

SUMMARY
Cancer cell-of-origin is difficult to identify by analyzing cells within terminal-stage tumors, whose identity could be concealed by the acquired plasticity. Thus an ideal approach to identify the cell-of-origin is to analyze proliferative abnormalities in distinct lineages prior to malignancy. Here we use Mosaic Analysis with Double Markers (MADM) in mice to model gliomagenesis by initiating concurrent p53/Nf1 mutations sporadically in neural stem cells (NSCs). Surprisingly, MADM-based lineage tracing revealed significant aberrant growth prior to malignancy only in oligodendrocyte precursor cells (OPCs), but not in any other NSC-derived lineages or NSCs themselves. Upon tumor formation, phenotypic and transcriptome analyses of tumor cells revealed salient OPC features. Finally, introducing the same p53/Nf1 mutations directly into OPCs consistently led to gliomagenesis. Our findings suggest OPCs as the cell-of-origin in this model even when initial mutations occur in NSCs, and highlight the importance of analyzing pre-malignant stages to identify the cancer cell-of-origin.
INTRODUCTION
Cancer is a disease of genetic mosaicism because cancerous cells harbor genetic mutations that are absent in normal cells within the same individual. In familial cancer patients, even though initial mutations exist in every cell, in most cases only specific cell types can progress into malignancy. Those cell types are called cancer cell-of-origin. Such a cell type-specific susceptibility implies the existence of a permissive or even synergizing signaling context in the cell-of-origin for particular genetic mutations to cause cancer formation. Therefore, identification of the cancer cell-of-origin would provide critical insights for understanding tumorigenic mechanisms and for designing rational therapeutic strategies.
Despite such importance, identification of the cell-of-origin for most cancers has been a daunting task (Visvader, 2011). The reliability of revealing cell identity solely based on molecular and cellular analyses of late stage tumors are often confounded not only by infiltrated bystander cells but also by the acquired plasticity often found in terminal cancerous cells. To circumvent these issues, genetically engineered mouse models have been widely used to determine the tumorigenic potential of a specific cell type by initiating mutations with a cell-type-specific Cre transgene. However, it is critical to note that cells initially acquiring mutations (cell-of-mutation) may not be the cell-of-origin. When mutations are introduced in stem/progenitor cells, it is extremely difficult to distinguish whether initial mutant cells directly transform, or if they merely pass on mutations to more restricted progeny that then transform. In the latter scenario, the mutated stem/progenitor cell is simply the cell-of-mutation while the transforming progeny is the actual cell-of-origin (Visvader, 2011).
The cell-of-origin for malignant glioma, a type of deadly brain cancer, remains controversial. Successful isolation of tumor cells with stem cell features (known as cancer stem cells) from human gliomas (Singh et al., 2004) implies neural stem cells (NSCs) as the cell-of-origin. However, such NSC-like features of malignant glioma cells could be acquired during transformation rather than reflect the nature of the original cell type (Visvader, 2011). Further evidence supporting the NSC-origin of glioma was obtained from mouse genetic studies. For example, the inactivation of tumor suppressor genes (TSGs) p53 and neurofibromatosis 1 (NF1) or the expression of a mutant form of p53 in NSCs consistently led to glioma formation in mouse models; and the physical locations of tumors appeared to associate with the subventricular zone (SVZ), where adult NSCs reside (Alcantara Llaguno et al., 2009; Wang et al., 2009; Zhu et al., 2005). However, other studies suggest that NSC-derived progeny such as astrocytes or oligodendrocyte precursor cells (OPCs) might directly transform (Bachoo et al., 2002; Lindberg et al., 2009; Persson et al., 2010). This unresolved controversy partially stems from the distinct oncogenic mutations used in these models that make the direct comparison difficult, and more importantly from the lack of high-resolution analyses of cellular aberrations during the transforming process.
MADM (Mosaic Analysis with Double Markers), a mouse genetic mosaic system (Zong et al., 2005), could in principle be used to analyze aberrations in individual cell lineages prior to the final transformation, and should thus be suitable for identifying cancer cell-of-origin. Via Cre/loxP-mediated mitotic inter-chromosomal recombination, MADM generates a small number of homozygous mutant cells thus mimicking the sporadic loss-of-heterozygosity (LOH) of TSGs in human cancers (Knudson, 1971). MADM also permanently labels these mutant cells with green fluorescent protein (GFP) and their sibling wild type (WT) cells with red fluorescent protein (RFP) within an otherwise unlabeled heterozygous mouse (Figures 1A and S1A). The single cell resolution benefited from the sparse labeling (0.1–1%, or much lower) (Zong et al., 2005) enables one to track mutant cells throughout the entire process of tumorigenesis. The sibling red WT cells serve as internal controls for green mutant cells, thereby greatly facilitating detailed analyses of cellular aberrations of all lineages in their native environment. MADM can provide features that are indispensable for a robust analytical paradigm to identify a cell-of-origin.
Figure 1. MADM-based glioma model allows phenotypic analysis at single cell resolution.

(A) Scheme of MADM-based glioma modeling. Inset illustrates how MADM concurrently mutates and labels cells (for full details, see Figure S1 and (Hippenmeyer et al., 2010; Zong et al., 2005)).
(B) Representative confocal images show hGFAP-Cre-induced MADM labeling of four NSC-derived cell types in a 2-month old WT-MADM mouse. Arrows point to MADM-labeled cells expressing corresponding markers. Scale bars: 20μm; inset, 5μm.
See also Figure S1.
Here we report the application of MADM to glioma modeling. After initiating p53/NF1 mutations sporadically in neural stem cells (NSCs), we analyzed mutant NSCs and all their progeny at pre-malignant stages. We found significant overexpansion and aberrant growth specifically in OPCs but not NSCs or other lineages. Upon tumor formation, marker staining and transcriptome analysis confirmed the OPC nature of tumor cells. Finally, introducing the same mutations into OPCs consistently led to gliomagenesis. Our findings reveal OPCs as the cell-of-origin for glioma even when initial p53/NF1 mutations occur in NSCs, thus resolving the current controversy by distinguishing cancer cell-of-mutation from cell-of-origin. Importantly, although our studies focused on glioma, the analytical paradigm with MADM developed here could be applied to identifying cellular origins for many other cancers.
RESULTS
Establishment of a MADM-based genetic mosaic model for glioma
MADM-based cancer modeling needs three prerequisites: cancer-causing gene mutation(s), MADM cassettes that reside centromerically to the prospective mutated genes, and a Cre transgene that expresses in a certain tissue or organ. To establish a MADM-based glioma model, we decided to inactivate p53 and NF1, both of which are among the most frequently mutated genes in human glioma patients (McLendon et al., 2008; Parsons et al., 2008) and have been used to model glioma in mice (Reilly et al., 2000; Zhu et al., 2005). We also engineered MADM cassettes into the Hipp11 genomic locus (Figures 1A and S1A, B) proximal to the p53 and NF1chromosomal locations on mouse Chr. 11 (Hippenmeyer et al., 2010). To induce MADM-based recombination, we chose hGFAP- or Nestin-Cre transgenes (termed NSC-Cre hereafter) that are expressed in both embryonic and adult NSCs (Petersen et al., 2002; Zhuo et al., 2001), thereby generating MADM-labeled cells in all NSCs-derived lineages: neurons, astrocytes, oligodendrocytes, and OPCs (Figure 1B). With the ability to identify MADM-labeled cell types, we can readily analyze the aberrant growth of GFP+ mutant cells by directly comparing to their RFP+ WT counterparts within each lineage in the same mouse brain.
MADM-based glioma model offers the opportunity to trace the entire tumorigenic process
After recombining mutant alleles of p53 and NF1 with MADM alleles to generate a mutant-MADM mouse model (Figure S1C), we analyzed overall expansion of green mutant cells at different ages. In contrast to the comparable distribution of green and red cells in WT-MADM brains in which all labeled cells are wild type (Figure 2A, left column), we observed a progressive over-representation of green mutant cells in mutant-MADM brains from postnatal day 5 (P5) to P60 (Figure 2A, top three of the right column), and eventually the formation of GFP+ full-blown tumors at ~5 months of age (Figure 2A, bottom right).
Figure 2. MADM-mediated sporadic concurrent inactivation of p53 and NF1 in embryonic NSCs reveals the entire process of gliomagenesis.

(A) Sagittal sections from brains of MADM mice at indicated ages. In WT-MADM mice (left column), both green and red cells are WT. In mutant-MADM mice (right column), green cells are p53 and NF1 double-null, red cells are WT. Tumor boundary is demarcated with dashed line. Scale bars: 2mm; Insets, 100μm. Ki67 staining shows that tumor cells are highly proliferative.
(B) Systematic quantification of G/R ratios in mutant-MADM brains from P5 to P60. Total cell numbers counted are shown in parenthesis. Each number is the sum from three brains.
(C) The percentage of BrdU+ cells in WT and mutant cell populations in the brain parenchyma of mutant-MADM mice at indicated ages.
(D) The proportion of BrdU+ cells with genotypes “−/−“, “+/−“, and “+/+” in the brain parenchyma of mutant-MADM mice at indicated ages. “+/−“ includes both double-colored and colorless BrdU+ cells. In (C and D) BrdU was administered 1.5 hours prior to sacrifice. Error bars +/− SEM; N = 3 mice. See Experimental procedures for sampling scheme. * P<0.05, ** P<0.01, paired T-test.
See also Figure S2.
Since mutant (green) and wild-type (red) sibling cells originate from the same mother cell in equal numbers initially (Figure 1A), the ratio of green to red cell numbers (referred to as the G/R ratio hereafter) allows us to quantitatively evaluate the extent of mutant cell expansion. A G/R ratio equal or close to 1 indicates no growth advantage of the mutant population, whereas a G/R ratio >1 indicates a growth advantage of mutant over WT cells (referred to as “over-expansion” hereafter). From P5 to P60, the average G/R ratio in mutant-MADM brains increased from ~4 to ~30 (Figure 2B), indicating a continuous over-expansion of mutant cells. However, the increase of G/R ratio appeared to reach a plateau between P30 and P60, suggesting that over-expansion of mutant cells had largely stopped between those ages. In addition to quantifying cell numbers, we also compared the proliferative status between mutant and WT cells with a brief BrdU pulse to label cells undergoing DNA replication. At all time points analyzed, the percentage of BrdU+ cells in the mutant population was significantly higher than that in the WT population (Figure 2C). Furthermore, while WT cells largely ceased to proliferate at early postnatal age (P10), some mutant cells (~ 0.3% among all mutant cells population) continued to divide and contributed to the entire population of BrdU+ cells in the brain parenchyma at P60 (Figure 2D). Thus, mutant cells showed not only elevated but also prolonged proliferative capacity compared to WT cells at a stage before tumor formation (termed the “pre-transforming” stage hereafter).
At later ages (4–5 months), all mutant-MADM mice developed brain tumors with strong Ki67 staining (Figure 2A, bottom right). Tumors were invariably GFP+ (N=28), indicating that they originated from MADM-induced mutant cells. Although transcriptome profiles of these tumors were very similar (see below), pathological analysis revealed great heterogeneity in their appearance: some showed typical astrocytic features; some showed malignant glioma features such as necrosis, multinuclear giant cells, perivascular and perineuronal satellitosis; most were highly anaplastic (Figures S2, 4B–D and 5D). Taken together, MADM allows us to trace mutant cells at all gliomagenic stages and provides the analytical accessibility between initial mutations and the final transformation.
Figure 4. MADM-generated glioma cells exhibit many OPC features.

(A) Representative image of a mutant-MADM brain carrying a GFP+ glioma.
(B–D) Adjacent H&E staining of tumor regions shows typical glioma features, including necrotic areas (“N “ in B), multinucleated giant cells (C), and peri-vascular satellitosis (D). Scale bars: (A), 2mm; (C and D), 200μm. Magnification in (B), 400×.
(E) Representative low magnification images show elevated expression of a panel of well-established glioma markers in tumor regions. All staining was done with adjacent sections from the same tumor. Tumor boundary is demarcated by dashed lines. T, tumor mass. Scale bars: 100μm except for CD9 staining, where it is 50μm.
(F) Quantitative RT-PCRs confirm the over-expression of OPC markers in MADM-generated glioma. (G–L) Confocal images at high magnification show that proliferating (Ki67+) green tumor cells express markers for OPCs (G–I, pointed by arrows) but not for other cell types (J–L). The signals of cell-type marker staining in the right column of (J–L) were converted to red for better examination of their co-localization with GFP. Some Ki67-negative green cells were CC1+ (circled in L). Scale bars: 20μm. (M) Transcriptome comparison between tumor samples and four neuroglial cell types (top four rows), and the four subtypes of human GBMs defined by TCGA (bottom four rows) with the single sample Gene Set Enrichment Analysis (ssGSEA) method. N and O represent tumor samples from mutant-MADM mice induced by NSC-Cre (Nestin-Cre or hGFAP-Cre) and by NG2-Cre, respectively; PDGFRα+ indicates primary tumor cells enriched by anti-PDGFRα immunopanning method. Color key: red to blue indicates significantly similar to dissimilar.
See also Figure S4.
Figure 5. Spatial analyses of early lesions based on perineuronal cytoarchitecture as a landmark implicate that glioma initiate at brain regions away from the SVZ.
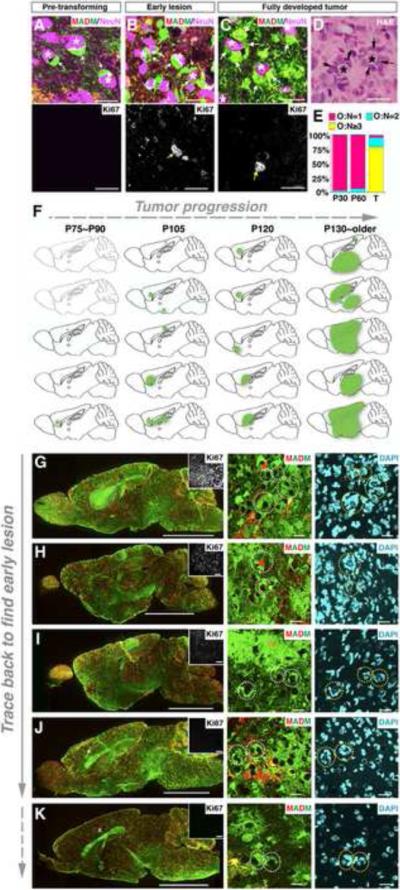
(A–C) Immunofluorescent staining of mutant-MADM brains at distinct tumorigenic stages. Neuronal nuclei were stained with NeuN and are marked as “*”. Arrows point to perineuronal pre-transforming OPCs or tumor cells. The proliferating status of perineuronal mutant cells is shown by Ki67 staining (yellow arrows in the bottom row).
(D) H&E staining of the adjacent section of (C) shows perineuronal satellitosis.
(E) Proportion of perineuronal structures with distinct mutant OPC-to-neuron ratios (O:N) either in pre-transforming MADM mutant brains (P30 and P60, N=3 brains each) or in tumors (T, N=4).
(F) Schematic summary of lesion sites (green spots), which are defined by O:N≥3 perineuronal structure together with MADM labeling, Ki67 staining, and also pathology in most cases. Brains devoid of any detectable lesions are shown in light gray. The analysis is based on a cohort of mutant-MADM mice induced by hGFAP-Cre.
(G–K) Representative brain images from (F) with gliomas from medium to small sizes. Insets show Ki67 staining of the tumor regions. The glioma identity in these brains was confirmed by pathological criteria except for (K), in which the perineuronal structure of ≥ 3 mutant OPCs suggests that it should be a lesion at its early stage.
Scale bars: (A–D), 20μm; (G–K): left column, 2mm; middle and right columns, 50μm.
See also Figure S5.
Mutant OPCs manifest dramatic over-expansion in pre-transforming MADM brains
We next sought to determine the identity of glioma cell-of-origin by separately determining the G/R ratio of NSCs and of each NSC-derived cell types at P60, when mutant cell expansion had largely ceased but tumors were yet to arise. We predicted that, at this ostensibly dormant stage in tumorigenesis, the mutant cell type capable of transformation should manifest significant over-expansion and maintain sustained proliferative activity. We first quantified the G/R ratios of all four NSC-derived cell types (Figure 3A). The average G/R ratio from different brain regions showed slight reduction of mutant neurons and only minor expansion of mutant astrocytes and oligodendrocytes. In stark contrast, the G/R ratio of OPCs was >130, significantly higher than those of the other three cell types (Figure 3A and S3). Notably, mutant OPCs appeared to have impaired differentiation potentials to give rise to mature oligodendrocytes since the G/R ratio of oligodendrocytes was ~10 times less than that of OPCs (Figure 3A). Moreover, brief BrdU pulses at P60 exclusively labeled mutant OPCs in brain parenchyma although the total number was low, suggesting that mutant OPCs were progressing but were still relatively dormant at this age (Figure 3B, left). Remarkably, despite such a dramatic over-representation of mutant OPCs, we did not observe any pathological features in these brains by conventional pathological analysis (Table S2 and data not shown), demonstrating the capability of MADM to probe into a previously inaccessible stage during tumor development.
Figure 3. Analysis at a pre-transforming stage of gliomagenesis suggests that OPCs rather than NSCs serve as the cell-of-origin.

(A) Average G/R ratios of each cell type in brain parenchyma of P60 mutant-MADM mice. * P<0.05, ** P<0.01, paired T-test. “#” P<0.0001, one-way ANOVA.
(B) Left chart: with a single BrdU injection (1.5 hours prior to sacrifice) at P60, BrdU-positive mutant (−/−) cells in the brain parenchyma consist entirely of OPCs (PDGFRα+). Right chart: upon BrdU administration by drinking water for one week, all BrdU-positive mutant (−/−) cells in the brain parenchyma belong to the oligodendrocytic lineage (Olig2+), and the majority of them are OPCs (PDGFRα+).
(C–E) “4+1” channel staining shows that all BrdU+ mutant OPCs in a P60 mutant-MADM mouse brain belong to the oligodendrocyte lineage. (C) Without Olig2 staining, all MADM labeled BrdU+ cells are mutant (green). Notably, some BrdU+ mutant cells are PDGFRα negative (marked with cyan circles).
(D) An adjacent section stained with Olig2 together with MADM, BrdU, and PDGFRα, shows that all mutant BrdU+ cells have red nuclei, indicating positive staining of Olig2. (E) Representative magnified confocal images show: mutant cells without Olig2 staining (left), mutant cells with Olig2 staining in red channel (middle), and heterozygous yellow cells (right). The orthogonal Z-axis is shown on the side of each panel. Scale bars: (C and D): 20μm, insets, 5 μm; (E): 5μm.
(F) G/R ratios of BrdU+ cells in the SVZ and NeuN+ cells in the olfactory bulb (OB) from P60 mutant-MADM mice. BrdU was given in drinking water for 7 days.
(G) Quantification of cells with indicated genotypes among all BrdU+ cells in the non-SVZ brain parenchyma (left chart) or the SVZ (right chart) after one week of BrdU administration. N=3 mice for all quantification in (A, B, F and G). See Figure S3 and Extended experimental procedures for detailed systematic sampling schemes. Error bars represent +/− SEM. Total cell numbers counted in (A, B, F and G) are shown in parenthesis. Each number is the sum from 3 brains.
See also Figure S3.
Mutant OPCs constitute the majority of the proliferation pool in pre-transforming MADM brains
It seems paradoxical that we found nearly full-penetrance of tumor formation in our model at 4–5 months of age (Table S2) while dividing cells at P60 were so rare (Figures 2C and 3B left). Considering that pre-transforming cells could undergo a prolonged cell cycle, we evaluated the slow-dividing cell population by extending the duration of BrdU administration and observed 30% of all mutant OPCs were able to incorporate BrdU (Figure 3B right, and data not shown), suggesting that many mutant OPCs were dividing slowly. We noticed that a small fraction of BrdU-positive mutant cells in the brain parenchyma did not express the OPC marker PDGFRα (Figure 3B right, and 3C). To determine if these were cells from other lineages or oligodendrocytes differentiated from recently divided OPCs, we co-stained brain sections adjacent to those shown in Figure 3C with antibodies against BrdU, PDGFRα Olig2, a “panoligo” marker that expresses in both OPCs and oligodendrocytes. Using a “4+1 channel” staining scheme (Figure 3C–E), we found that all BrdU+, PDGFRα-negative cells examined expressed Olig2 (Figure 3D), suggesting that they were newly differentiated oligodendrocytes from initially BrdU-labeled mutant OPCs. Taken together, our detailed analysis reveals that, among all mutant cell types, OPCs are the only proliferative population in the brain parenchyma at pre-transforming stages.
Although we did not observe the same extent of over-expansion in other mutant cell types as that in mutant OPCs at P60, it could be a result of increased cell death in neuronal and astrocytic lineage following the over-expansion of NSCs. Therefore it is critical to directly analyze the proliferative activity of adult NSCs within the SVZ (Doetsch et al., 1999). After 7 days of BrdU feeding of mutant-MADM mice, we quantified all BrdU+ cells in the SVZ, which include type B, C and A cells ((Doetsch et al., 1999), also see Figure S6A). In stark contrast to our findings in the brain parenchyma, where 85% of BrdU-positive cells were mutant (Figure 3G, left), only ~1% of BrdU-positive cells were mutant in the SVZ (Figure 3G, right). More importantly, the G/R ratio of BrdU+ cells in adult SVZ was not significantly different from 1 (p=0.39), indicating that there was no over-expansion of mutant adult NSCs (Figure 3F). Furthermore, we did not observe any over-expansion of mutant granule neurons in the olfactory bulb, which are the main progeny generated by adult NSCs (Figure 3F). Therefore, p53 and NF1 mutations seemed unable to enhance the proliferative activity of adult NSCs. Taken together, detailed comparison between mutant and WT cell behaviors in NSCs and all progeny lineages strongly suggests that NSCs function as the cell-of-mutation but fail to directly transform, while OPCs function as the cell-of-origin for glioma.
Tumor cells in fully developed gliomas exhibit salient OPC features
Following the investigation at pre-transforming stages, we next analyzed the expression pattern of cell-specific markers in malignant tumors generated in the MADM-glioma model (Figure 4A–D). Under low magnification, we observed prominent enrichment of NSC markers Nestin and Sox2, and the astrocytic marker GFAP, but not neuronal or oligodendrocyte markers in the tumor region (Figure 4E). This observation is consistent with previous findings in both mouse models and human patients (Alcantara Llaguno et al., 2009; Louis et al., 2007; Wang et al., 2009; Zhu et al., 2005) and has been considered as evidence to support the NSC origin of gliomas. However, it is important to note that these tumors also showed enriched expression of OPC markers, such as Olig2, PDGFRα, NG2, CD9 and O4 (Figure 4E and data not shown). The expression of some OPC makers was further confirmed by quantitative RT-PCR from crude tumor samples (Figure 4F).
To clarify whether these markers were expressed by tumor or bystander cells, we analyzed the tumor sections at higher magnification and found that GFP+ proliferating tumor cells expressed many markers for OPCs (Figure 4G–I) but not for neurons or astrocytes (Figure 4J and 4K). In some tumors, there were a few GFP+ cells expressing oligodendrocyte marker CC-1 but not Ki67 (Figure 4L), which could be either residential cells or oligodendrocytes differentiated from tumor cells. To extend the molecular characterization of the cellular identity, we next performed global transcriptome comparison between tumor samples and all four neuroglial cell types (Cahoy et al., 2008) and confirmed that tumor cells closely resembled OPCs but not any other neuroglial cell types (Figure 4M). Importantly, regardless of diverse pathological features (Figure S2), all tumor samples shared almost identical molecular profiles (Figure 4M and data not shown), suggesting a common cell-of-origin for these morphologically heterogeneous tumors.
In line with immunofluorescent staining (Figures S2 and 4E), we observed elevated Nestin level in some but not all tumors by transcriptome and qRT-PCR analyses (data not shown). However, it is unclear whether Nestin is expressed in OPC-like tumor cells or in bystander cells. To clarify this problem, we enriched for OPC-like tumor cells by using an immuno-panning method widely used to purify WT OPCs based on their surface expression of PDGFRα (Figure S4A–C) (Cahoy et al., 2008). The transcriptome of purified OPC-like tumor cells displayed an even higher extent of similarity to that of normal OPCs (Figure 4M), demonstrating that these cells contributed to the OPC-signatures of glioma samples. qRT-PCR results showed that, in addition to consistent expression of OPC markers, purified OPC-like cells from some tumors expressed Nestin at a comparable level to NSC-derived neurospheres (Figure S4D). This finding was further confirmed by marker co-staining of the tumor mass, showing that some GFP+ dividing tumor cells co-expressed both PDGFRα and Nestin (Figure S4E). Importantly, regardless of their Nestin expression levels, these panned-tumor cells, when orthotopically allografted into NOD-SCID mouse brains, effectively initiated secondary tumors (Figure S4F and Table S4) that recapitulated both histological (Figure S4G–K) and molecular features (Figures 4F, 4M, and S4D) of their primary tumors. Taken together, these data demonstrate that highly proliferating tumor cells manifested salient OPC features. Although Nestin was expressed in some tumor cells, the fact that primary tumor cells with either undetectable or high level of Nestin expression could efficiently initiate new tumors implies that Nestin expression appears irrelevant to tumorigenicity and is likely acquired during transformation. Therefore, analyses of tumor cells further support OPCs, rather than NSCs, neurons or astrocytes, as the cell-of-origin for glioma in p53, NF1 mutation-driven glioma model.
Localization of earliest neoplastic lesions suggests a grey matter origin of glioma
The analyses of lesion locations at early stages of transformation could provide great insights for understanding the cancer cell-of-origin. We first analyzed morphological or cyto-architectural changes of mutant cells in fully developed tumors, reasoning that some features could serve as landmarks of transformation, thereby helping determine the locations of early lesions. In all malignant tumors, even in the smallest lesions identifiable by classic pathology, MADM staining revealed a prominent cyto-architectural feature in which multiple glioma cells wrapped around a single neuronal cell body (Figure 5C), a salient pathological feature known as perineuronal satellitosis in human glioma (Figure 5D). It should be noted that normal OPCs are often juxtaposed to neuron cell bodies (Figure S5A), implicating that perineuronal satellitosis in tumors might stem from this unique cyto-architecture of OPCs.
When we compared OPC::neuron association from pre-transforming to tumor-bearing brains, we found that the number of mutant OPCs for each accompanying neuron changes from mostly one (occasionally two) prior to transformation (Figures 5A, 5E and S5B) to frequently three or higher in any Ki67-positive lesions (Figure 5B, 5C and 5E). Therefore, we used “three or more mutant OPCs per neuron” as a landmark to examine a cohort of brains for initiating lesions that cover a period from right after the dormant stage to around the time when tumors can readily be identified (Figure 5F and 5G–5K). In 2.5–3.5-month old mice, we observed the earliest detectable lesions in cortical gray matters (Figure 5F, P75–P90 and top of P105 column). In further developed lesions, tumor cells were more frequently found to overlap with white matter tracks and the SVZ (Figure 5F, bottom of P105, P120 and >P120 columns). This observation indicates that previous findings of glioma in white matter tracks and the SVZ (Persson et al., 2010; Zhu et al., 2005) might be the path or endpoint, rather than the starting point, of tumor cell migration; and further argues against NSCs and for OPCs as the cell of origin.
OPCs can be directly transformed into malignant glioma by p53/NF1 mutations
Our results thus far point toward the following scenario for glioma development in our model: NSCs carrying p53/NF1 mutations give rise to mutant OPCs, which multiply, progress, and eventually transform into malignancies. However, it remains unclear whether mutations must occur in NSCs, or whether OPCs can be directly transformed by the same set of mutations. To address this question, we next used NG2-Cre that has been reported to specifically express in OPCs but not NSCs (Komitova et al., 2009; Zhu et al., 2008) to perform MADM analysis. We first verified that NG2-Cre exclusively labels OPCs and oligodendrocytes but not mature astrocytes or neurons in brain parenchyma of WT-MADM mice (Figure 6A–D). Next we examined the entire SVZ of WT-MADM mice and never found any labeled cells co-expressing GFAP (Figure S6B–C). Lastly, we thoroughly examined the olfactory bulbs (OB) from four WT-MADM mice and did not find any NeuN+ MADM-labeled green or red cells (Figure S6D–E), further supporting the absence of NG2-Cre expression in NSCs.
Figure 6. OPCs can be directly transformed into malignant glioma.
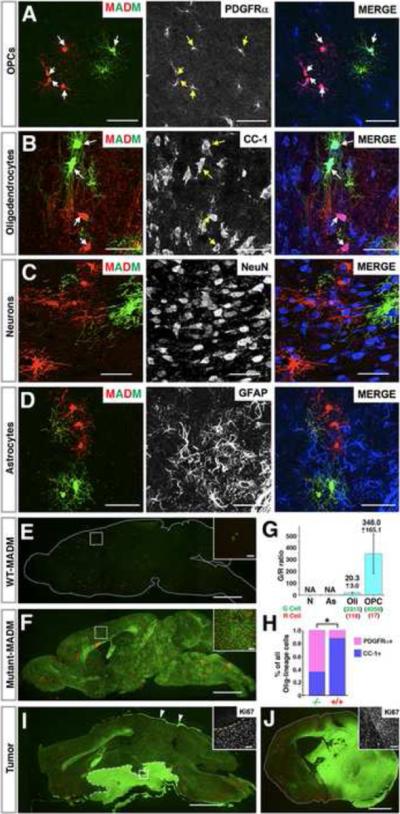
(A–D) In the MADM system NG2-Cre transgene labels OPCs and oligodendrocytes but not astrocytes or neurons. Arrows point to MADM-labeled cells expressing indicated markers.
(E–H) Mutant OPCs over-expand at pre-transforming stages. Brain sections from P60 WT-MADM (E) or mutant-MADM (F) mice induced by NG2-Cre. (G) G/R ratios within each cell lineage in P60 Mutant-MADM brains induced by NG2-Cre. NA: not applicable. Error bars represent +/− SEM. Total cell number being counted are shown in parenthesis. (H) Percentage of OPCs (PDGFRα+) versus oligodendrocytes (CC1+) within mutant and WT cell populations. N=3 mice in (G) and (H). * P<0.05, T-test.
(I and J) Representative gross images of malignant glioma in NG2-Cre induced mutant-MADM mice, either locating around hypothalamus with invasion into the subarachnoid space (I) or residing within the brain parenchyma (J). Arrows in (I) point to tumor cells spreading along the meninges. Ki67 Staining in insets shows that tumor cells are highly proliferative. Scale bars: 2mm; Inset, 100μm.
See also Figure S6.
In stark contrast to the extremely sparse labeling in WT-MADM mouse brains induced by NG2-Cre (Figure 6E), the green mutant OPCs in mutant-MADM mice populated the entire brain (Figure 6F) and reached an average G/R ratio of more than 300 (Figure 6G) at P60. Consistent with findings from the NSC-Cre based MADM model, the differentiation process of mutant OPCs was hampered (Figure 6G–H). These data demonstrate that introduction of NF1 and p53 gene mutations directly into OPCs is sufficient to drive over-proliferation and to impair OPC differentiation.
After 8 months of age, we found GFP+ tumors in brains of NG2-Cre-induced mutant-MADM mice at nearly full penetrance (Figures 6I–J, S7A and Table S3). These tumors showed marker staining and transcriptome profiles indistinguishable from NSC-Cre-induced malignant gliomas (Figures S7C–H, 4F and 4M). Dissociated cells from these tumors could effectively initiate secondary tumors in allografting assays (Figure S7B, S7I–N and Table S4), demonstrating the malignancy of these OPC-initiated tumors.
Despite their indistinguishable marker expression patterns, transcriptome profiles and tumor-initiating ability, we observed that tumors induced by NSC-Cre tended to reside in the brain parenchyma, while those induced by NG2-Cre were more frequently found around the ventral hypothalamic region and often invaded into the subarachnoid space (Figures 6I, 7B, and Tables S3). However, tumors induced by the two Cre lines at equivalent locations showed indistinguishable pathological features (Figures 7A and 7B). Moreover, although tumor cells with subarachnoid invasion were morphologically distinct from those in the brain parenchyma, transplanting them into the brain parenchyma of NOD-SCID mice resulted in secondary tumors morphologically indistinguishable from those primary tumors in the brain parenchyma (Figure 7C). These data strongly suggest that the variation in tumor pathology is merely an effect of tumor location. The differences in latency and spatial distribution between NG2-Cre and NSC-Cre induced tumors most likely are attributed to technical reasons, such as the much lower expression level of NG2-Cre transgene or possible spatial biases of these Cre lines. It is also possible that these two models have distinct non-cell autonomous cues for tumor formation. For instance, NSC-Cre generates heterozygous astrocytes or neurons while NG2-Cre does not, which might function as special niches to facilitate tumor formation and to bias the tumor location. Nevertheless, all lines of evidences, including marker expression patterns (Figures 4F–L and S7), transcriptome profiles (Figure 4M), tumorigenic capacity and pathology (Figures S4, 7 and S7), indicate that tumors from both models are intrinsically identical. Therefore, our data demonstrate that OPCs can be directly transformed into malignant glioma as a result of concurrent p53/NF1 mutations.
Figure 7. Comparative pathological analyses of NSC- and NG2-Cre induced tumors suggest that tumor cell morphology is highly dependent on the location rather than initially mutated cell types.

(A and B) Regardless of the Cre lines used, tumors at the same location exhibit indistinguishable pathological features.
(C) Transplantation of tumor cells from NG2-Cre induced glioma with subarachnoid invasion (top row) into the brain parenchyma of NOD/SCID mice to generate secondary tumors (bottom row). Pathological features of the primary and the secondary tumors mimic tumor features in (B) and (A) respectively. Tumor boundaries are demarcated by dashed lines. The magnification of images in the middle columns is 400×. Images from the right column are 2.5× digital zoom-in of the corresponding middle-column ones.
Finally, we investigated the human relevance of our studies by comparing the transcriptome profile of tumor cells in our model with the molecular signatures of human glioma samples. Among four subtypes of human malignant glioma revealed by recent studies from the Cancer Genome Atlas (TCGA) (Verhaak et al., 2010), both NSC-Cre- and NG2-Cre-initiated glioma models match well with the proneural subtype (Figure 4M), which has the poorest responses to chemo- and radiotherapies (Verhaak et al., 2010). Therapeutic strategies designed according to the intrinsic OPC nature should have promising effectiveness for proneural or even other subtypes of glioma.
DISCUSSION
In this study, we used the mouse genetic mosaic system termed MADM to track the entire tumorigenic process with the aim of identifying the cell-of-origin for glioma when initial concurrent mutations of p53 and NF1 occur in embryonic NSCs. Taking advantage of the in vivo cellular resolution and WT internal control cells afforded by MADM, we analyzed lineage-specific cellular aberrations at pre-malignant stages and observed dramatic over-expansion and elevated proliferative activity in the OPC lineage but minimal abnormalities in NSCs and all other progeny lineages. Furthermore, marker staining, cyto-architecture, and transcriptome analyses all point to OPCs as the transforming cell type for malignant glioma. Finally, direct introduction of p53/NF1 mutations into OPCs led to the formation of glioma indistinguishable from those initiated from NSCs, based on both transcriptome and pathological analyses. Therefore, our studies clarified that, for the glioma model induced by p53 and NF1, while NSCs could serve as the cell-of-mutation, OPCs serve as the actual cell-of-origin.
MADM as a tool to distinguish cancer cell-of-origin and cell-of-mutation
The identification of the cancer cell-of-origin is crucial but often controversial, even with the use of genetic modified mouse models (Visvader, 2011). Conceptually, caution must be taken that cells acquiring initial mutations (cell-of-mutation) may not directly transform into malignancy, because their transforming potential could be manifested by their progeny cell types. For example, when Patched mutation was introduced into NSCs in a mouse model of medulloblastoma, malignant transformation only occurred after NSCs gave rise to lineage-restricted granular neuron precursors (GNPs) (Schuller et al., 2008; Yang et al., 2008). In this scenario, the transforming potential of NSCs (cell-of-mutation) is only manifested by mutant progeny GNPs, the cell-of-origin that possesses the conducive signaling context. Therefore, to unambiguously identify cancer cell-of-origin, one has to genetically dissect the transforming potentials of all progeny derived from the initial mutated cell type. This could be feasible if highly specific Cre transgenic lines are available, such as in the case of medulloblastoma studies (Schuller et al.; Yang et al., 2008). However, such a strategy cannot be generalized to other cancer models due to the lack of specific Cre transgenic mice in every lineage for most tissues. An alternative approach is to analyze aberrant growth in all cell lineages derived from initial mutant cells during pre-transforming stages (Visvader, 2011), which is now possible with MADM-based cancer models.
Our study extends rather than contradicts previous reports. Limited by the cellular resolution at pre-transforming stages, previous studies demonstrated the transforming potential of NSCs, but did not clearly distinguish cell-of-mutation from cell-of-origin (Alcantara Llaguno et al., 2009; Wang et al., 2009; Zhu et al., 2005). MADM, on the other hand, offers a robust analytical paradigm for the identification of the cell-of-origin with both permanently GFP-labeled mutant cells and RFP-labeled WT internal control cells immediately after the initial mutations occurred. In principle, MADM could be used for all types of cancer, as long as oncogenic mutations for a particular type of cancer, a tissue-specific stem cell Cre transgenic line, and cellular markers of each lineage within that tissue are available.
Our studies underline the importance of intersection between genetic mutations and the signaling context within the cell-of-origin. We show here that OPCs are particularly sensitive to p53/NF1 mutations while NSCs and other brain cell types are much less responsive. Interestingly, it has been reported that the NF1 mutation alone can promote OPCs to proliferate in mice and zebrafish (Bennett et al., 2003; Lee et al., 2010), suggesting that NF1 plays a pivotal role in regulating OPC proliferation with conserved mechanisms across species. Such intersections should be further exploited to help understanding of the molecular mechanism of OPC transformation, which should provide critical insights for developing effective therapeutic strategies. Notably, while OPCs appear to be particularly responsive to NF1 mutation, other cell types including NSCs could be specifically vulnerable to other genetic lesions. It would be very interesting to investigate whether different mutations could transform distinct cell-of-origins, which might account for the great heterogeneity of human glioma.
OPC is an important glioma cell-of-origin with under-appreciated proliferative and plastic potentials
One of the notions that support the prevalent view of NSCs as the glioma cell-of-origin is the persistent proliferative activity of NSCs in the brain during the entire life, which makes them susceptible to oncogenic mutations. Additionally, NSCs share many cellular properties with glioma cancer stem cells, such as the capability to self-renew, the potential to differentiate into multiple cell lineages, and the expression of some common cellular markers such as Nestin and Sox2. However, a large body of work has revealed that many of these features are also shared by OPCs. First, OPCs in fact represent the largest proliferative pool in the brain parenchyma for both rodents and human (Dawson et al., 2003; Geha et al., 2010). Second, OPCs at early developmental stages have been found to express many commonly used stem cell markers, including Nestin and Sox2. Third, OPCs isolated from rat optic nerve can be readily reprogrammed into a multipotent NSC-like status under in vitro conditions, which become self-renewable and can differentiate into astrocytes and neurons in addition to oligodendrocytes (Kondo and Raff, 2000). Intriguingly, OPCs from human subcortical white matter intrinsically behave like NSCs without reprogramming (Nunes et al., 2003), raising the possibility that primate OPCs are more plastic than their rodent counterparts. Taken together, the intrinsic nature of OPCs renders them great susceptibility to oncogenic mutations, which could be harnessed to devise effective therapeutic strategies.
Previously, it has been reported that the injection of PDGF-BB-expressing virus into the corpus callosum, the expression of PDGF-BB under CNPase promoter, and the over-expression of v-erbB under human S100β promoter all led to glioma formation with OPC features (Assanah et al., 2006; Lindberg et al., 2009; Persson et al., 2010). Although providing circumstantial evidence, these studies have faced challenges to unequivocally pinpoint OPCs as the cell-of-origin for malignant glioma (Visvader, 2011) due to non-cell-autonomous effects caused by the secretion of PDGF-BB (Assanah et al., 2006; Lindberg et al., 2009) and the use of non-OPC-specific promoter to drive the initial tumorigenic event (Persson et al., 2010). By circumventing all these caveats, our studies now firmly establish that OPCs serve as the cell-of-origin for malignant glioma with relevant genetic mutations. For clinical applications, intrinsic OPC properties should be fully exploited to design effective therapies for glioma, especially for the proneural subtype. For example, understanding the unique proliferative capacity of OPCs could help devise treatments to stall tumor progression; deciphering the migration mechanism of OPCs could enhance the effectiveness of surgery by preventing the infiltration of tumor cells into the entire brain; and probing into the differentiation process of OPCs could facilitate the design of differentiation therapy strategies.
The application of MADM beyond cancer cell-of-origin studies
Compared to conventional models, the MADM-based cancer model bears significant advantages for exploring critical cancer biology problems beyond the cell-of-origin. Its reliable labeling of tumor cells at the single cell resolution should help distinguish bona fide tumor cells from bystander cells in a complex tumor mass, which is highly valuable for dissecting tumor architecture, tracking metastasized tumor cells, and studying tumor-niche interactions. Importantly, the availability of a built-in internal control enables quantitative comparison between mutant and WT sibling cells. Such rigorous phenotypic analyses within the same animal largely remove variations often introduced by comparing phenotypes between individual animals, thereby helping identify subtle but important phenotypes that could be easily missed by using conventional approaches.
Using Drosophila genetic mosaic system, combined with the application of positive marking with gene manipulation at the mosaic level helped elucidate many fundamental questions in cancer biology, such as identifying important oncogenes and TSGs by forward genetic screening (Potter et al., 2001; Tapon et al., 2001) and deciphering the complicated tumorigenic processes such as metastasis (Pagliarini and Xu, 2003). Along this same principle, we expect that a further modified MADM system could address even more sophisticated problems in cancer biology beyond the capability of the current one. For instance, by replacing fluorescent reporters with a transcription factor such as tTA or rtTA to specifically manipulate gene expression in mosaic mutant cells, one could model “second-hit” events or therapeutic strategies and gain deep insights into the molecular network for cancer formation. By incorporating novel magnetic resonance imaging reporters such as H-ferritin (Genove et al., 2005) into MADM cassettes, one could non-invasively trace tumor initiation, progression and metastasis in real time. Also by combining with transposon-based mutagenesis such as Sleeping Beauty (Collier et al., 2005) or PiggyBac (Ding et al., 2005), the MADM system should be able to perform forward genetic screening for novel recessive TSGs in mouse somatic cells without involving large number of animals.
Concluding remarks
In summary, our study demonstrates the importance of analyzing the entire process of tumor development for identifying cancer cell-of-origin. MADM-based tumor modeling can be applied in principle to any other tumor type, and should help resolve many important problems in cancer biology. Broadly, the ability to perform sporadic single-cell genetic manipulations in unambiguously labeled cells should make MADM an invaluable analytical tool for fields such as developmental biology and neuroscience that rely heavily on in vivo analyses.
EXPERIMENTAL PROCEDURES
Mouse lines and genotyping methods
All animal procedures were based on animal care guidelines approved by the Institutional Animal Care and Use Committee. Mouse lines and genotyping methods used in this study can be found in Extend Experimental procedures.
Tissue preparation and histology
After anesthesia mice were perfused with 4% ice-cold paraformaldehyde (PFA) following the standard procedure. Brains were isolated, post-fixed (overnight at 4°C), cryoprotected in 30% sucrose (overnight at 4°C) and embedded into optimal cutting temperature (O.C.T.) prior to cryo-sectioning with a cryostat.
Imaging
All confocal images were collected by an Olympus FV-1000 upright laser confocal microscope and analyzed with Fluoview 1000 software. Adobe Photoshop CS3 was used for image processing.
Immunohistochemistry
Immunohistochemistry was performed using standard methodology. Details about antibodies and working solutions can be found in Extend Experimental procedures and Table S1.
Quantification
All quantification described in this work was performed based on the systematic sampling scheme described in Extend Experimental procedures and Figure S3.
Purification of WT OPC and OPC-like glioma cells
WT OPCs from P8 cortical caps, or OPC-like tumor cells from fresh glioma samples, were purified as previously described (Cahoy et al., 2008) with minor modification. Detailed procedures can be found in Extend Experimental procedures and Figure S4.
Tumor cell grafting
Tumor cells were suspended into Neurobasal medium with a density of 50,000 viable cells /μl. 2–3 μl of tumor cell suspension was injected into right striata of NOD-SCID mice (JAX laboratory) as previously described (Ligon et al., 2007). Mice were monitored daily and sacrificed at the onset of neurological symptoms or once moribund, or based on designed time-point cohort.
Microarray and gene expression analysis
44K Mouse Development Oligo Microarrays (Agilent Technologies) were used for microarray analysis. Transcriptome comparison of tumor samples with WT neuroglia cell types (Cahoy et al., 2008) or human GBM subtypes was preformed as previously described (Verhaak et al., 2010). Array data are available at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE26676. Detailed procedures are described in Extend Experimental procedures.
Supplementary Material
Highlights.
Modeling of human cancers using MADM (Mosaic Analysis with Double Markers)
Probing into pre-transforming stages for lineage-specific aberrant growth
Oligodendrocyte precursor cells (OPCs) as a cell-of-origin for glioma
Conceptual distinction between cancer cell-of-mutation and cell-of-origin
ACKNOLEDEGEMENT
We thank A. Henner and M. Valle for technical support, J. Dugas for his help for the immunopanning method, and B. Bowerman, C. Doe, R. Galvao, W. Hong, M. Kohwi, M. Raff, R. Read, B. Tasic, X. Wu, and Y. Zhu for critical comments on the manuscript. This work is supported by NIH grants (R55-CA136495, R01-CA136495). M.R.M was supported by NSF pre-doctoral fellowship. S.H. was supported by postdoctoral fellowships from the European Molecular Biology Organization (ALTF 851-2005), Human Frontier Science Program Organization (LT00805/2006-L) and Swiss National Science Foundation (PA00P3_124160). H.Z. is a Pew Scholar in Biomedical Sciences, supported by The Pew Charitable Trusts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006;26:6781–6790. doi: 10.1523/JNEUROSCI.0514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Rizvi TA, Karyala S, McKinnon RD, Ratner N. Aberrant growth and differentiation of oligodendrocyte progenitors in neurofibromatosis type 1 mutants. J Neurosci. 2003;23:7207–7217. doi: 10.1523/JNEUROSCI.23-18-07207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, Chneiweiss H, Daumas-Duport C, Varlet P. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol. 2010;20:399–411. doi: 10.1111/j.1750-3639.2009.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genove G, DeMarco U, Xu H, Goins WF, Ahrens ET. A new transgene reporter for in vivo magnetic resonance imaging. Nat Med. 2005;11:450–454. doi: 10.1038/nm1208. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Youn YH, Moon HM, Miyamichi K, Zong H, Wynshaw-Boris A, Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komitova M, Zhu X, Serwanski DR, Nishiyama A. NG2 cells are distinct from neurogenic cells in the postnatal mouse subventricular zone. J Comp Neurol. 2009;512:702–716. doi: 10.1002/cne.21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science. 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- Lee JS, Padmanabhan A, Shin J, Zhu S, Guo F, Kanki JP, Epstein JA, Look AT. Oligodendrocyte progenitor cell numbers and migration are regulated by the zebrafish orthologs of the NF1 tumor suppressor gene. Hum Mol Genet. 2010;19:4643–4653. doi: 10.1093/hmg/ddq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266–2275. doi: 10.1038/onc.2009.76. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4 edn International Agency for Research on Cancer (IARC); 2007. [Google Scholar]
- McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis M, Olson JJ, Mikkelsen T, Lehman N, Aldape K, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–1231. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18:669–682. doi: 10.1016/j.ccr.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, Lee EY, Zhu Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15:514–526. doi: 10.1016/j.ccr.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.