration in Huntington's disease (Graham et al., 2006). Caspase-6 can cleave tau, affecting its ability to stabilize microtubules (Horowitz et al., 2004), and caspase-6-mediated cleavage of tau may play a role in AD pathogenesis (Guo et al., 2004; Klaiman et al., 2008). In our models of cerebral ischemia, active caspase-6 colocalized with axonal markers, implicating this caspase in the degeneration of neuronal axons. Although present in the same process, some axonal regions with active caspase-6 lacked the process marker, suggesting that caspase-6 was either cleaving the marker or leading to its destabilization. In support of this function for caspase-6 in stroke, we have observed a reduction in tau in wild-type mice subjected to tMCAo relative to caspase-6−/− mice. Further proteomic analysis of tissue lysate from infarcted brains from caspase-6−/− and wild-type mice could be used to reveal a broader spectrum of proteins cleaved by caspase-6 during stroke and potentially many that regulate axon stability.
Moreover, caspase-6 is involved in process degeneration in dissociated DRG neurons subjected to trophic factor deprivation (Nikolaev et al., 2009); that study proposed that caspase-6 is responsible for only process degeneration, not for neuronal death. Our studies find that caspase-6 is mediating both process degeneration and neuronal death during ischemia (Fig. 5). The temporal activation of caspase-6 in the stroke penumbra corresponds with the progression of axonal degeneration. For other forms of neurodegeneration, axon degeneration is a major contributor to cell death and may instigate death via removal of target-derived trophic factors (Ferri et al., 2003; Fischer et al., 2004; Stokin et al., 2005). In these instances, axon degeneration preceded cell death. In clinical cases of cerebral ischemia, axon degeneration is observed as early as 2 d postischemia (Thomalla et al., 2004); however, the molecular events triggering axon degeneration may begin earlier. In the penumbral region, we found that axon loss preceded neuronal loss, which indirectly implies that axon degeneration precedes neuronal loss after an ischemic event.
Our collective results suggest that the caspase-9/caspase-6 pathway is an excellent target for stroke therapy. This pathway is active in the neurons and is responsible for a significant amount of axon loss and neuron death after ischemia (Fig. 7). Moreover, the temporal course of activation provides for an expanded therapeutic window when targeting these molecular events.
Figure 7.

Schematic representation of tMCAo mechanistic and functional timeline. A, Molecular and functional effects of tMCAo. B, Intervention with Pen1-XBIR3 prophylactically at −3 h inhibits active caspase-9, blocks activation of caspase-6, and prevents process and neuronal loss. Intervention with Pen1-XBIR3 therapeutically at 4 hpr provides functional recovery up to 21 d.
Footnotes
C.M.T. was supported by the American Heart Association and National Institutes of Health (NIH) Grants NS035933 and NS43089. G.S.S. and S.J.S. were supported by NIH Grant CA69381. E.S.C. was supported by NIH Grant NS40409. We thank Rebecca Goldstein for technical support, Sharon Pierre-McClain for critical reading of the manuscript, and Leah Hanson for assistance with the intranasal technique.
References
- Benchoua A, Guégan C, Couriaud C, Hosseini H, Sampaïo N, Morin D, Onténiente B. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001;21:7127–7134. doi: 10.1523/JNEUROSCI.21-18-07127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Bruce JN, Falavigna A, Johnson JP, Hall JS, Birch BD, Yoon JT, Wu EX, Fine RL, Parsa AT. Intracerebral clysis in a rat glioma model. Neurosurgery. 2000;46:683–691. doi: 10.1097/00006123-200003000-00031. [DOI] [PubMed] [Google Scholar]
- Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res. 1997;19:641–648. doi: 10.1080/01616412.1997.11740874. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–531. doi: 10.1097/00006123-199603000-00021. discussion 532. [DOI] [PubMed] [Google Scholar]
- Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. Highly efficient small interfering RNA delivery to primary mammalian neurons induces microRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denault JB, Salvesen GS. Human caspase-7 activity and regulation by its N-terminal peptide. J Biol Chem. 2003;278:34042–34050. doi: 10.1074/jbc.M305110200. [DOI] [PubMed] [Google Scholar]
- Dhuria SV, Hanson LR, Frey WH., 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gómez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- Fan YF, Lu CZ, Xie J, Zhao YX, Yang GY. Apoptosis inhibition in ischemic brain by intraperitoneal PTD-BIR3-RING (XIAP) Neurochem Int. 2006;48:50–59. doi: 10.1016/j.neuint.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Feinstein-Rotkopf Y, Arama E. Can't live without them, can live with them: roles of caspases during vital cellular processes. Apoptosis. 2009;14:980–995. doi: 10.1007/s10495-009-0346-6. [DOI] [PubMed] [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Guégan C, Braudeau J, Couriaud C, Dietz GP, Lacombe P, Bähr M, Nosten-Bertrand M, Onténiente B. PTD-XIAP protects against cerebral ischemia by anti-apoptotic and transcriptional regulatory mechanisms. Neurobiol Dis. 2006;22:177–186. doi: 10.1016/j.nbd.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaiman G, Petzke TL, Hammond J, Leblanc AC. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics. 2008;7:1541–1555. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komotar RJ, Kim GH, Sughrue ME, Otten ML, Rynkowski MA, Kellner CP, Hahn DK, Merkow MB, Garrett MC, Starke RM, Connolly ES. Neurologic assessment of somatosensory dysfunction following an experimental rodent model of cerebral ischemia. Nat Protoc. 2007;2:2345–2347. doi: 10.1038/nprot.2007.359. [DOI] [PubMed] [Google Scholar]
- Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA. Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A. 2002;99:15188–15193. doi: 10.1073/pnas.232473399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie C, Hirsch JG, Rossmanith C, Hennerici MG, Gass A. Clinicotopographical correlation of corticospinal tract stroke: a color-coded diffusion tensor imaging study. Stroke. 2004;35:86–92. doi: 10.1161/01.STR.0000106912.09663.EB. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Hanson LR, Frey WH., 2nd The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin-like growth factor-I in rats. J Stroke Cerebrovasc Dis. 2004;13:16–23. doi: 10.1016/j.jstrokecerebrovasdis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Thorne RG, DeFor TA, Frey WH., 2nd Intranasal administration of insulin-like growth factor-I bypasses the blood–brain barrier and protects against focal cerebral ischemic damage. J Neurol Sci. 2001;187:91–97. doi: 10.1016/s0022-510x(01)00532-9. [DOI] [PubMed] [Google Scholar]
- McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- Mitsios N, Gaffney J, Krupinski J, Mathias R, Wang Q, Hayward S, Rubio F, Kumar P, Kumar S, Slevin M. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem Biophys. 2007;47:73–86. doi: 10.1385/cbb:47:1:73. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Park KJ, Grosso CA, Aubert I, Kaplan DR, Miller FD. p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat Neurosci. 2010;13:559–566. doi: 10.1038/nn.2513. [DOI] [PubMed] [Google Scholar]
- Petullo D, Masonic K, Lincoln C, Wibberley L, Teliska M, Yao DL. Model development and behavioral assessment of focal cerebral ischemia in rats. Life Sci. 1999;64:1099–1108. doi: 10.1016/s0024-3205(99)00038-7. [DOI] [PubMed] [Google Scholar]
- Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabacchi SA, Friedman WJ, Shelanski ML, Troy CM. Divergence of the apoptotic pathways induced by 4-hydroxynonenal and amyloid beta-protein. Neurobiol Aging. 2004;25:1057–1066. doi: 10.1016/j.neurobiolaging.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamás A, Lengvári I. Examination of sensorimotor performance following middle cerebral artery occlusion in rats. Brain Res Bull. 2003;59:459–466. doi: 10.1016/s0361-9230(02)00962-0. [DOI] [PubMed] [Google Scholar]
- Ribe EM, Serrano-Saiz E, Akpan N, Troy CM. Mechanisms of neuronal death in disease: defining the models and the players. Biochem J. 2008;415:165–182. doi: 10.1042/BJ20081118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC, Fesik SW. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem. 2000;275:33777–33781. doi: 10.1074/jbc.M006226200. [DOI] [PubMed] [Google Scholar]
- Thomalla G, Glauche V, Koch MA, Beaulieu C, Weiller C, Röther J. Diffusion tensor imaging detects early Wallerian degeneration of the pyramidal tract after ischemic stroke. Neuroimage. 2004;22:1767–1774. doi: 10.1016/j.neuroimage.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Pronk GJ, Padmanabhan V, Frey WH., 2nd Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Tizon B, Ribe EM, Mi W, Troy CM, Levy E. Cystatin C protects neuronal cells from amyloid-beta-induced toxicity. J Alzheimers Dis. 2010;19:885–894. doi: 10.3233/JAD-2010-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ. Mechanisms of p75-mediated death of hippocampal neurons: role of caspases. J Biol Chem. 2002;277:34295–34302. doi: 10.1074/jbc.M205167200. [DOI] [PubMed] [Google Scholar]
- Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8:72–77. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- Wagner DC, Riegelsberger UM, Michalk S, Härtig W, Kranz A, Boltze J. Cleaved caspase-3 expression after experimental stroke exhibits different phenotypes and is predominantly non-apoptotic. Brain Res. 2011;1381:237–242. doi: 10.1016/j.brainres.2011.01.041. [DOI] [PubMed] [Google Scholar]
- Wahl F, Allix M, Plotkine M, Boulu RG. Neurological and behavioral outcomes of focal cerebral ischemia in rats. Stroke. 1992;23:267–272. doi: 10.1161/01.str.23.2.267. [DOI] [PubMed] [Google Scholar]
- Watanabe C, Shu GL, Zheng TS, Flavell RA, Clark EA. Caspase 6 regulates B cell activation and differentiation into plasma cells. J Immunol. 2008;181:6810–6819. doi: 10.4049/jimmunol.181.10.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]