

Abstract
Several neurodegenerative diseases are influenced by the innate immune response in the central nervous system (CNS). Microglia, have pro-inflammatory and subsequently neurotoxic actions as well as anti-inflammatory functions that promote recovery and repair. Very little is known about the transcriptional control of these specific microglial behaviors. We have previously shown that in HIV associated neurocognitive disorders (HAND), the transcription factor p53 accumulates in microglia and that microglial p53 expression is required for the in vitro neurotoxicity of the HIV coat glycoprotein gp120. These findings suggested a novel function for p53 in regulating microglial activation. Here we report that in the absence of p53, microglia demonstrate a blunted response to interferon-γ, failing to increase expression of genes associated with classical macrophage activation or secrete pro-inflammatory cytokines. Microarray analysis of global gene expression profiles revealed increased expression of genes associated with anti-inflammatory functions, phagocytosis and tissue repair in p53 knockout (p53−/−) microglia compared with those cultured from strain matched p53 expressing (p53+/+) mice. We further observed that p53−/− microglia demonstrate increased phagocytic activity in vitro and expression of markers for alternative macrophage activation both in vitro and in vivo. In HAND brain tissue, the alternative activation marker CD163 was expressed in a separate subset of microglia than those demonstrating p53 accumulation. These data suggest that p53 influences microglial behavior, supporting the adoption of a pro-inflammatory phenotype, while p53 deficiency promotes phagocytosis and gene expression associated with alternative activation and anti-inflammatory functions.
Keywords: Neuroinflammation, Macrophages, CNS Ischemia, Phagocytosis, Alternative Activation
INTRODUCTION
CNS inflammation is observed in a variety of chronic neurodegenerative diseases and in response to acute CNS injury. Modulation of the inflammatory response is currently a target for therapeutic development in a number of diseases including stroke, amyotrophic lateral sclerosis (ALS), Parkinson disease (PD) and Alzheimer disease (AD) (Henkel et al. 2004; Imamura et al. 2003; Moreau et al. 2005; Nagatsu and Sawada 2005; Tarkowski et al. 2003; Tarkowski et al. 1997). In addition, the CNS innate immune system modulates neural stem cell proliferation and differentiation (Barish et al. 1991; Baron et al. 2008; Bernardino et al. 2008; Chiu et al. 2008; Clarke et al. 2008; Deboy et al. 2006; Wong et al. 2004). For example, stimulating microglia, the resident macrophages of CNS tissue, with interleukin 4 (IL-4) results in the release of factors that promote neurogenesis (Butovsky et al. 2006; Choi et al. 2008). Thus, microglia can exert a unique repertoire of responses that may impact CNS function during disease or injury. Nevertheless, the molecular regulation of microglial behavior is not well understood.
Macrophages respond to specific environmental stimuli by adopting functionally distinct activation phenotypes, including but not limited to classical and alternative activated states (Goerdt et al. 1999; Gordon 2003). Classical activation is induced by exposure to T helper type 1 (TH1) cytokines such as interferon gamma (IFNγ) or bacterial lipopolysacharride (LPS). Classical activation results in macrophages capable of releasing proinflammatory cytokines and reactive oxygen species (ROS) such as nitric oxide (NO) (Goerdt et al. 1999; Gordon 2003). T helper type 2 (TH2) cytokines such as IL-4 trigger alternative activation, leading to increased phagocytosis, release of tissue remodeling molecules and secretion of anti-inflammatory cytokines (Martinez et al. 2008; Varin and Gordon 2009). Some extracellular signals and subsequent intracellular signaling pathways that promote acquisition of a particular activation type have been described, but the transcriptional regulation of monocyte plasticity is not well characterized.
Microglia actively patrol their local environment and are poised to respond to perturbations in the CNS (Hanisch and Kettenmann 2007; Nimmerjahn et al. 2005). Like macrophages, activated microglia may exhibit features associated with classical or alternative activation (Colton et al. 2006; Ponomarev et al. 2007). Classically activated microglia release neurotoxic factors including proinflammatory cytokines, prostanoids, ROS, and excitatory amino acids (Colton 2009). Conversely, factors that promote alternative activation have been shown to stimulate neuroprotection in CNS disease models(Chiu et al. 2008; Clarke et al. 2008; Deboy et al. 2006). Thus, the relative influence of the different microglial activation phenotypes may have a very important impact on CNS disease outcome.
Despite the potential importance of microglial behavior in neurologic diseases little is known about transcriptional regulation of microglial activation phenotype. Microglia are characterized by their functional plasticity and reactivity to their environment, manifesting characteristics distinct from most tissue resident macrophages (Carson et al. 1998; Davoust et al. 2008). In addition to typical macrophage functions of phagocytosis and immune surveillance, microglia also contribute to regulation of synaptogenesis, neurogenesis and neuronal apoptosis (Elkabes et al. 1996; Marin-Teva et al. 2004). Thus, given their repertoire of unique functions, microglia may be subject to transcriptional regulation of phenotype(s) that are distinct from other circulating or tissue resident macrophages.
The transcription factor p53 is an apical mediator of apoptosis, growth arrest and DNA repair pathways (Vogelstein et al. 2000). In response to cellular stresses like oxidative injury or DNA damage, p53 is stabilized by post-translational modifications. Stabilized p53 translocates to the nucleus and alters transcription (Appella and Anderson 2001). p53 accumulates in neuronal nuclei in several human neurodegenerative diseases and p53 transcriptional activity has been implicated in the pathogenesis of others (Bae et al. 2005; de la Monte et al. 1997; Eve et al. 2007; Garden et al. 2004; Mogi et al. 2007).
In HAND, p53 accumulates in the nuclei of neurons and non-neuronal cells including microglia (Jayadev et al. 2007). Using mixed cerebrocortical cultures exposed to the HIV coat protein gp120 as an in vitro model of HAND pathogenesis, we previously reported that p53 expression in microglia is required for HIV-gp120 induced neurotoxicity (Garden et al. 2004). In addition to identifying a novel role for p53, we observed that the number of apoptotic p53+/+ neurons co-cultured with p53−/− microglia decreased, suggesting that p53−/− microglia may have neurotrophic actions and/or are more effective phagocytes than p53+/+ microglia. Taken together, these findings suggested the hypothesis that p53 may participate in the regulation of microglial activation phenotype.
To address this issue we examined cultured p53−/− microglial behaviors and global gene expression profiles compared with p53 expressing murine microglia. We further studied the effect of p53 deficiency on microglial activation phenotype in vivo by examining immunoreactivity for CD206 (macrophage mannose receptor), a marker of macrophage alternative activation, in ischemic brain tissue sections. Finally, to address the possibility that p53 influences microglial behavior in human disease, tissue sections from HAND patients were co-labeled for p53 and the alternative activation marker CD163 to determine if these two proteins segregate into distinct populations of microglia.
Materials and Methods
Mice
Mice deficient in p53 and strain matched p53 expressing mice (Donehower et al. 1992) were maintained in a specific pathogen free facility under guidance of an IACUC approved protocol.
Cell Culture
The BV2 microglia cell line was grown in DMEM supplemented with 25U/ml penicillin, 25mg/ml streptomycin, 10% FBS and. 2mM L-Glutamine). Primary murine microglia cultures were obtained from P 3–4 neonatal mice as described (Moller et al. 2000). Pure microglia cultures were obtained by harvesting floating cells from the mixed glial cell cultures. Neuron-microglia co-cultures were generated using previously described methods (Garden et al. 2004) by plating microglia harvested from mixed glial cultures on top of established primary neuron cultures.
Immunolabeling
Co-cultured microglia were labeled for nuclear p53 immunoreactivity (Antibody 1C12, Cell Signaling, 1:1000) and microglia specific tomato lectin (Vector Labs) according to previously published methods (Garden et al. 2004; Uo et al. 2009). Mouse brains following middle cerebral artery occlusion (MCAO) were fixed in 4% paraformaldehyde and cryoprotected in 30% sucrose solution prior to being cut into 6 micrometer thick sections using a cryostat. Sections were mounted on slides and immunolabeled with an antibody against CD206 (ABD Serotec, 1:300) according to previously reported methods (Choi et al. 2010). Human brain sections were deparaffinized and rehydrated using standard methods. Antigen retrieval was performed in citrate buffer at pH 6.0. Co-labeling for p53 and CD68 (DAKO KP1 clone, 1:100) was performed using a previously published protocol (Jayadev et al. 2007). Co-labeling for p53 and CD163 was performed by adapting a previously published protocol (Fischer-Smith et al. 2008) using a monoclonal antibody to CD163 (Vector 1:50) and co-labeling with antibodies to p53 using the same method as that developed to double label for p53 and CD68. Immunoreactivity for p53 was visualized by reaction with the peroxidase substrate diaminobenzadine (DAB:Vector) and CD68 and CD163 were identified by reactions with very intense purple peroxidase substrate (VIP:Vector). Co-labeled sections were analyzed by counting the total number of cells labeled for CD68 or CD163 and the number of p53/CD68 or p53/CD163 co-labeled cells in ten randomly selected fields of view per section using a 40X objective.
Reporter Assay
Transcriptional activity of endogenous p53 in cortical neurons was evaluated via transduction of lentivirus carrying a firefly luciferase expression unit containing either four copies of the p53 responsive element BS2 derived from the human PUMA promoter or mutant versions of this element as previously described (Uo et al. 2007). Microglia were incubated with control or mutant reporter lentivirus at 2.5 multiplicity of infection (MOI) and plated at 5×103/well. Forty-eight hours after infection, cells were treated with 5μM camptothecin or control (DMSO) for 16 hours. Luciferase activity was then measured per manufacturer’s protocol using the Bright-Glo System (Promega).
ELISA assays
Microglia from both genotypes were plated in poly-D-lysine coated wells at a density of 1×105 cells per well of a 96 well plate and cultured in 200μl D10C media. Twenty-four hours after plating, cells were treated with 10 U/ml IFNγ or bovine serum albumin (BSA) for an additional 24 hours. Microglia-conditioned media was harvested and analyzed for IL-12 and TNFα concentration by multiplex ELISA using the Bio-Plex system (BioRad, Hercules, CA) with reagents obtained from BioSource (Invitrogen, Carlsbad, CA).
Real Time Quantitative Reverse Transcriptase (RT) Polymerase Chain Reaction (PCR)
Microglia of both genotypes were plated on poly-D-lysine coated 35mm dishes at a density of 1×106 cells per dish in D10C media and treated with IL-4 (10ng/ml), IFNγ (10u/ml) or BSA at 10ng/ml for 24 hours. RNA was extracted using the RNeasy mini protocol (Qiagen, Valencia, CA). Quantitative RT-PCR was performed using Taqman Onestep Master Mix and validated Assay-on-Demand™ probe sets for mouse Ym1, FIZZ1, IL-1α, IL-1β and MARCO using an ABI PRISM 7000 (ABI, Foster City, CA). RT-PCR experiments were performed using a housekeeping gene (actin or GAPDH) as an internal control and the results reported as percent change from the ratio of the gene in question to the housekeeping gene or as a fold change calculated using the 2−(Δ−ΔCT) formula.
Microarray Studies
Purified microglia were cultured at a density of 1×106 cells per 35mm dish in isolation for 24 hours, then treated with vehicle control or 400pM recombinant glycosylated gp120 (SF162, NIH AIDS Reagent Program) or equal protein concentration of BSA for 24 hours. RNA was extracted using the RNeasy mini protocol (Qiagen, Valencia, CA) and pooled from three separate culture preparations for each genotype and treatment. Pooled RNAs were employed for hybridization with Affymetrix whole mouse genome microarrays. The hybridizations, intensity measurements and bioinformatics analyses were performed at the University of Washington Functional Genomics and Bioinformatics Core Laboratory. Microarray results were normalized using the GCRMA method. Gene ontology analysis was performed using the GOStats program in the Bioconductor software package.
Phagocytosis Assay
Fluorescently-labeled apoptotic cells were generated by labeling of BV-2 cells with red fluorescent PKH26 (Sigma, St. Louis, MO), followed by UV treatment for 60 minutes. UV treated cells were incubated at 37°C for 48 hours. Rounded apoptotic bodies that lifted off the tissue culture dish were then isolated, washed and added to green fluorescent PKH67 (Sigma, St. Louis, MO) labeled BV2 cells or primary microglia pretreated with 10ng/ml tumor necrosis factor-α (TNFα), 10 U/ml IFNγ or 10 ng/ml IL-4 or equal concentration of BSA for 24 hours. After incubation for 6 hours at 37°C, the cultures were washed to remove remaining apoptotic cells and fixed with 4% paraformaldehyde. The percent microglia that internalized apoptotic cells was determined using fluorescence microscopy. A blinded observer with no knowledge of the genotype or treatment group performed all counting of phagocytic cells. Control cultures were incubated at 4°C for 2 hours prior to fixation to assess the rate of association unrelated to active phagocytosis. All control cultures demonstrated < 1% of microglia associated with a fluorescent apoptotic cells (data not shown).
Induction of 45 minute transient cerebral ischemia in mice
Sixteen16–20 week old p53+/+ and p53−/− male mice were anesthetized and prepared for induction of experimental CNS ischemia by MCAO as previously described (Gibson and Murphy 2004). Cerebral blood flow was evaluated by Doppler monitoring to insure that the MCAO was sufficiently effective to generate ischemic injury. Six days following MCAO mice were killed under anesthesia by cardiac perfusion with cold saline and 1 ml of 4% paraformaldehyde followed by cervical dislocation.
Statistical Analysis
Results were compared using t-tests, one- or two- way analysis of variance (ANOVA) with Prism 4.0 statistical software (GraphPad, San Diego, CA). The choice of test depended on the number of comparisons and whether an experiment was designed to assess a single effect of genotype, treatment or both genotype and treatment. Post-hoc comparisons were performed using the Tukey multiple comparison test for one-way ANOVA and the Bonferroni post-test for two-way ANOVA. CD68/p53 and CD163/p53 co-labeled microglia were compared using a paired t-test on the values obtained from adjacent sections.
Results
Inflammatory stimuli influence p53 transcriptional activity in cultured microglia
We previously showed that p53−/− primary microglia in neuron/microglia co-cultures are functionally distinct from wild-type primary microglia in response to HIV gp120 coat protein (Garden et al. 2004). This effect could have resulted from suppression of microglia behaviors that require p53 activation by gp120 or from an effect of p53 deficiency that impacts microglia behavior without regard to gp120 exposure. In order to determine if p53 is activated in microglia by gp120 treatment specifically in the neuron/microglia co-culture paradigm, we performed immunofluorescent labeling for nuclear p53 and co-labeling using a microglia specific tomato lectin. We observed that a subset of microglia demonstrate detectable nuclear p53 in basal conditions. Treatment with gp120 leads to an increase in the number of microglia with nuclear p53 accumulation that is equal to the level seen in cultures treated with the DNA damage inducer camptothecin (Fig. 1A). Thus, stabilized p53 is present in primary microglia under basal conditions and augmented by an inflammatory stimulus. We previously reported that microglia in these conditions do not undergo apoptosis (Garden et al. 2004). Thus, this finding suggests that p53 may have a function in microglia unrelated to the induction of apoptosis that exists in basal culture conditions and is amplified following exposure to a pro-inflammatory stimulus.
Figure 1.
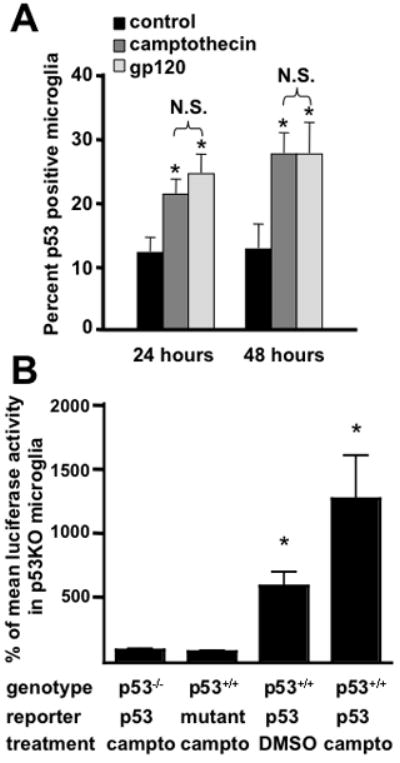
Microglia exposed to gp120 undergo p53 stabilization. A) The percent of cultured primary microglia co-labeled for nuclear p53 accumulation and microglia specific tomato lectin 24 and 48 hours after treatment with HIV-gp120, camptothecin or vehicle control. There is a significant increase in the number of microglia with p53 accumulation by 24 hours of gp120 treatment. Using this method, gp120 treatment lead to the same level of p53 activation as camptothecin, a positive control compound that activates p53 by causing DNA damage. B) Transcriptional activity of p53 measured in cultured primary microglia by reporter gene assay. Microglia from p53+/+ and p53−/− mice were cultured and infected with a lentivirus expressing a p53 luciferase reporter. Treatment with camptothecin resulted in an approximate doubling of luciferase activity over that observed in untreated p53+/+ cultures. Basal p53 transcriptional activity was also detectable above the background luciferase activity observed in p53−/− microglia or p53+/+ microglia expressing a mutant p53 promoter driven luciferase construct. *=p<0.01 by ANOVA with post-hoc comparison. N.S.=not significant.
To confirm that nuclear p53 in microglia was transcriptionally active we used a luciferase reporter assay (Fig. 1B). Cultured primary microglia infected with lentivirus expressing a luciferase reporter controlled by a p53 responsive promoter revealed both basal and camptothecin-inducible p53 transcriptional activity. No expression of the luciferase reporter construct was observed in p53−/− microglia or when p53+/+ microglia were infected with a lentivirus expressing luciferase under the control of a mutant promoter. Taken together, these findings demonstrate that p53 is transcriptionally active in cultured microglia.
Classical activation is suppressed in p53−/− microglia
Since p53−/− microglia were unable to promote HIV-gp120 neurotoxicity (Garden et al. 2004), we considered the possibility that p53−/− microglia is less capable of responding to a classical activation signal. Cultured p53+/+ and p53−/− microglia were treated with IFNγ for 24 hours. We evaluated expression of two cytokine genes, IL-1α and IL-1β, typically induced during classical activation. In addition, we analyzed the expression of MARCO, a scavenger receptor up-regulated during classical activation (Mukhopadhyay et al. 2006). We observed that IFNγ treatment results in increased mRNA for IL-1α, IL-1β in p53+/+ microglia, which was prevented in p53−/− microglia (Fig 2A). MARCO expression was induced by IFNγ treatment even in p53−/− microglia, but significantly less so than in microglia expressing p53, suggesting that p53−/− microglia are capable of responding to IFNγ treatment, but the magnitude of activation of some genes associated with a pro-inflammatory response involves p53 dependent signaling. Interestingly, MARCO expression was also significantly reduced in p53−/− microglia under basal conditions (Fig. 2A), suggesting that p53 also has some effect on microglia gene expression even in the absence of a pro-inflammatory signal. To determine if p53 genotype influences the ability of microglia to secrete pro-inflammatory cytokines associated with classical activation, we analyzed IFNγ induced IL-12 and TNFα release by Luminex ELISA. We observed that p53−/− microglia release significantly less pro-inflammatory cytokines in response to IFNγ treatment compared with identically treated p53+/+ microglia (Fig. 2B).
Figure 2.
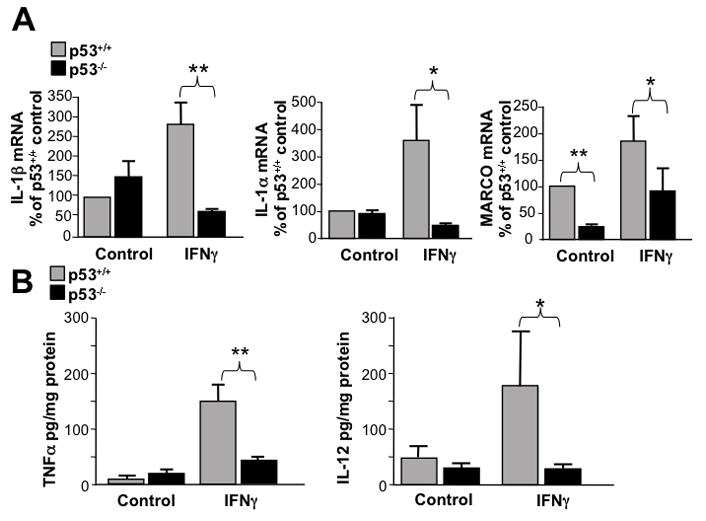
Classical activation is suppressed in p53−/− microglia. A) Q-PCR results from p53+/+ and p53−/− microglia treated with IFNγ or control for interleukin-1α (IL-1α), interleukin-1β (IL-1β) and the murine scavenger receptor MARCO. All three classical activation mRNA’s were significantly lower following IFNγ stimulation in p53−/− microglia (*=p<0.05, **=p<0.01 by two-way ANOVA post-hoc comparison, n=4). B) Secretion of the pro-inflammatory cytokines TNFα and interleukin-12 (IL-12) was enhanced by IFNγ treatment in p53+/+ microglia, but p53−/− microglia failed to respond to IFNγ stimulation by increasing release of these pro-inflammatory cytokines. (*=p<0.05, **=p<0.01 by two-way ANOVA post-hoc comparison, n=3)
The presence of p53 strongly influences microglial gene expression and the transcriptional response to HIV gp120
To delineate the mechanism by which p53 influences the ability of microglia to undergo classical activation and cause neurotoxicity following HIV-gp120 treatment we evaluated global gene expression profiles of p53+/+ and p53−/− microglia. We observed a profound effect of p53 genotype on microglial gene expression with approximately 5% of the probes displaying a two-fold difference in the level of expression between genotypes (Fig. 3A). This represents approximately 10% of the probes in the whole mouse genome array with detectable expression in cultured microglia. Internal validation of the microarray data set was obtained by identifying known targets of p53-mediated transcription (Table 1). We observed above threshold hybridization for 14 known p53 target genes in p53+/+ microglia. Of these 14 p53 targets, 10 had a greater than two-fold reduction of hybridization in p53−/− microglia. External validation was performed by evaluating expression by alternate means for a selection of genes that demonstrated large changes in mRNA hybridization intensity including MARCO (Fig. 1A), SDF-1 and mannose receptor (data not shown).
Figure 3.
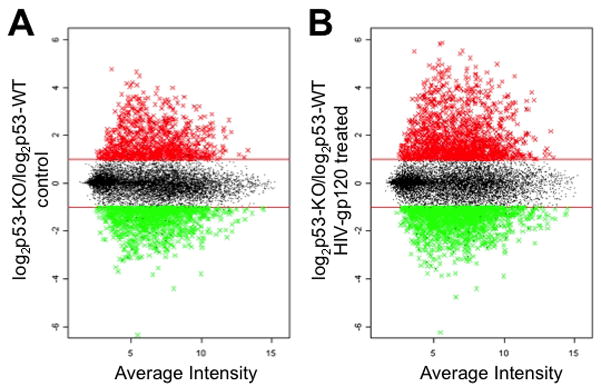
The impact of p53 genotype on microglial gene expression patterns. A) The relative intensity of hybridization to microarray probes between mRNA’s from p53−/− and p53+/+ microglia 24 hours after the addition of vehicle control is displayed on the Y-axis using log2 scale. The probes are distributed along the X-axis according to average intensity. Horizontal lines signify the cut off value of a two fold difference between genotypes. Probes with more than a 2 fold increase in expression in p53−/− microglia are displayed in red, and probes with a greater than 2 fold decrease in expression in p53−/− microglia are displayed in green. B) The results of a microarray experiment performed using mRNA extracted from p53+/+ and p53−/− microglia 24 hours following gp120 exposure are displayed according to the same format described for A.
Table 1.
p53 target genes are consistently down-regulated in p53−/− microglia. N.C.= No Change, N.D.=Not Detected
| Gene Name | Function | Fold Change |
|---|---|---|
| Cyclin-dependent kinase inhibitor 1A (P21) | Cell cycle regulator/p53 mediated growth arrest | −21.1 |
| Proline/serine-rich coiled-coil 1 | p53 mediated growth arrest | −11.6 |
| Trp53 | Cell cycle regulator, pro-apoptotic transcriptional regulator | −8.6 |
| Cyclin G1 | Cell cycle regulator | −7.4 |
| Transformation related protein 53 inducible nuclear protein 1 | Promotes apoptosis and cell cycle arrest | −5.9 |
| Tgf-alpha | Mitogenic growth factor | −4.2 |
| Wig1/Zmat3 | Growth arrest promoting p53 target | −3.8 |
| Mdm2-Transformed mouse 3T3 cell double minute 2 | Regulator of p53 | −3.5 |
| Bax | Regulator of apoptosis | −2.7 |
| Cyclin D1 | Cell cycle regulator | −2.0 |
| Sestrin 1 | Response to ROS/DNA damage | −1.8 |
| GADD45a | Cell cycle regulator | −1.7 |
| Proliferating cell nuclear antigen | Cofactor of DNA polymerase delta | N.C. |
| Wip1/Ppm1D | Stress response regulator of p38 MAP Kinase | N.C. |
| DR5/KILLER | Pro-apoptotic p53 target gene | N.D. |
| Dickkopf-1 | Inhibition of Wnt signaling | N.D. |
To evaluate whether the muted response to IFNγ by p53−/− microglia was secondary to a change in expression of known IFNγ signaling factors we probed the microarray data set for differential expression of genes that signal the IFNγ response (Ifnrg1, Ifnrg2, Jak1, Jak2, Stat1, Irf1 and Irf9). All but Irf9 were expressed at levels above threshold and none demonstrated significant differences in expression between genotypes. We also probed the microarray data for genes that are known to inhibit IFNγ signaling (Socs1, Socs3, Srf2, and Shp-2/Ptpn2). We found that only Socs3 and Shp-2/Ptpn2 were expressed above threshold and there was no effect of p53 genotype on the level of expression. These findings suggest that p53−/− microglia have the required genes for IFNγ signaling but do not initiate classical activation pathways presumably secondary to some effect of p53 deficiency on the ability of microglia to initiate a normal pro-inflammatory response.
The effect of p53 genotype on microglia treated with HIV-gp120 was even larger, with approximately 15% of the expressed microglia genome differentially regulated between p53+/+ and p53−/− microglia (Fig. 3B). To further evaluate the impact of p53 on gene expression changes stimulated by HIV-gp120 we identified genes differentially expressed following gp120 treatment that were specific to each genotype. While this was a much smaller number of differentially regulated mRNAs, we discovered that there was almost no overlap between the two genotypes in these gene lists. In fact less that 2% of the probes with > 2-fold change in expression following gp120 treatment demonstrated concurrent responses across genotypes. We performed a gene ontology analysis on the microarray data set in order to determine if there was any overlap in functional groups with altered expression following HIV-gp120 treatment. We found statistically significant alterations in 121 functional groups in p53−/− microglia and 134 functional groupings in p53+/+ microglia. Of the functional groups identified as having altered expression following HIV-gp120 treatment only five groups overlapped between genotypes and none of the overlapping groups had a p value <0.02. This observation suggests that p53 has a very strong determining effect on how gene expression will be regulated following exposure to HIV-gp120.
Microglia from p53−/− mice demonstrate increased expression of genes associated with alternative activation
The divergent response between wild-type and p53−/− microglia to HIV-gp120 is consistent with a model in which p53 deficiencyleads to a differentially polarized response to an activation stimulus. When evaluating the microarray data set to identify genes associated with classical or alternative activation, we observed that p53−/− microglia demonstrate upregulation of a large number of genes associated with alternative activation. The list of these genes, the fold-increase in expression in p53−/− compared with p53+/+ microglia, and their proposed functional roles are shown in Table 1. In addition to the genes listed in Table 1, mRNA for many procollagens, collagen metabolizing enzymes and enzymes involved in collagen cross linking were increased in p53−/− microglia compared with wildtype. While not all proteins that have been associated with alternative activation are listed in Table 1, the list of genes upregulated in the absence of p53 does span a variety of functions associated with alternative macrophage activation including extracellular matrix remodeling, tissue repair and down regulation of the inflammatory response. In contrast, no genes reportedly associated with alternative macrophage activation were decreased in p53−/− microglia. To further validate this finding, we examined the expression of Ym1 and FIZZ1, two genes frequently used as markers of alternative activation (Gordon 2003). Since the microarray platform we employed had only one probe for both of these mRNAs and neither was observed to be expressed above the limits of detection, we analyzed expression of these marker genes using the more sensitive method of real time quantitative RT-PCR. Both Ym1 and FIZZ1 are dramatically upregulated in p53−/− compared with p53+/+ microglia (Fig. 4).
Figure 4.
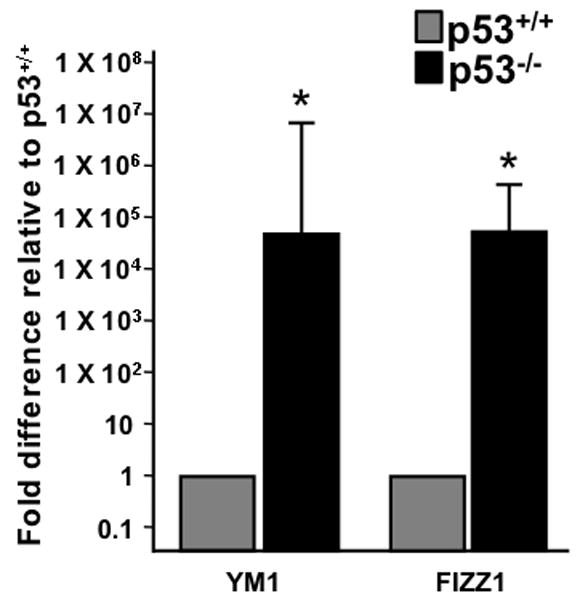
Microglia from p53−/− mice express markers of alternative activation. Real-Time quantitative RT-PCR (Q-PCR) results on microglia from p53+/+ and p53−/− mice are shown for two genes upregulated following alternative activation of macrophages, Ym1 and FIZZ1 (n=3 from separate culture experiments and Q-PCR runs, p<0.001 by t-test).
Microglia from p53−/− mice demonstrate increased internalization of apoptotic cells
In order to assess whether p53−/− microglial function as alternatively activated microglia we examined their ability to internalize apoptotic cells. We hypothesized that if microglia develop alternative activation functions similar to macrophages, then IL-4 should stimulate microglia to internalize apoptotic cells. To test this hypothesis, we developed a phagocytosis assay using the BV2 microglia cell line. We observed that pre-treatment with IL-4 promotes increased internalization of apoptotic cells by BV2 cells while cytokines associated with the induction of classical activation (TNFα and IFNγ) inhibit phagocytosis of apoptotic cells (Fig. 5A). To determine if p53 genotype impacts the ability of microglia to internalize apoptotic cells, cultured primary microglia from p53+/+ and p53−/− mice were treated with IL-4 for 24 hours, then exposed to fluorescently labeled apoptotic cells. Microglia deficient in p53 were more likely to internalize apoptotic cells both at baseline and in response to IL-4 treatment (Fig. 5B). This finding suggests that in the absence of p53, microglia are more likely to possess one of the functional characteristics associated with alternative activation. Nevertheless, p53 deficient microglia remain responsive to IL-4, demonstrating the expected increase in phagocytic activity following exposure to this cytokine.
Figure 5.
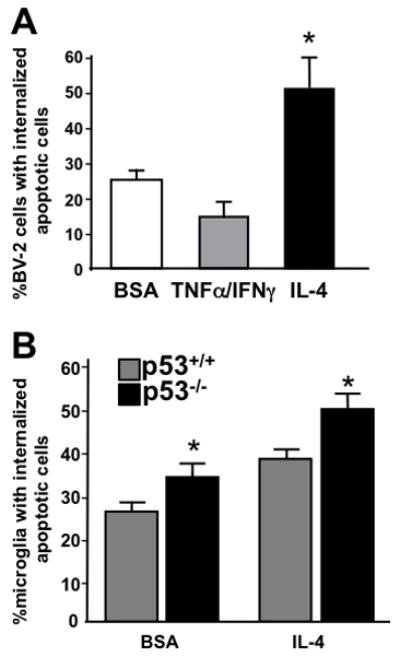
The phagocytosis of apoptotic cells is enhanced in alternatively activated microglia as well as p53−/− microglia. A) BV-2 cells were exposed to tumor necrosis factor α (TNFα 10ng/ml) and interferon γ (IFNγ 10u/ml) cytokine stimulation to induce classical activation or interleukin-4 (IL-4) to promote alternative activation. Phagocytosis of apoptotic cells was measured 24 hours following stimulation. Treatment with IL-4 induced a significant (*=p<0.05 by ANOVA, n=5) increase in the portion of BV-2 cells that internalized apoptotic cells. B) Primary microglia from p53+/+ and p53−/− mice were treated with IL-4 or bovine serum albumin (BSA) as a protein control for 24 hours and the portion of cells internalizing apoptotic cells determined. Microglia from p53−/− mice were significantly more likely (*p<0.05 by two-way ANOVA post-hoc comparison, n=3) to participate in the phagocytosis of apoptotic cells than were p53+/+ microglia in both the control and IL-4 treated conditions.
Microglia from p53−/− mice demonstrate increased alternative activation in vivo
To determine if the results observed using cultured microglia could be recapitulated during neuroinflammation in vivo, we employed ischemic injury to induce an inflammatory response and evaluated brains during the delayed tissue repair phase when features of alternative activation should appear. CD206 immunoreactivity is an established marker of alternatively activated macrophages (Porcheray et al. 2005). The expression of CD206 (macrophage mannose receptor) on parenchymal microglia following ischemia induced by MCAO was revealed by immunohistochemistry. Adult p53+/+ and p53−/− mice underwent MCAO confirmed by cerebral blood flow monitoring. Forebrain sections through the region of ischemia from animals sacrificed 6 days following MCAO demonstrated increased CD206 immunoreactive microglia in the penumbral region compared with a matched anatomical region on the contralateral non-ischemic side (Fig. 6). This increase in CD206 labeled microglia was significantly greater in p53 deficient mice than in mice expressing p53. This finding suggests that p53 may influence the probability that microglia will express markers of alternative activation in vivo during the phase of neuroinflammation associated with tissue repair. An alternate hypothesis supported by this finding is that in the absence of p53, more invading myeloid cells transit from the periphery into the CNS and appear as CD206 immunoreactive cells that cannot be differentiated from microglia in histological sections.
Figure 6.
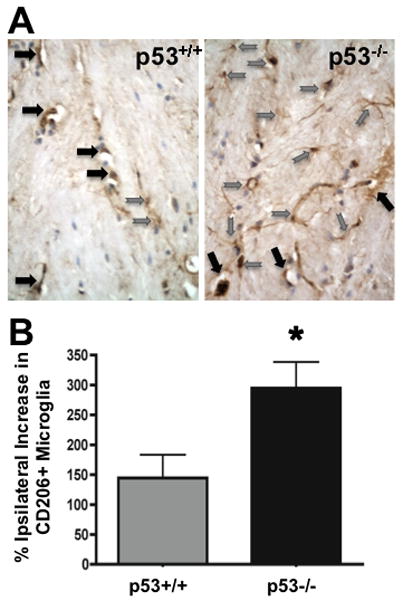
Alternative activation of microglia in vivo is influenced by p53. A) Tissue sections from the ischemic penumbra region of p53+/+ and p53−/− mice labeled for CD206 reveal an increased number of ramified parenchymal microglia in p53 deficient mice compared with p53+/+ control, but no change in the number of CD206 positive perivascular macrophages (Grey arrows=microglia, black arrows=perivascular macrophages). B) The percent increase in CD206 labeled microglia ipsilateral to the MCAO is significantly increased (*p<0.05 by t-test, n=4) in p53 deficient mice compared with the increase observed in p53 expressing mice.
Stabilized nuclear p53 is less frequently observed in the alternatively activated subset of microglia in cortical tissue from patients with HAND
Microglia in HAND patients demonstrate nuclear p53 accumulation (Garden et al. 2004; Jayadev et al. 2007) as well as increased expression of markers for both classical and alternative activation (Fischer-Smith et al. 2008). If p53 is important for initiating the classical activation phenotype, accumulation of nuclear p53 should be less prevalent in the population of microglia expressing markers for alternative activation. The scavenger receptor CD163 is associated with alternative activation in macrophages and has been previously reported as upregulated in microglia of HIV infected humans and in a primate model of neuroAIDS (Fischer-Smith et al. 2008; Roberts et al. 2004). To determine if the association between lack of p53 expression and alternative activation in microglia is also present in HAND cases, adjacent cortical tissue sections from six HAND patients were co-labeled with antibodies recognizing all microglia (CD68) and p53 or CD163 and p53 (Fig 7). We observed that the mean percent of all CD68 labeled cells co-labeled for p53 was significantly higher than the portion of CD163 and p53 co-labeled cells (37% +/− 5% vs. 9% +/− 2.5%, p<0.0001). Tissue sections from HIV negative controls show no immunoreactivity for p53 or CD163. The observed lack of association between nuclear p53 and CD163 immunoreactivity in HAND microglia suggests that p53 accumulation in vivo occurs in a specific population of microglia that are more likely to adopt a classical rather than alternative activation phenotype. These observations suggest an overarching and novel role for p53 in regulating microglial behavior that is not an artifact specific to p53 deficient mice. Additionally, taken together with the finding that p53 genotype affects the likelihood of microglia demonstrating alternative activation in vivo, these findings also suggest that the association between microglial p53 activity and the classical activation phenotype is not an artifact of the primary microglia culture environment.
Figure 7.
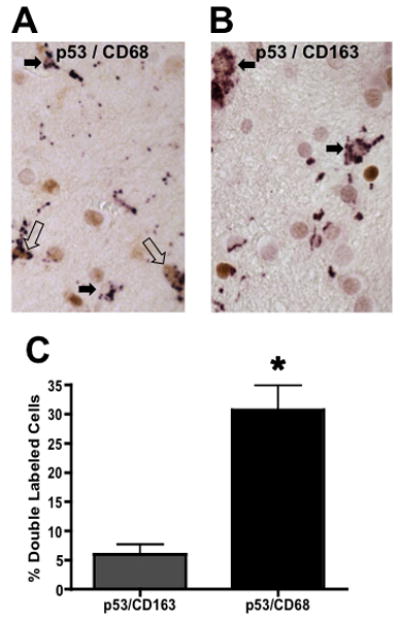
Separate populations of microglia demonstrate immunoreactivity for a marker of alternatively activated and deactivated macrophages, CD163 or p53 activation in HAND cases. A) Human cortical tissue sections immunolabeled for p53 (brown) and CD68 (purple) reveal that a portion of microglia are immunoreactive for both markers. Open arrows point to double labeled cells and closed arrows identify CD68 single labeled cells. B) A section adjacent to the one in A is shown labeled with antibodies to p53 (brown) and CD163 (purple). Closed arrows point to CD163 labeled microglia. C) Quantification of double labeled microglia in adjacent sections from twelve HAND patients (*=p<0.0001 using a paired t-test).
Discussion
In this study we observed that p53 is active in non-apoptotic microglia and that the presence of p53 influences microglial gene expression. Microglia derived from p53−/− mice have increased expression of genes associated with alternative activation, exhibit increased internalization of apoptotic cells, have a blunted response to IFNγ and are more likely to adopt an alternative activation phenotype in vivo. These findings provide evidence that p53 supports IFNγ-induced classical activation and that p53 deficient microglia are skewed towards alternative activation. The molecular mechanism by which p53 regulates microglial gene expression to produce this effect is not known. Nevertheless, data presented here suggest that p53 does not directly prevent microglia from responding to pro-or anti-inflammatory stimuli. Expression of mRNA coding for proteins involved in IFNγ signal transduction or the suppression of IFNγ responses were not altered in p53−/− microglia, suggesting that even though p53 has a dramatic impact on global gene expression patterns, the ability to respond to IFNγ is not directly effected. Furthermore, p53 deficient microglia are capable of responding to IFNγ by increasing the expression of the pro-inflammatory gene MARCO, even though the magnitude of the response was less than that observed in wild-type microglia. The ability to respond to IL-4 by increasing phagocytic activity and expression of alternative activation marker genes was also preserved in p53−/− microglia in spite of the finding that these activities were significantly increased under basal conditions. Thus, the mechanism by which p53 influences microglial behavior is likely to be downstream of cytokine receptors and their signal transduction pathways, potentially involving transcriptional regulation of specific cellular behaviors.
Prior studies are consistent with a model in which classical activation is required for neurotoxicity following exposure to the HIV gp120 coat protein in vitro (Bezzi et al. 2001; Garden et al. 2002; Kaul et al. 2001). Thus, it is likely that p53is required for neurotoxicity following exposure to gp120 due to its role in promoting the classical activation phenotype in microglia (Garden et al. 2004). Furthermore, in HAND cortical tissue there is significantly less p53 accumulation in the subpopulation of microglia demonstrating expression of an alternative activation marker than in the total microglial population. This finding suggests that the observations reported here are relevant to the regulation of microglial activation within a human disease.
Evidence is emerging that p53 may play a role in innate immunity beyond its established functions in the induction of apoptosis or cell cycle arrest. The impact of p53 mediated transcription on myeloid cells has not been well characterized, however it has been shown that the p53 inhibitor pifithrin-alpha can decrease the neurotoxicity elicited from rat microglia exposed to an inflammatory stimulus (Davenport et al. 2010).
As a transcription factor, p53 could directly influence innate immune function through regulation of gene expression. For example, vascular macrophages lacking p21, a p53 canonical transcriptional target, are more phagocytic and express lower levels of pro-inflammatory cytokines compared with wild-type (Merched and Chan 2004). Furthermore, p53 and the inflammation pathway transcription factor NFκB have been shown to cross-talk and are capable of interfering with each other’s transcriptional activity (Dijsselbloem et al. 2007; Komarova et al. 2005; Webster and Perkins 1999). The findings reported here in conjunction with these prior reports, suggest that p53 may exert influence on the innate immune response. It remains to be determined however, if that influence is unique to the CNS environment or a more generalized effect in other tissues.
In the CNS microglia exhibit both neurotoxic and neuroprotective actions. Microglia perform protective roles by expressing neurotrophic factors (Batchelor et al. 2002; Nakajima et al. 2001), removing extracellular toxins such as β-amyloid (Koenigsknecht-Talboo and Landreth 2005; Mandrekar et al. 2009) and supporting neurogenesis through the secretion of insulin like growth factor-1(IGF-1) (Butovsky et al. 2006; Choi et al. 2008). Most of the protective functions ascribed to microglia are associated with the alternative activation phenotype. The promotion of anti-inflammatory phenotypes by Th2 cytokines has been well documented in macrophages and more recently in microglia (Michelucci et al. 2009). IL-4 induces an anti-inflammatory phenotype in microglia, suppressing NO production, TNFα release and neurotoxicity (Butovsky et al. 2005; Chao et al. 1993; Zhao et al. 2006).
Alternative activation patterns in microglia have been documented in both human and animal models of human neurodegenerative disease (Colton et al. 2006; Hung et al. 2002) as well as in microglia activated in vitro (Ponomarev et al. 2007). Microglia in culture induce the alternative activation markers Ym1, FIZZ1, arginase 1 and mannose receptor (CD206) in response to IL-4 treatment (Colton et al. 2006). Expression of alternative activation mRNAs was also upregulated in brain tissue from Alzheimer disease (AD) patients compared with age matched controls as well as in an AD mouse model (Colton et al. 2006). Alternative activation of microglia has also been demonstrated using the experimental autoimmune encephalomyelitis (EAE) mouse model for multiple sclerosis. During EAE, microglia upregulate Ym1 expression in an IL-4 dependent manner and do not induce production of NO when IL-4 is present. However, with IL-4 deficiency the pathology of EAE is exacerbated, supporting the hypothesis that alternative activation of microglia has an important role in limiting the pathology of EAE (Ponomarev et al. 2007). Additionally, in an in vitro model of ALS using primary motor neuron-microglia co-cultures, alternative activation by IL-4 treatment suppresses the LPS induced release of NO and superoxide while maintaining IGF-1 release by microglia (Zhao et al. 2006).
Classically activated microglia are thought to exacerbate pathogenesis in a variety of CNS disease and injury states. Typically the neurotoxic actions of microglia are mediated by the release of factors associated with classical activation. Both pro-inflammatory cytokines like TNFα and small molecule neurotoxins, some of which may interact with the N-methyl-D-aspartate (NMDA) type glutamate receptor, mediate pro-inflammatory neurotoxicity (Garden and Moller 2006). Release of excitotoxins and ROS from classically activated microglia have been shown to promote p53 mediated neuronal death (Garden and Morrison 2005; Morrison et al. 2003). Accumulation of ROS leads to DNA damage and initiation of cell injury pathways that promote accumulation of p53 (Harms et al. 2004; Martindale and Holbrook 2002; Sohal and Weindruch 1996).
We have shown that in HAND, which has been associated with all of the neurotoxicity mechanisms described above, p53 is activated in multiple cell types including neurons and microglia (Garden et al. 2004; Jayadev et al. 2007). If p53 influences microglial phenotype by promoting classical activation, it is possible that in the milieu of an HIV infected brain, p53 may perpetuate a positive feedback loop of cytotoxicity. In this scheme, ROS generation initiated by viral infection could lead to microglia p53 stabilization. This in turn would promote microglial acquisition of a classically activated phenotype, leading to release of neurotoxic cytokines and small molecules capable of over-stimulating NMDA receptors. It is also possible that other neurodegenerative diseases associated with inflammation and oxidative injury including AD, PD and Huntington’s disease would be associated with a similar effect of p53 on microglial function, which in turn could exacerbate the process of neurodegeneration and/or suppress the ability of microglia to perform the neurotrophic functions associated with the alternative activation phenotype. Thus, it is possible that determining which genes are regulated by p53 in microglia could identify potential therapeutic targets for modulating CNS inflammation in a variety of diseases including HAND.
Table 2.
Genes associated with alternative macrophages are upregulated in p53−/− microglia under basal conditions.
| Gene Name | Function | Fold Change |
|---|---|---|
| PDGF-RB | Trophic factor receptor | 13.4 |
| EGF-containing fibulin-like ECM protein 1 | ECM remodeling | 12.2 |
| scavenger receptor class F, member 2 | Endocytosis and Internalization | 11.4 |
| interleukin 1 receptor-like 1 | Anti-Inflammatory Decoy receptor | 10.5 |
| Histocompatibility-28 | Antigen Presentation | 9.4 |
| prostaglandin-endoperoxide synthase 2 | PGE2 synthesis | 9.3 |
| mannose receptor, type 2 | Endocytosis and Internalization | 9.3 |
| MMP 2 | ECM remodeling | 9.00 |
| EGF-R | Trophic factor receptor | 8.5 |
| FGF7 | Trophic Factor | 7.8 |
| VEGFC | Trophic Factor | 7.5 |
| TGFβ receptor III | Anti-Inflammatory receptor | 6.7 |
| Thrombospondin 2 | ECM, angiogenesis | 6.2 |
| GULP, engulfment adaptor | Endocytosis and Internalization | 6.0 |
| Thrombospondin 1 | ECM, promotes TGFβ expression | 5.7 |
| scavenger receptor class A, member 3 | Endocytosis and Internalization | 5.1 |
| MMP 3 | ECM remodeling | 4.5 |
| PGI2 synthase | PGI2 synthesis | 4.5 |
| TGFβ 1 induced transcript 1 | Anti-inflammatory responsive gene | 4.3 |
| Histocompatibility-2, Q8 | Antigen presentation | 3.8 |
| FLT1 | VEGF Receptor | 3.8 |
Acknowledgments
We thank Drs. Thomas Moeller and Nephi Stella for helpful discussions, Dr. Theodore Bammler and Richard Beyer for assistance interpretation of microarray data and Annette Zawalinsky, Weiqun Guo, Huy Nguyen and Mei Deng for technical assistance. This work was supported by NS055652 (S.J.), NS35533 (R.S.M.), NS045528, NS062269 and the Friends of Alzheimer’s Disease Research (G.A.G.), with facilities support from P30-HD02774 to the UW Center for Human Development and Disability and P30 NS055088 NINDS Insitutional Center Core Grant-Neuroproteomics. Human tissue was obtained with thanks to the National NeuroAids Tissue Consortium supported by NIH grant N01MH32002.
Footnotes
Disclosures: The authors have no potential conflicts of interest.
References
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268(10):2764–72. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47(1):29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Barish ME, Mansdorf NB, Raissdana SS. Gamma-interferon promotes differentiation of cultured cortical and hippocampal neurons. Dev Biol. 1991;144(2):412–23. doi: 10.1016/0012-1606(91)90433-4. [DOI] [PubMed] [Google Scholar]
- Baron R, Nemirovsky A, Harpaz I, Cohen H, Owens T, Monsonego A. IFN-gamma enhances neurogenesis in wild-type mice and in a mouse model of Alzheimer’s disease. FASEB J. 2008;22(8):2843–52. doi: 10.1096/fj.08-105866. [DOI] [PubMed] [Google Scholar]
- Batchelor PE, Porritt MJ, Martinello P, Parish CL, Liberatore GT, Donnan GA, Howells DW. Macrophages and Microglia Produce Local Trophic Gradients That Stimulate Axonal Sprouting Toward but Not beyond the Wound Edge. Mol Cell Neurosci. 2002;21(3):436–53. doi: 10.1006/mcne.2002.1185. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Agasse F, Silva B, Ferreira R, Grade S, Malva JO. Tumor Necrosis Factor-alpha Modulates Survival, Proliferation and Neuronal Differentiation in Neonatal Subventricular Zone Cell Cultures. Stem Cells. 2008;26(9):2361–71. doi: 10.1634/stemcells.2007-0914. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4(7):702–10. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29(3):381–93. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31(1):149–60. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22(1):72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. J Immunol. 1993;151(3):1473–81. [PubMed] [Google Scholar]
- Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH, Jr, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008;105(46):17913–8. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, Shea Donohue T, Bauer AJ, Linden DR, Szurszewski JH, Gibbons SJ, et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138(7):2399–409. 2409, e1. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59(4):568–80. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RM, Lyons A, O’Connell F, Deighan BF, Barry CE, Anyakoha NG, Nicolaou A, Lynch MA. A pivotal role for interleukin-4 in atorvastatin-associated neuroprotection in rat brain. J Biol Chem. 2008;283(4):1808–17. doi: 10.1074/jbc.M707442200. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport CM, Sevastou IG, Hooper C, Pocock JM. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J Neurochem. 2010;112(2):552–63. doi: 10.1111/j.1471-4159.2009.06485.x. [DOI] [PubMed] [Google Scholar]
- Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29(5):227–34. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Sohn YK, Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer’s disease. J Neurol Sci. 1997;152(1):73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- Deboy CA, Xin J, Byram SC, Serpe CJ, Sanders VM, Jones KJ. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp Neurol. 2006;201(1):212–24. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Dijsselbloem N, Goriely S, Albarani V, Gerlo S, Francoz S, Marine JC, Goldman M, Haegeman G, Vanden Berghe W. A critical role for p53 in the control of NF-kappaB-dependent gene expression in TLR4-stimulated dendritic cells exposed to Genistein. J Immunol. 2007;178(8):5048–57. doi: 10.4049/jimmunol.178.8.5048. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16(8):2508–21. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eve DJ, Dennis JS, Citron BA. Transcription factor p53 in degenerating spinal cords. Brain Res. 2007;1150:174–81. doi: 10.1016/j.brainres.2007.02.088. [DOI] [PubMed] [Google Scholar]
- Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008;14(4):318–26. doi: 10.1080/13550280802132857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22(10):4015–24. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. Faseb J. 2004;18(10):1141–3. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1(2):127–37. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Garden GA, Morrison RS. The multiple roles of p53 in the pathogenesis of HIV associated dementia. Biochem Biophys Res Commun. 2005;331(3):799–809. doi: 10.1016/j.bbrc.2005.03.185. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Murphy SP. Progesterone enhances functional recovery after middle cerebral artery occlusion in male mice. J Cereb Blood Flow Metab. 2004;24(7):805–13. doi: 10.1097/01.WCB.0000125365.83980.00. [DOI] [PubMed] [Google Scholar]
- Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, Hakiy N, Klemke CD, Dippel E, Kodelja V, et al. Alternative versus classical activation of macrophages. Pathobiology. 1999;67(5–6):222–6. doi: 10.1159/000028096. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 2004;61(7–8):822–42. doi: 10.1007/s00018-003-3304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55(2):221–35. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Hung SI, Chang AC, Kato I, Chang NC. Transient expression of Ym1, a heparin-binding lectin, during developmental hematopoiesis and inflammation. J Leukoc Biol. 2002;72(1):72–82. [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (Berl) 2003;106(6):518–26. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison RS, Garden GA. The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J Neuroimmune Pharmacol. 2007;2(4):359–70. doi: 10.1007/s11481-007-9095-x. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25(36):8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, et al. p53 is a suppressor of inflammatory response in mice. Faseb J. 2005;19(8):1030–2. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29(13):4252–62. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–47. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu Rev Immunol. 2008 doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Merched AJ, Chan L. Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation. 2004;110(25):3830–41. doi: 10.1161/01.CIR.0000148681.01282.89. [DOI] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210(1–2):1741–53. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Mogi M, Kondo T, Mizuno Y, Nagatsu T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci Lett. 2007;414(1):94–7. doi: 10.1016/j.neulet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Moller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. J Neurochem. 2000;75(4):1539–47. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology. 2005;65(12):1958–60. doi: 10.1212/01.wnl.0000188907.97339.76. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochem Res. 2003;28(1):15–27. doi: 10.1023/a:1021687810103. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Chen Y, Sankala M, Peiser L, Pikkarainen T, Kraal G, Tryggvason K, Gordon S. MARCO, an innate activation marker of macrophages, is a class A scavenger receptor for Neisseria meningitidis. Eur J Immunol. 2006;36(4):940–9. doi: 10.1002/eji.200535389. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11(8):999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Honda S, Tohyama Y, Imai Y, Kohsaka S, Kurihara T. Neurotrophin secretion from cultured microglia. J Neurosci Res. 2001;65(4):322–31. doi: 10.1002/jnr.1157. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27(40):10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, Dormont D, Gras G. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481–9. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ES, Masliah E, Fox HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE) J Neuropathol Exp Neurol. 2004;63(12):1255–64. doi: 10.1093/jnen/63.12.1255. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(9):1200–5. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C, Ekholm S, Tarkowski A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol. 1997;110(3):492–9. doi: 10.1046/j.1365-2249.1997.4621483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J Neurosci. 2007;27(45):12198–210. doi: 10.1523/JNEUROSCI.3222-05.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Veenstra TD, Morrison RS. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29(9):2824–32. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varin A, Gordon S. Alternative activation of macrophages: Immune function and cellular biology. Immunobiology. 2009;214(7):630–41. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19(5):3485–95. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G, Goldshmit Y, Turnley AM. Interferon-gamma but not TNF alpha promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp Neurol. 2004;187(1):171–7. doi: 10.1016/j.expneurol.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Zhao W, Xie W, Xiao Q, Beers DR, Appel SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem. 2006;99(4):1176–87. doi: 10.1111/j.1471-4159.2006.04172.x. [DOI] [PubMed] [Google Scholar]