

Abstract
Emerging evidence has shown that oxidation of RNA including mRNA is elevated in several age-related diseases, although investigation of oxidized levels of individual RNA species has been limited. Recently we reported that an Aldehyde Reactive Probe (ARP) quantitatively reacts with oxidatively modified depurinated/depyrimidinated (abasic) RNA. Here we report a novel method to isolate oxidized RNA using ARP and streptavidin-beads. An oligo RNA containing abasic sites which were derivatized with ARP was pulled down by streptavidin-beads, whereas a control oligo RNA was not. In vitro oxidized RNA as well as total cellular RNA isolated from oxidatively stressed cells was also pulled down dependent on oxidation level, and concentrated in the pull-down fraction. Quantitative RT-PCR using RNA in the pull-down fraction demonstrated that several gene transcripts were uniquely increased in the fraction by oxidative stress. Thus, our method selectively concentrates oxidized RNA by pull-down and enables the assessment of oxidation levels of individual RNA species.
Keywords: ARP, RNA oxidation, streptavidin, pull down, abasic site
Introduction
Oxidation of RNA has been shown to occur with increasing age in muscle [1] and brain [2], and in several age-related diseases including atherosclerosis [3], dementia with Lewy bodies [4], Parkinson’s disease [5], Amyotrophic lateral sclerosis [6], and Alzheimer’s disease [7-8]. RNA oxidation is also elevated in mild cognitively impaired individuals who may be predisposed to Alzheimer’s disease, suggesting that oxidized RNA is not merely a byproduct of the pathological milieu, but one of the primary causes of disease pathogenesis [9-11].
Despite our understanding of the potential importance of RNA oxidation, there have been few studies that assess oxidative damage to a specific RNA sequence. Rhee et al. reported that the oxidative damage sites in RNA could be detected by primer extension termination of reverse transcription [12]. Furthermore, it was found that after generation of oxidative lesions in RNA by photosensitization with several dyes, reverse transcription tended to be terminated on purines rather than pyrimidines. In addition, a method for quantifying RNA oxidative damage has been reported using RT-PCR based on the fact that full-length cDNA synthesis is hampered by oxidative damage to RNA [13]. In the latter study, the incomplete cDNA production rate was assessed by differential RT-PCR with two primer sets corresponding to the 3’-region or 5’-region. As a result, RT-PCR amplification of the 5’-region was relatively decreased by RNA oxidation. Thus, these two methods are based on inhibition of reverse transcription mediated by oxidative damage. Although the detection limit is reportedly very sensitive, these techniques are not appropriate for comparing the extent of damage among different RNA transcripts.
Comprehensive analyses of mRNA oxidation were originally reported using an antibody against 8-hydroxyguanosine (8-OHGuo). Oxidized mRNA isolated from Alzheimer’s disease patients [8, 14] or Familial Amyotrophic Lateral Sclerosis model mice (B6SJL-Tg[SOD1-G93A] 1Gur) [6] was selectively immunoprecipitated and highly oxidized mRNAs were identified. It was reported that some of oxidized mRNAs were transcripts implicated in neurodegenerative diseases, although it remained unclear whether these changes were the result of the disease process itself or the result of technical artifacts. Therefore, it is critical to develop alternative approaches to assess oxidative damage to individual RNAs.
Depurination/depyrimidination has been known to be a major oxidative modification of DNA, which is induced chemically by DNA damaging or oxidizing agents such as alkylating agents or ionizing radiation [15-16]. As a result, a sugar moiety without its corresponding base is generated and termed an abasic site. Recently we found that an Aldehyde Reactive Probe (ARP), which has been developed for assay of DNA abasic sites [17] also reacts with RNA containing abasic sites as well. ARP reactivity was quantitatively increased in RNA after several different types of in vitro oxidations by reactive oxygen species produced by the Fenton reaction, γ-radiation, or reactive nitrogen species using peroxynitrite. Moreover, ARP reactivity was also increased in cellular RNA under oxidative stress conditions by hydrogen peroxide or peroxynitrite. Thus, RNA abasic sites were confirmed to be a sensitive RNA oxidation marker [18].
In this study, we developed a method to isolate oxidized RNA using ARP and streptavidin-magnetic beads. We show that synthetic oligo RNA containing abasic sites followed by derivatization with ARP is pulled down by streptavidin-magnetic beads. Taking advantage of both chemical and thermal stability of streptavidin, highly specific binding of ARP-derivatized RNA has been achieved. Oxidized RNA is selectively concentrated by this method since the pull-down fraction is enriched with abasic sites. Furthermore, we use this method to quantify oxidation level of individual mRNA species using qRT-PCR.
Materials and Methods
Cell culture and reagents
All buffer solutions for RNA synthesis, isolation and oxidation were pretreated with chelex 100 (BioRad, Hercules CA) or contained 1 mM of EDTA. All the reagents used were purchased from Sigma-Aldrich unless specified. HeLa cells and Neuro 2a cells were purchased from ATCC, and maintained in DMEM plus 10% fetal bovine serum (Clonetech, Mountain View CA) in 5% CO2 at 37°C. For oxidative stress, the cells under subconfluent conditions were treated with hydrogen peroxide with indicated concentrations for 30 min.
Preparation of RNA
In vitro synthesis and oxidation of RNA were carried out according to our previous report [19]. Briefly, RNA was synthesized using T7 RNA polymerase (Ambion, Austin TX) using a DNA template encoding luciferase2 (Promega, Madison WI), in the presence of 50μCi of 32P-uridine triphosphate (PerkinElmer, Waltham MA). RNA was treated with DNase I, followed by purification using a QIAGEN RNeasy kit (QIAGEN, Valencia CA). Two hundred fifty (250) μg/ml RNA was reacted with the same amount of hydrogen peroxide and a 1:1 Fe(II)-ascorbate mixture in 10 mM HEPES buffer (pH7.4) at 37 °C for 30 min. The metal catalyzed oxidation was terminated by mixing with 10mM of EDTA, then oxidized RNA was precipitated with ethanol at -20 °C. Total cellular RNA derived from cultured cells was isolated with TRIzol (Invitrogen, Carlsbad CA) according to the manufacturer’s protocol except for homogenization in the presence of 10mM deferroxiamine to efficiently protect from oxidation [20]. The final RNA preparation was suspended in 5mM Tris-HCl (pH7.5), 1mM EDTA (TE buffer). Synthetic oligoribonucleotides, 5’-(6-FAM)AGUUCCACGGUAACGCUUAGUC-3’ and 5’-(6-FAM)AGUUCXACGGUAACGCXUAGUC-3’ (X: abasic site) were prepared according to the previous report [18].
Pull down fractionation
ARP derivatization
Two hundred (200) μg/ml RNA was incubated with 2mM of N’-aminooxymethylcarbonylhydrazino D-biotin (Dojindo, Rockville MD) in 50mM sodium acetate (pH5.0) buffer for 1h at 37°C. The reaction was terminated by mixing with 5mM formaldehyde. ARP derivatized RNA was precipitated by mixing with 0.1 volume 3M sodium acetate and 3 volumes of ethanol at -20°C for 1h, followed by washing (2×) with chilled 75% ethanol, then resuspended with TE buffer. The RNA was precipitated with ethanol once again. These repeated precipitation steps allow complete elimination of residual ARP from the RNA suspension.
Reaction with streptavidin-beads
A T1 streptavidin-magnetic bead suspension (Invitrogen) was washed (2×) with 2 volumes of 0.1N NaOH, 0.05M NaCl, then 2 volumes of 0.1M NaCl (2×) using a magnetic stand. The beads were suspended with Solution A (1M NaCl, 20mM Tris-HCl (pH7.5), 5mM EDTA, 1% NP-40) and incubated with 0.5mg/ml of E. coli transfer RNA (Sigma-Aldrich) for 30min at room temperature. After incubation, the beads were mixed with ARP-derivatized RNA at a final concentration of 50μg/ml at 70°C for 20min with mild vortexing. The supernatant fraction was carefully removed using a magnetic stand and precipitated with ethanol as described above. The beads were washed (2×) with 20 volumes of Solution A, then washed (3×) with 20 volumes of solution B (2mM Tris-HCl adjusted pH7.5, 0.5mM EDTA, 1% NP-40), washed (3×) with 20 volumes of Solution C (4M urea, 10mM Tris-HCl adjusted pH7.5, 1mM EDTA, 1% NP-40), then (2×) with Solution D (2mM Tris-HCl adjusted pH7.5, 1mM EDTA) at room temperature. After discarding solution D completely, the beads were suspended with 2 volumes of biotin solution (2.5mM biotin, 1 mM EDTA at pH 7.0 adjusted with Tris base), and heated to 95°C for 4min. The suspension was carefully transferred to a new tube and the pellet was suspended with the same volume of solution D. Both supernatants of the pull-down fraction were combined and precipitated at -20°C with ethanol and 2μl glycogen (Ambion) as described above. The RNA precipitate was rinsed (2×) with cold 75% ethanol, then resuspended with TE buffer. For RNA quantification, absorbance of the suspension at 260nm was measured using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington DE). Alternatively, an aliquot of the radioactive RNA suspension was spotted onto a Hybond N+ positive charged membrane (GE Healthcare, Piscataway, NJ), cross-linked with 120 mJ/cm2, followed by an overnight exposure and analyzed using a Storm 840, PhosphorImager (GE Healthcare).
cDNA synthesis and realtime RT-PCR
Five hundred (500) ng to 1μg ARP-RNA in the pull-down fraction was subjected to reverse transcription with 1μl SuperScript III reverse transcriptase (Invitrogen), 1μl RNaseOUT recombinant ribonuclease inhibitor (Invitrogen) 0.1mM random hexamer or 25ng/μl oligo(dT)12-18 primer (Invitrogen), and 0.5mM dNTPs at 50°C for 1h, followed by 70°C for 15min according to the manufacture’s protocol.
For labeling cDNA derived from poly(A) RNA, 0.1 mM of oligo dT20 conjugated with 3’-triethyleneglycol biotin (Eurofins MWG Operon, Des Moines IA) instead of the oligo(dT) primer was used for the reverse transcription reaction. The cDNA was incubated with 1μl of RNaseH (Invitrogen) and 1μl of RNase cocktail (Ambion, Austin TX) containing RNaseA and RNaseT1 at 37°C for 30min to digest ARP-RNA, followed by ethanol precipitation. Precipitated cDNA was resuspended with formaldehyde sample loading buffer (Ambion), heat denatured at 65°C for 5 min, and loaded onto an agarose gel for electrophoresis in 0.5×TAE buffer. cDNA was transferred onto a positive charged membrane (Whatman, Piscataway NJ). Detection of the biotin-labeled cDNA was followed by ARP assay as described in the previous report [18].
For quantitative RT-PCR, 0.5μl of the cDNA reaction mixture was mixed with Power SYBR Green master mix (AppliedBiosystems, Calrsbad CA) together with 250nM of primers for mouse β-actin or eukaryotic elongation factor 2 (eEF2) which were designed using ABI Primer Express software (Table 1) according to the manufacturer’s protocol. Real-time PCR was performed using a 7900HT sequence detection system (ABI) with 95°C for 10min, followed by 40 cycles of 95°C×15sec and 60°C × 1min. To assess the amplification rate, standard curves for each primer set were performed using serially diluted cDNA.
Table 1.
PCR primers
| gene | primer location | forward | reverse |
|---|---|---|---|
| β-actin | 31 | GCTTCTTTGCAGCTCCTTCGT | GCGCAGCGATATCGTCATC |
| 426 | CGTGAAAAGATGACCCAGATCA | CACAGCCTGGATGGCTACGT | |
| 1035 | GCTCTGGCTCCTAGCACCAT | GCCACCGATCCACACAGAGT | |
|
| |||
| eEF2 | 69 | CCATCCGCCACCATGGT | CCGGATGTTGGCTTTCTTGT |
| 1670 | TGACCCTATGGTGCAGTGCAT | ACCGGCGCCAGCAATAA | |
| 2441 | CCTGCCTGTCAATGAGTCCTTT | CTGGCCGCCGGTGTT | |
Data analyses
Real-time PCR data were processed for quantification using the SDS software 2.0 and RQ Manager software (Applied Biosystems). Relative quantity of the RNA was based on the Ct values. The mean and standard deviation values were calculated by Excel or SigmaPlot software.
Results
Pull-down of abasic RNA
In our previous report, we found that RNA derivatized with ARP (ARP-RNA) increased dose-dependently after oxidation of RNA. These results, using a chemilluminescent assay and streptavidin conjugated with horseradish peroxidase [18], suggested that ARP-RNA could be reliably separated by streptavidin-magnetic beads (strep-beads). To examine this further, RNA oligonucleotides containing abasic sites were derivatized with ARP. Different amounts of abasic ARP-RNA were incubated with strep-beads followed by washing buffer according to the manufacturer’s protocol (Figure 1). Abasic ARP-RNA was pulled down more than 98% when RNA levels were below the binding capacity of the strep-beads. The estimated capacity of strep-beads binding to ARP-RNA was consistent with the manufacturer’s description. The pull-down recovery of control derivitized RNA was low (less than 17%). These results indicate that abasic ARP-RNA is separated and quantitatively pulled down by strep-beads.
Figure 1.
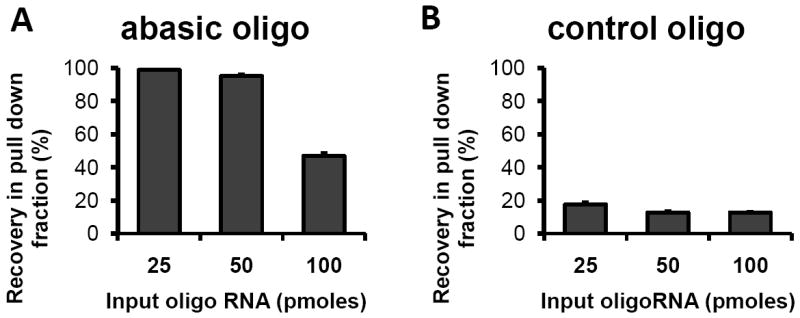
Abasic RNA is selectively pulled down by strep-beads. Fluorescent oligo RNAs with (A) or without abasic sites (B) were derivatized with ARP. Various amounts of ARP-RNA were incubated with 0.1mg of strep-beads for 15min at room temperature. After repeated washing with 1M NaCl buffer salt, the beads were heated at 65°C in formamide solution to dissociate ARP-RNA. The dissociated ARP-RNA was quantified based on fluorescence.
Assessment of pull-down conditions
The experiment in Figure 1 was conducted according to the manufacturer’s protocol, however we modified the protocol for messenger RNA using luciferase encoded RNA (1.8kb). To reduce non-specific binding, strep-beads were washed with a buffer containing a high concentration of urea (Figure 2A). RNA without ARP derivatization was effectively eliminated in the pull-down fraction, whereas oxidized ARP-RNA was only slightly decreased (95%). This result indicated that 4M urea effectively reduced non-specific binding. The reaction between ARP-RNA and strep-beads was tested at 70°C to disrupt RNA secondary structure since streptavidin is known as a protein with extreme thermal stability [21]. Recovery of oxidized ARP-RNA at 70°C was more than 2-fold compared to that at 37 °C incubation, whereas oxidized RNA without ARP deivatization was not pulled down in either condition (Figure 2B). Thus, reaction at high temperature was effective for isolation of ARP-RNA.
Figure 2.
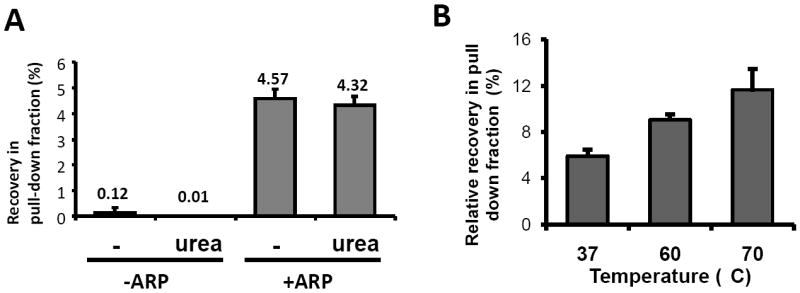
Effects of washing and reaction temperature. A. In vitro oxidized or non-oxidized radiolabeled RNA (1.8 kb) with or without derivatization, was incubated with strep-beads at 37°C for 15min. The beads were washed with several buffer solutions including 4M urea, and then heat denatured to dissociate the bound RNA. Radioactivity in the pull-down fraction as well as initial input RNA was quantified using a PhosphorImager. B. In vitro oxidized radiolabeled ARP-RNA was incubated with strep-beads at different temperatures for 30 min. The beads were washed several times with salt buffers including 4M urea buffer solution and heat denatured. Radioactivity in the pull-down fraction and initial input RNA was determined. The results are represented as means ± SD from triplicate samples.
Assessment using in vitro oxidized RNA
We investigated the characteristics of the pull down experiment using in vitro oxidized 32P-labeled RNA. The ARP reactivity of input RNA oxidized by Fe(II)/ascorbate/H2O2 increased dose dependently (Figure 3A). Furthermore, the ARP-streptavidin pull-down yield of the in vitro oxidized RNA was also found to increase dose-dependently and corresponded to its ARP reactivity, which indicated that oxidized RNA was quantitatively pulled down by the method (Figure 3B). To obtain quantitative recovery, it was necessary to eliminate unreacted ARP completely by repeated ethanol precipitation or spin column purification after derivatization with ARP. When ARP reactivity in the pull-down fraction was assayed, it was found that specific ARP reactivity was concentrated by approximately 3 fold compared to the starting RNA material and greater than 10-fold compared to the supernatant fraction (Figure 3C).
Figure 3.

Assessment of pull-down using in vitro oxidized RNA. In vitro synthesized radiolabeled RNA (1.8 kb) was oxidized in vitro in the presence of the same amounts of H2O2 and Fe(II)-ascorbate (0, 2, or 5 μM) at 37 °C for 30 min. Oxidation of the RNA was examined using ARP reactivity (A). Then, the oxidized RNA was subjected to pull-down isolation using strep-beads. The total amount of recovery in the pull-down fraction is shown in (B). RNAs in all fractions (including the supernatant fraction) were assayed using ARP reactivity using a constant amount of RNA. Panel C shows the relative specific ARP reactivity in each fraction. The results are means±SD from tests in triplicate.
Assessment using cellular RNA under oxidative stress
Total cellular RNA was isolated from HeLa cells treated with H2O2 for 30min. As expected, ARP reactivity of total RNA increased dose-dependently based on the oxidative stress conditions (Figure 4A). The amount of total RNA recovered in the pull-down fraction was also increased after oxidative stress treatment, although the increment was moderate compared to ARP reactivity of the initial RNA (Figure 4B). We assessed the recovery of poly(A) RNA in the pull-down fraction by cDNA synthesis using an oligo(dT) primer conjugated with biotin at the 3’-end. RNA in the pull-down fraction was reverse transcribed to synthesize cDNA, followed by treatment with a mixture of RNaseH, RNAseA, and RNaseT1 to digest ARP-RNA. Aliquots of cDNA from the pull-down fraction were blotted, and the membrane was developed by streptavidin-HRP and chemilluminescent reagent (Figure 4C). The intensity of cDNA was increased in the pull-down fraction derived from oxidative stressed cells, indicating that poly(A) RNA was also increasingly oxidized in response to oxidative stress. In addition, the cDNA size distributions were found to be quite similar to that of total RNA (Figure 4C far left lane). These results suggest that cDNA synthesis of poly(A) RNA in the pull-down fraction was not inhibited by derivatization with ARP.
Figure 4.
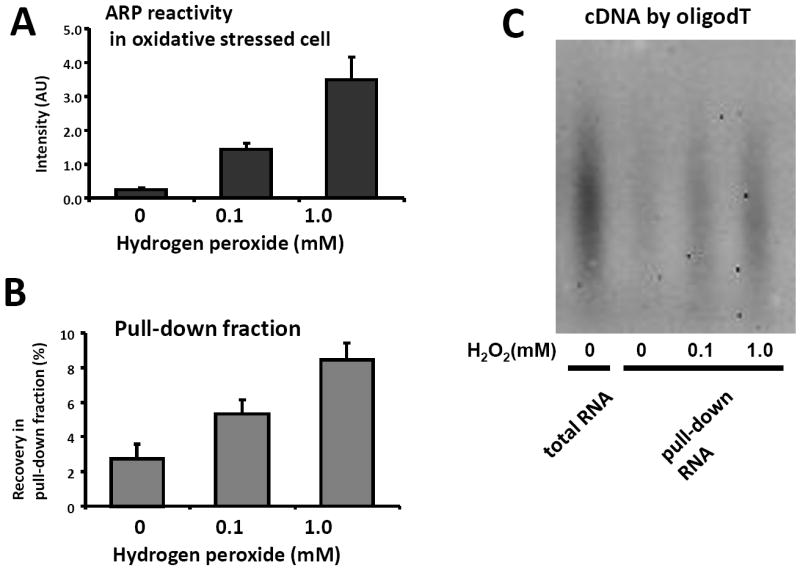
Assessment of pull-down using total cellular RNA. Total cellular RNA was isolated from HeLa cells treated with 0.1mM or 1mM H2O2 for 30min. The isolated RNA was examined using ARP reactivity (A) and RNA pulled-down using strep-beads. The yield of RNA in the pull-down fraction was determined by absorbance at 260nm after the fraction was concentrated by ethanol precipitation (B). The same amount of RNA in the pull down fraction was reverse-transcribed in the presence of oligo dT primers conjugated with biotin to synthesize cDNA derived from poly(A) RNA. After rigorous RNase digestion of ARP-RNA, the cDNA was blotted on membranes and detected using strepavidin-HRP and chemilluminescence (C).
Assessment of RNA oxidation levels using quantitative RT-PCR
We assessed whether individual mRNA derivatized with ARP could be accurately quantified by qRT-PCR based on the location of the PCR primers. Total RNA was isolated from Neuro 2a cells treated with H2O2 for 30 min, and pulled down after derivatization with ARP. Complementary DNA derived from the ARP-RNA was synthesized using either oligo(dT) primers or random hexamers. These cDNAs were used for real-time PCR with three sets of PCR primers for the β-actin gene (Figure 5A) or the eEF2 gene (Figure 5B) to assess relative RNA quantities (RQs). The RQ values of oligo(dT) primed cDNA or random primed cDNA were decreased as the primer location was positioned farther from the poly(A) region. This was particularly evident for oligo(dT) primed cDNAs. However, the decreasing rate toward 5’-end was comparable between the control cell group and the oxidized cell group in each panel. Moreover, the decrement of RQ values using 5’-end probes was comparable among the pull-down RNA fractions. These results suggest that incomplete termination of reverse transcription was not primarily due to oxidative damage or ARP-derivatization under the oxidation conditions tested.
Figure 5.
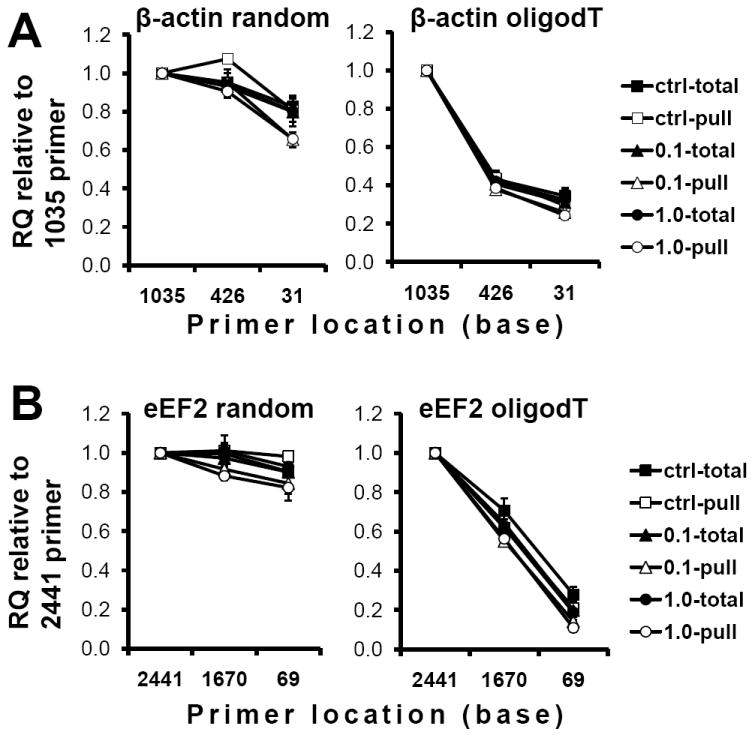
Quantitative RT-PCR for total and pull-down RNA fraction. Total cellular RNA was isolated from Neuro-2a cells treated with 0.1mM or 1mM H2O2 for 30min. ARP-RNA (1μg of total ARP-derivatized RNA) was reverse transcribed with random hexamers (left panels) or oligo dT primers (right panels). The cDNA was subjected to real time PCR using three different sets of primers specific for β-actin (A) or eEF2 (B). Based on the Ct value, quantities of the gene transcripts were determined. X-axis is the 5’position of the forward primer. The graphs are represented as relative quantity compared to the 1035 primer (A) or 2441 primer (B), respectively and expressed as means±SD from tests in triplicate.
Using the RNA quantities from Figure 5, the oxidized level of the gene was calculated as the ratio of RQ in the pull-down RNA to RQ in total RNA. Figures 6A and B demonstrate oxidized levels of β-actin mRNA and eEF2 mRNA in three different oxidative stress conditions, respectively. As expected, the oxidation level of cells treated with H2O2 was higher than that in control cells, which is the case using either random primed cDNA (left panels) or oligo(dT) primed cDNA (right panels). Thus, the calculated oxidation levels of the mRNA using ARP pull-down isolation coupled with qRT-PCR remained relatively constant using oligo(dT) primed cDNA or random primed cDNA regardless of the PCR primer location, even though the RQ values varied substantially depending on locations of PCR primers.
Figure 6.
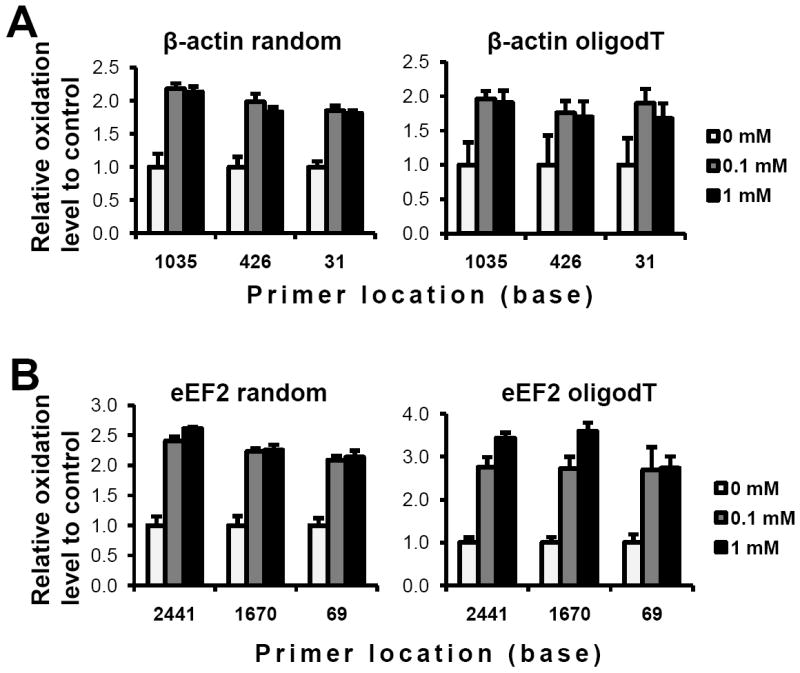
Oxidation levels of mRNA. Based on qRT-PCR data, the ratio of RNA quantities in the pull-down fraction compared to total RNA quantities was determined in response to three different oxidation conditions. The graphs are represented as relative ratio to control condition in each group using the same primers in each category: β-actin (A) or eEF2 (B) based on random primed cDNA (left panels) or oligo dT primed cDNA (right panels).
Discussion
There have been a number of microarray and proteomic studies to investigate expression changes in RNA transcripts and proteins under oxidative stress conditions. Evidence has emerged that the expression of mRNA and protein are not always related primarily due to post-transcriptional regulation. For example, translational regulation by RNA binding proteins or microRNAs is thought to be required for optimal gene expression in response to various internal or external stimuli [22-23]. Alternatively, it is also possible that protein synthesis is adversely modulated due to oxidative damage of mRNA or RNA that constitutes part of the protein synthetic machinery (e.g. rRNA, tRNA). Our group and others previously demonstrated that oxidation of mRNA reduces protein production [19, 24]. Furthermore, moderately oxidized mRNA generated aberrant polypeptides including prematurely terminated peptides due to translational errors. These findings suggest that oxidative modifications to mRNA affect translational activity and fidelity under pathological disease conditions.
Recent reports using immunoprecipitation after treatment of mRNA with an antibody against 8-OH-Guo have indicated selective oxidation of mRNA in brains from Alzheimer’s patients [8, 14] or FALS mice [6]. Remarkably, several gene transcripts associated with age-related diseases, including APP, SOD1, and presenilin appeared to be highly oxidized. Therefore, it is important to confirm these seminal findings, since oxidative dysfunction of those selective mRNAs may be directly involved in disease pathogenesis. However, the immunoprecipitation method using the 8-OHGuo antibody does not appear appropriate for quantitative analysis due to technical limitations. This may be the result of low binding capacity of the antibody and small yields after immunoprecipitation. To address this concern, we needed to use a small quantity of radiolabeled RNA or purified poly(A)RNA instead of using total RNA (unpublished data). Unfortunately poly(A)RNA tends to result in a high background since the molecule is susceptible to oxidation during purification and immunoprecipitation due to the presence of dissolved oxygen and trace amounts of metal ions [19]. To overcome these potential issues, we developed a method to isolate oxidized RNA quantitatively from the total cellular RNA pool using ARP and streptavidin-beads. Pull down experiments showed efficient recovery of abasic oligo RNA but not of control oligo RNA (Figure 1). Taking advantage of the extreme thermo- and chemical-stability of streptavidin/biotin interaction, reduced non-specific binding and increased recovery of oxidized RNA were achieved.
Our results indicate that the amount of pulled down RNA from cell culture was increased by oxidative stress, however the increment of the recovery was not directly proportional to ARP reactivity. There could be several possible reasons, but it is most likely that the RNA was subject to oxidation in multiple sites following H2O2 treatment. We found that reverse transcription with oligo(dT) in the pull-down fraction was not inhibited by oxidative damage despite derivatization with ARP. Although it is unknown how the enzyme crosses over the ARP-derivatized sites, it has been reported that modified ribonucleotides with bulky fluorescent dyes do not result in inhibition of reverse transcription [25]. Although we previously reported that RNA abasic sites inhibited reverse transcription extension using some reverse transcriptases [26], it is possible that other reverse transcriptases are capable of ‘reading through’ ARP-derivitized sites depending on the reaction conditions.
Real-time RT-PCR or cDNA microarray is currently the most sensitive and accurate method to identify and quantify individual RNA levels. Therefore, our goal was to establish a method that could be utilized with these experimental tools. In this study, we analyzed the utility of either oligo(dT) primers or random primers for cDNA synthesis, and investigated the consequences of RNA oxidation and interactions with PCR primer location. In most microarray systems cDNA or cRNA is synthesized using oligo(dT) primers. Oxidative damage, including the incorporation of abasic sites, could perturb reverse transcription extension, which in turn may cause changes in RNA levels depending upon primer location. Our results demonstrated that the cDNA synthesis rate was not significantly inhibited in response to oxidized ARP-RNA, since the decreased RQ values toward the 5’-end were comparable between control cell total RNA (which contains less ARP-derivatized RNA) and oxidized cell total RNA (which contains more ARP-derivatized RNA). In addition, inhibition of cDNA synthesis by the pull-down procedure that included high temperature incubations and repetitive washing steps was minimal, since the location dependent decrease of RQ values were comparable between total RNA and the corresponding pull-down RNA. As a result, assessment of the levels of oxidized mRNAs were independent of primer location in the cDNA using either oligo dT primers or random primers. mRNA concentrations were constant using cDNA prepared with either oligo(dT) primers or random primers, which indicates that our method is also applicable to any microarray systems. Using microarray analysis coupled with pull-down isolation, we also observed that oxidation of mRNA is a selective rather than a random process; some mRNAs are highly oxidized whereas others appear resistant to oxidation partly dependently on guanosine content (unpublished data). Thus, oxidation profiles of individual RNA species derived from any RNA source are available to be quantitatively investigated using this method. We expect that this technology may reveal the potential molecular mechanisms for disease onset/progression as it relates to RNA oxidation, identification of oxidation susceptible gene transcripts and prediction of their dysfunctional protein products in each disease.
Acknowledgments
This research was supported by NIH grants AG11370 and NS056218. The equipments and screen for PhosphorImager were provided from University of Oklahoma, Department of Cell Biology and Biochemistry. The washing steps for ARP-RNA/strep-beads complex were refined based on the protocols derived from manufacturers and a web site developed by Dr. David Wilson in 1998, http://genetics.mgh.harvard.edu/szostakweb/protocols/biotin-avidin/index.html
Abbreviations
- ARP
Aldehyde Reactive Probe
- ARP-RNA
RNA derivatized with ARP
- eEF2
eukaryotic elongation factor-2
- RQ
relative quantity
- qRT-PCR
quantitative RT-PCR
- strep-beads
streptavidin-magnetic beads
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hofer T, Marzetti E, Xu J, Seo AY, Gulec S, Knutson MD, Leeuwenburgh C, Dupont-Versteegden EE. Increased iron content and RNA oxidative damage in skeletal muscle with aging and disuse atrophy. Exp Gerontol. 2008;43:563–570. doi: 10.1016/j.exger.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetylL-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci USA. 2002;99:2356–2361. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinet W, de Meyer GR, Herman AG, Kockx MM. Reactive oxygen species induce RNA damage in human atherosclerosis. Eur J Clin Invest. 2004;34:323–327. doi: 10.1111/j.1365-2362.2004.01343.x. [DOI] [PubMed] [Google Scholar]
- 4.Nunomura A, Chiba S, Kosaka K, Takeda A, Castellani RJ, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport. 2002;13:2035–2039. doi: 10.1097/00001756-200211150-00009. erratum appears in Neuroreport. 2003 Feb 10;14(2):293. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang Y, Kong Q, Shan X, Tian G, Ilieva H, Cleveland DW, Rothstein JD, Borchelt DR, Wong PC, Lin CL. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE. 2008;3:e2849. doi: 10.1371/journal.pone.0002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nunomura A, Perry G, Hirai K, Aliev G, Takeda A, Chiba S, Smith MA. Neuronal RNA oxidation in Alzheimer’s disease and Down’s syndrome. Ann N Y Acad Sci. 1999;893:362–364. doi: 10.1111/j.1749-6632.1999.tb07855.x. [DOI] [PubMed] [Google Scholar]
- 8.Shan X, Tashiro H, Lin CL. The identification and characterization of oxidized RNAs in Alzheimer’s disease. J Neurosci. 2003;23:4913–4921. doi: 10.1523/JNEUROSCI.23-12-04913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. J Neurosci Res. 2007;85:3036. doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]
- 11.Kong Q, Shan X, Chang Y, Tashiro H, Lin CL. RNA oxidation: a contributing factor or an epiphenomenon in the process of neurodegeneration. Free Radic Res. 2008;42:773–777. doi: 10.1080/10715760802311187. [DOI] [PubMed] [Google Scholar]
- 12.Rhee Y, Valentine MR, Termini J. Oxidative base damage in RNA detected by reverse transcriptase. Nucleic Acids Res. 1995;23:3275–3282. doi: 10.1093/nar/23.16.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong X, Tao R, Li Z. Quantification of RNA damage by reverse transcription polymerase chain reactions. Anal Biochem. 2006;357:58–67. doi: 10.1016/j.ab.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 14.Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol Aging. 2006;27:657–662. doi: 10.1016/j.neurobiolaging.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 15.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Burrows CJ, Muller JG. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem Rev. 1998;98:1109–1152. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 17.Ide H, Akamatsu K, Kimura Y, Michiue K, Makino K, Asaeda A, Takamori Y, Kubo K. Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry. 1993;32:8276–8283. doi: 10.1021/bi00083a031. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka M, Song H, Kupfer PA, Leumann CJ, Sonntag WE. An assay for RNA oxidation induced abasic sites using the Aldehyde Reactive Probe. Free Radic Res. 2011;45:237–247. doi: 10.3109/10715762.2010.535529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci USA. 2007;104:66–71. doi: 10.1073/pnas.0609737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez M, Argarana CE, Fidelio GD. Extremely high thermal stability of streptavidin and avidin upon biotin binding. Biomol Eng. 1999;16:67–72. doi: 10.1016/s1050-3862(99)00041-8. [DOI] [PubMed] [Google Scholar]
- 22.Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem. 2008;389:243–255. doi: 10.1515/BC.2008.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 24.Shan X, Chang Y, Lin CL. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J. 2007;21:2753–2764. doi: 10.1096/fj.07-8200com. [DOI] [PubMed] [Google Scholar]
- 25.Stone MD, Mihalusova M, O’Connor C M, Prathapam R, Collins K, Zhuang X. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature. 2007;446:458–461. doi: 10.1038/nature05600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kupfer PA, Crey-Desbiolles C, Leumann CJ. Trans-lesion synthesis and RNaseH activity by reverse transcriptases on a true abasic RNA template. Nucleic Acids Res. 2007;35:6846–6853. doi: 10.1093/nar/gkm767. [DOI] [PMC free article] [PubMed] [Google Scholar]