

Abstract
Systemic loss-of-function studies have demonstrated that Pax3 transcription factor expression is essential for dorsal neural tube, early neural crest and muscle cell lineage morphogenesis. Cardiac neural crest cells participate in both remodeling of the pharyngeal arch arteries and outflow tract septation during heart development, but the lineage specific role of Pax3 in neural crest function has not yet been determined. To gain insight into the requirement of Pax3 within the neural crest, we conditionally deleted Pax3 in both the premigratory and migratory neural crest populations via Wnt1-Cre and Ap2α-Cre and via P0-Cre in only the migratory neural crest, and compared these phenotypes to the pulmonary atresia phenotype observed following the systemic loss of Pax3. Surprisingly, using Wnt1-Cre deletion there are no resultant heart defects despite the loss of Pax3 from the premigratory and migratory neural crest. In contrast, earlier premigratory and migratory Ap2α-Cre mediated deletion resulted in double outlet right ventricle alignment heart defects. In order to assess the tissue-specific contribution of neural crest to heart development, genetic ablation of neural crest lineage using a Wnt1-Cre-activated diphtheria toxin fragment-A cell-killing system was employed. Significantly, ablation of Wnt1-Cre-expressing neural crest cells resulted in fully penetrant persistent truncus arteriosus malformations. Combined, the data show that Pax3 is essential for early neural crest progenitor formation, but is not required for subsequent cardiac neural crest progeny morphogenesis involving their migration to the heart or septation of the outflow tract.
Keywords: mouse embryo, Pax3, cardiac neural crest, congenital heart defects, lineage mapping, conditional knockout, genetic cell abaltion
INTRODUCTION
Disruption of multipotent migratory neural crest (NC) lineage development, either cell autonomously or non-cell-autonomously, results in numerous forms of human birth defects, including Waardenburg, DiGeorge, Neurofibromatosis Type-1, Hirschsprung, Treacher-Collins and Leopard syndromes (Read & Newton, 1997; Hutson and Kirby, 2003; Epstein and Parmacek, 2005; Ismat et al., 2006; Trainor et al., 2009; Stewart et al., 2010). The cardiac NC (CNC) subpopulation is essential for vertebrate cardiovascular development and in utero survival (Kirby et al., 1983; Conway et al., 1997a; Jiang et al., 2000; Stoller and Epstein, 2005; Hutson and Kirby, 2007; Snider et al., 2007; Rosenquist & Finnell, 2007). Among the most common CNC-related congenital birth defects are the conotruncal malformations (Kirby, 2008), including persistent truncus arteriosus (PTA) and pulmonary atresia (PAt). Additional defects such right-sided outflow tract (OFT) defects, aortic ring and double outlet right ventricle (DORV) have also been associated with aberrant CNC morphogenesis (Stoller and Epstein, 2005; Hutson and Kirby, 2007) and this may depend upon interactions with secondary heart field derivatives (Waldo et al., 2005; Goddeeris et al., 2007; Li et al., 2010). Often associated with these life-threatening defects are interrupted aortic arch and pharyngeal arch (PA) patterning anomalies (Waldo et al., 1996; Conway et al., 2003). The range of CNC-associated phenotypes suggests that these defects lie along a continuum representing relative severity of CNC-dependent deficiency.
The CNC originates from precursors within the lower hindbrain neural tube (NT) between the otic placode and 4th somite of the developing mammalian embryo and cannot be substituted via adjacent cranial or trunk NC (reviewed in Creazzo et al., 1998). After delaminating from the dorsal region of the neuroepithelial tube, the mesenchymal CNC progeny migrates ventrolaterally along stereotypical routes and are induced to differentiate through reciprocal signaling with neighboring cells (Trainor & Krumlauf, 2002; Hutson & Kirby, 2007; Sauka-Spengler & Bronner-Fraser, 2008). NC cells derived from the CNC region populate the dorsal root and sympathetic ganglia, thymus, thyroid, as well as the pharyngeal arches (PAs) and heart itself (Creazzo et al., 1998). The CNC ensheathes the initially bilaterally symmetrical PA arteries (PAAs) and subsequently differentiates into smooth muscle once the vessels are repatterned to form the great arteries (Waldo et al., 1996; Choudhary et al., 2006; Snider et al., 2007; Yashiro et al., 2007). A subpolulation of the CNC continue to migrate and colonize the OFT of the heart and interact with neighboring cardiac progenitor cells derived from the second heart field (Waldo et al., 2005; Li et al., 2010). Although temporally defined via quail-chick chimeric analysis (LeDouarin & Teillet, 1974), chick microsurgical ablation of pre-migratory neural folds (Kirby et al., 1983) and mouse transgenic lineage mapping approaches (Yamauchi et al., 1999; Jiang et al., 2000; Epstein et al., 2000); identification of a CNC-specific inducing factor within the early NT has remained elusive. This has meant elucidation of the role played by CNC within the heart itself is poorly understood, even though their requirement is unquestioned. Although many signaling pathways and transcription factors involved in CNC development have been identified and their absence shown to cause congenital heart defects (Meulemans & Bronner-Fraser, 2004; Scholl & Kirby, 2009), the mechanism by which the NC cells themselves regulate PAA remodeling and OFT morphogenesis remains elusive (Snider & Conway, 2007).
Pax genes are expressed early during embryogenesis and are thought to have a role in establishing morphological boundaries and early regionalization (Mansouri, 1998). In conjunction with several other developmentally key transcription factors, Pax3 has been implicated in promoting NC induction, maintenance, migration and differentiation in several different model organisms (Conway et al., 1997a; Li et al., 1999; Epstein et al., 2000; Kwang et al., 2002; Tang et al., 2004; Bajolle et al., 2006; Morgan et al., 2008; Chang et al., 2008). Most evidence has come naturally occurring Splotch (Sp and Sp2H) allelic mutants and maternally induced diabetic models. As Pax3 is widely expressed within the early neural folds/NT, the emigrating cranial, cardiac, trunk and enteric NC and muscle lineages (Goulding et al., 1991; Engleka et al., 2005); Sp and Sp2H mouse mutants exhibit a myriad of lethal congenital defects. Significantly, the OFT defects in systemic Pax3-deficient mutants are thought to be due to a decrease in the number of migrating CNC that ultimately leads to a lack of CNC-derived cells in the OFT septum (Conway et al., 2000; Epstein et al., 2000). While these data indicate Pax3 plays a NC-specific role, there remains controversy as to whether Pax3 plays a cell-autonomous and/or environmental role and precisely what the spatiotemporal requirement of Pax3 is during CNC morphogenesis and heart development (Li et al., 1999; Conway et al., 2000; Mansouri et al., 2001; Kwang et al., 2002; Chan et al., 2004). Thus, the lineage-specific requirement for Pax3 activity in various aspects of mammalian NC development remains unknown.
Here, using a novel Pax3 conditional allele (Koushik et al., 2002) which allowed for the generation of both homozygous systemic and targeted lineage-restricted deletions, we demonstrate that Pax3 is indispensable for normal heart development. Further, Wnt1-Cre, Ap2α-IRESCre and P0-Cre mediated NC-restricted loss revealed that Pax3 plays an essential early role in NC progenitor formation and specification, but is not required for subsequent CNC progeny morphogenesis involving their migration to the heart. These phenotypes were also evaluated alongside embryos in which Wnt1-Cre was used to genetically ablate the NC lineage. Combined these lineage-specific approaches provide insight into understanding the role that Pax3 plays intrinsically within the CNC during NC formation and subsequent PAA remodeling and OFT morphogenesis.
MATERIALS AND METHODS
Genetically Modified Mice
The generation and characterization of C57Bl6 Pax3 conditionally targeted embryonic stem cells and mice, in which exon 5 was flanked with loxP sites, was described previously (Koushik et al., 2002). Pax3flox/+ mice were either crossed to female C57Bl6 Splicer allele (as Tie2-Cre transgene is expressed in female germ cells; Koni et al., 2001), to systemically remove exon5 in the germline; or to conditionally delete exon5 via Wnt1-Cre transgenic (Jiang et al. 2000), P0-Cre (Yamauchi et al., 1999), Ap2α-IRESCre knock-in (Macatee et al., 2003), Tie2-Cre transgenic (Koni et al., 2001), Nkx2.5Cre knock-in (Moses et al. 2001) or αMHC promoter-driven Cre (Agah et al., 1997) intercrossing. For timed pregnancies, the day of observed vaginal plug was designated embryonic day 0.5 (E0.5), and embryos and fetuses were dissected in cold 0.1M phosphate buffered saline (PBS). Resultant offspring amnion or tail biopsy genomic DNA were genotyped to detect wildtype (315bp), floxed (349bp) and/or Δ5 recombined (400bp) alleles (Koushik et al., 2002; Zhou et al., 2008). To assess genetic equivalence, Pax3Δ5/+ mice were intercrossed with Sp2H/+ mice and PCR genotyped as described (Conway et al., 2000). To examine CNC cell migration, systemic Pax3Δ5/+ mice were also intercrossed with Connexin 43-lacZ (Lo et al. 1997) and RA response element-lacZ transgenic reporter mice (Colbert et al., 1993). Pax3Δ5/+, Pax3flox/+ and Wnt1-Cre/R26rDTA mice were placed on R26R indicator background (Soriano, 1999) for lineage mapping and to assess Cre-mediated recombination efficiency. Wnt1-Cre-mediated genetic NC cell ablation was carried out using ROSA26-eGFP-DTA (R26rDTA; Ivanova et al. 2005) mice as described (Snider et al., 2009). Cre, lacZ or R26rDTA transgenes were detected via PCR genotyping as described (Snider et al. 2008a,b). All animal procedures were performed with the approval of the Institutional Animal Care and Use Committee of Indiana University School of Medicine.
Histologic analysis, Ink injection and X-gal Staining
Isolation of tissues, fixation, processing, and whole mount staining for β-galactosidase was performed as described (Snider et al. 2008a,b). Once staining was complete, tissues were refixed in 4% paraformaldehyde overnight and either cleared via use of increasing concentrations of glycerol (10–50%) for whole mount imaging or embedded in paraffin, cut into 10μm serial sections; and counterstained with hematoxylin/eosin. For gross examination of the cardiovascular system, E10–12 embryos were injected with Pelican India Ink diluted 50% in PBS in the left ventricle of the heart (straight through the body wall) or into the umbilical vessels, as described (Conway et al. 2003; Snider et al., 2008a). Immediately following injection, embryos were fixed in 4% paraformaldehyde overnight, processed through a series of methanol/PBS to 100 % methanol, and cleared for several hours in 2 volumes benzyl benzoate and 1 volume benzyl alcohol. For each assay, whole embryos and/or serial sections were examined for at least three individual embryos of each genotype at each stage of development. Wildtype littermates were always used as age-matched control samples.
Western blot analysis
For Western analysis, individual E10.5 embryos (n=5 of each genotype) were homogenized in 300μl protein lysis buffer as described (Zhou et al. 2008). For each sample, 50μl was resolved using 10% SDS-PAGE (Bio-Rad), transferred to nitrocellulose, blocked for 1 hour and probed with mouse monoclonal anti-Pax3 (1:2,000 dilution) or monoclonal anti-Pax7 (1:2,000 dilution) antibodies (both obtained from the Hybridoma Bank) in blocking solution. The signal was detected via ECLPlus (Amersham) with peroxidase-conjugated goat anti-mouse secondary antibody (1:5,000 dilution, Promega). To verify equal loading, all blots were subsequently stripped (0.2M NaOH for 5 minutes at room temperature), thoroughly washed, re-blocked and then probed with mouse anti-actin antibody (1;5,000 dilution, Sigma). X-ray films were scanned and signal intensity measured using ImageJ software (downloaded from wsr@nih.gov).
In situ hybridization and Immunohistochemical analysis
Embryos were fixed in 4% paraformaldehyde at 4°C overnight and processed for paraffin sections for in situ hybridization or immunohistochemistry. In situ hybridization of Wnt1, Ap2α and Pax3 exon5 and N-terminus Pax3 was performed as described (Zhou et al. 2008) using sense and anti-sense S35-labeled cRNA probes. Immunostaining was carried out using ABC kit (Vectorstain) with DAB and hydrogen peroxide as chromogens, as described (Zhou et al. 2008). For detection of Pax3, a primary goat anti-Pax3 antibody (1:200 dilution; Santa Cruz sc-7748) was used, TUNEL and α-smooth muscle actin (αSMA, dilution 1:3,000, Sigma) were performed as previously described (Lindsley et al., 2007; Zhou et al. 2008). For all of these assays, serial sections were examined using at least three individual E8, E10.5 and E12 embryos of each genotype.
Verification of conditional Pax3Δ5 deletion
The recombination efficiency of Pax3flox/flox allele was examined in purified Schwann cells by PCR. Schwann cells were isolated from wildtype and Pax3Δ5/flox/Wnt1-Cre mutant mouse embryo dorsal root ganglia (DRG) at E13.5 as described (Yang et al. 2003). Briefly, isolated embryonic DRGs were enzymatically dissociated from individual embryos (n=3 wildtype and 3 Pax3Δ5/flox/Wnt1-Cre mutants) and filtered to obtain a single cell preparation. Cells were grown in 10% FBS (HyClone Laboratories, Utah, USA) supplemented with 250ng/ml NGF (Harlan Bioproducts for Science Inc., Indiana, USA). The medium was changed after 24hours to serum-free defined N2 medium (Invitrogen Corp.) containing 250ng/ml NGF and penicillin/streptomycin (1 mM/ml) (BioWhittaker Inc., Walkersville, Maryland, USA). After 6 days, Schwann cells and neurons were separated from fibroblasts and the Schwann cells enzymatically dissociated from the neurons in 0.01% collagenase (Sigma-Aldrich, Missouri, USA). Cells were resuspended in DMEM with 10% FBS and given Schwann cell growth medium containing 10ng/ml recombinant human Glial Growth Factor-2 (rhGGF2) (Heregulin; Sigma-Aldrich), 1mM/ml penicillin/streptomycin, with 2mM forskolin (EMD Biosciences Inc., California, USA) added to suppress fibroblast growth. After 1 week, duplicate cells were either harvested or stained with an antibody directed against S-100 (1;500 dilution; Sigma-Aldrich), an acidic, calcium-binding protein present in Schwann cells but not fibroblasts, to verify Schwann cell purity. Genomic DNA was isolated and examined via PCR using forward and reverse PCR primers, as well as a 3rd primer flanking the 1st loxP site (Koushik et al., 2002). The combination of the forward primer and the 3rd primer amplifies the flox (349bp) and wildtype alleles (315bp) but not the recombined allele; whilst the forward and reverse primers amplify the recombined and floxed alleles (Supplemental Fig. 1).
DOPA Oxidase detection of NC-derived melanocytes
Neonatal skin biopsies (n=3 of each genotype) were cryoprotected, serially sectioned and fixed with 2% paraformaldehyde for 15min at room temperature. Frozen sections were then treated with 0.1% DOPA (Sigma, St Louis, MO, USA) in 0.1M phosphate buffer (pH 7.2) for 5h at 37°C. Stained sections were then post-fixed with 2% paraformaldehyde, and β-galactosidase co-staining performed as previously described (Snider et al. 2008a,b).
RESULTS
Pax3Δ5/Δ5 systemic knockout recapitulates Sp2H mutant embryos
Following Cre-mediated removal of the hypomorpohic neomycin selection cassette (Zhou et al. 2008), viable mice homozygous for Pax3 conditional allele (Pax3f/f) with homeo-domain encoding exon5 flanked by loxP-sites were generated and maintained on a C57Bl6 background. Crossing male Pax3f/f with Tie2-Cre transgenic females generated germline (via oocyte-expressing Cre activity) heterozygous deletion of exon5 (Pax3Δ5/+) in resultant offspring. Although Tie2-Cre is normally expressed in the endothelium, it is also transiently expressed in adult ovary oocytes (Koni et al., 2001). Thus, by crossing a Tie2-Cre-positive female with a Pax3f/f male, generation of heterozygous Pax3Δ5/+ systemic knockouts was possible. Pax3Δ5/+ mutant mice are viable and exhibit white spots on their forehead and belly (Fig. 1B), similar to those seen in Splotch heterozygotes. Intercrossing of Pax3Δ5/+ mutants resulted in expected Mendelian ratio of wildtype and heterozygous offspring at birth, but no Pax3Δ5/Δ5 mutants were present (n=36 litters). As expected, when fetuses were harvested at E14.5, all Pax3Δ5/Δ5 homozygotes were dead, indicating that the Δ5 allele is analogous to the Splotch (Sp2H) allele and that the presence of at least one copy of the Pax3 homeodomain is required for normal development and in utero survival. Deletion of exon5 creates a premature stop codon and loss of Pax3 homeodomain and its downstream sequence (Koushik et al., 2002). Western analysis using an antibody that recognizes the C-terminus of Pax3 (missing in Δ5 mutant protein) confirmed that wildtype Pax3 protein is reduced by ~50% in heterozygotes and absent in the homozygous mutant. Levels of the closely related Pax7 were slightly reduced due to hypoplastic somites which co-express Pax7 (Fig. 1A).
Figure 1. Characterization of Pax3Δ5 null allele.

(A) Western analysis of duplicate wildtype (+/+), heterozygous (+/Δ5) and homozygous (Δ5/Δ5) systemic Pax3 exon5-deleted (Δ5) mutant E10.5 whole embryos, probed for Pax3 and Pax7 protein. Note when the signal intensity was quantified using ImageJ software, Pax3 protein is reduced ~50% in heterozygote's and absent in Pax3Δ5 nulls, the closely-related Pax7 protein family member is reduced slightly due to loss of somites that co-express Pax7. The housekeeping Actin protein is unaffected. (B) PCR screening of the offspring revealed that none of the Pax3Δ5 nulls were born, and that heterozygous mutants exhibit characteristic white belly spots. (C) Gross examination of E13.5 wildtype and Pax3 null littermates. Nulls exhibit exencephaly (Ex) and spina bifida (Sb), and die ~E14. (D–I) Histological analysis of transverse H&E stained E13.5 sections reveals that Pax3 null NC-derived dorsal root ganglia (drg) are hypoplastic but that the neural tube (nt) is unchanged (E), when compared to normal littermate controls (D). Sections through the OFT and ventricles of the heart, reveals that Pax3 nulls exhibit pulmonary atresia (PAt in G) and a concomitant membranous interventricular septal defect (VSD in I), but that wildtype littermates have separate aorta (Ao) and pulmonary trunks (P) that each exit the heart (F) with an intact ventricular septum (H). Note that the length of the OFT is equivalent between mutant and wildtype (F,G).
Whole embryo and histological analyses revealed that Pax3Δ5 nulls recapitulates all aspects of the Sp2H allele (Conway et al., 1997a; 2000); including pigmentation defects in heterozygotes (Fig. 1B), NT closure defects (Fig. 1C), hypoplastic dorsal root ganglia (DRG) and thymus defects (Fig. 1E), somite/skeletal abnormalities (not shown), failure of cardiac OFT septation (Fig. 1G), concomitant interventricular septal defects (VSD; Fig. 1I), and 100% midgestational lethality by E14.0 (n=23 homozygotes compared to 70 controls - both wildtype and Pax3Δ5/+ - examined from 12 litters). Furthermore, analysis of Pax3Δ5/+ and Pax3Sp2H/+ compound mutants (i.e. Pax3Δ5/Sp2H) demonstrated that the Pax3Δ5 allele is genetically identical to the Sp2H allele (data not shown). Similar to the Sp2H allele, ~61% of the Pax3Δ5Δ5 nulls exhibited both exencephaly and spina bifida, whilst 21% exhibited exencephaly only and the remaining mutants exhibited spina bifida NT defects only. Further detailed histological analysis revealed that the E14 Pax3Δ5Δ5 hearts had 100% penetrant PAt and VSD (Fig. 2D; n=23/23 homozygotes examined). The fact that the mutant single vessel is comparable in length and width to wildtype littermate OFTs and contains a multicuspid semilunar valve with 3 well-proportioned cusps (as seen in each of the wildtype aortic and pulmonary valves), supports the diagnosis of PAt as recently defined by Kirby (2008). PAt is a congenital malformation of the pulmonary valve in which the valve orifice fails to develop, either in association with an intact ventricular septum or an accompanying VSD. The PAt defects seen in Pax3Δ5Δ5 hearts is PAt with VSD, and the VSD provides a way out for blood within the right ventricle. Failure of development of the OFT conal and/or truncal ridges, either via defective conal cushion transformation (Ya et al., 1998), absent subpulmonary myocardium (Theveniau-Ruissy et al., 2008), second heart field (SHF) deficiencies (Kirby et al., 2008) or truncal cushion NC colonization abnormalities (Hutson & Kirby, 2003; Snider et al., 2007; Tang et al., 2010) can all result in persistence of the common arterial trunk. While SHF-related OFT defects are usually associated with dysmorphic OFT length/width, myocardial cuff and valve leaflet number phenotypes; NC-related OFT defects rarely affect OFT length and/or leaflet number (Kirby, 2008; Li et al., 2010). Since these ridges are also necessary to complete the development of the ventricular septum, conotruncal defects in the mouse is always associated with a VSD and invariably communicates with both ventricles (Conway et al., 2003). Apart from the VSD, the Pax3Δ5Δ5 myocardium including the compact zone, trabeculae and subpulmonary myocardium were intact (Figs.2 and 3). Moreover, Pax3 itself is not thought to be expressed within the myocardium (Epstein et al., 2000; Engleka et al., 2005).
Figure 2. Examination of the Pax3 null OFT vasculature and neural crest gene expression during septation.
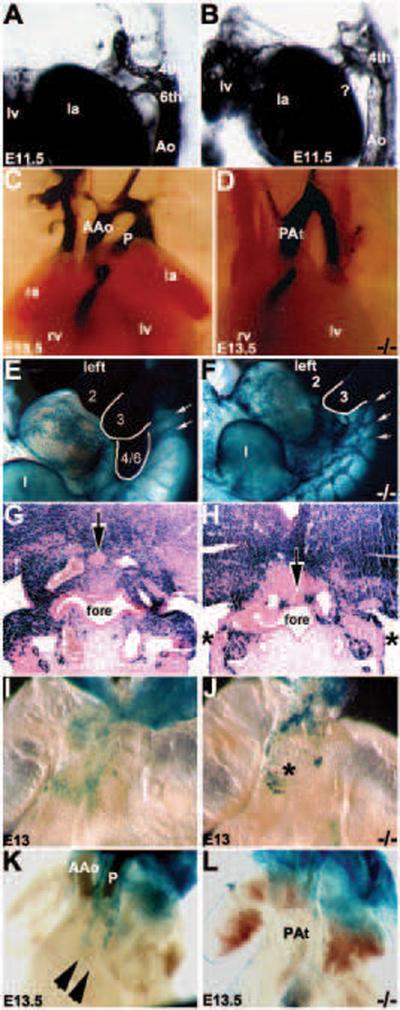
(A–D) Intracardiac ink injection analysis of pharyngeal arch artery (PAA) formation and remodeling during OFT morphogenesis. In E11.5 Pax3 nulls, the left 6th PAA has abnormally regressed (? in B) and there is an enlarged left 4th PAA (when compared to wildtype littermates, A). In wildtype E13.5 hearts (C), separate ascending aortic (AAo) and pulmonary trunks (P) exit the heart. However, in Pax3 null hearts there is only a single OFT (D). (E–H) Whole-embryo lacZ staining of Cx43 expressing cells in E11 wildtype/Cx43-lacZ (E) and Pax3 null/Cx43-lacZ (F) embryos viewed from left. Note there is a drastic down-regulation of Cx43-lacZ expression specifically within Pax3 null 4/6th arch region and there is disorganized streams of migrating NC (indicated by arrows in F). Histology confirms there is significantly less lacZ staining in both left and right Pax3 null 6th PAA region (indicated by *) and the thyroid primordium is abnormally located (arrow in H). (I,J) Wholemount lacZ staining of Cx43 expressing cells in E13.0 wildtype/Cx43-lacZ (I) and Pax3 null/Cx43-lacZ (J) hearts. Whilst Cx43-lacZlacZ-positive cells are located within the wildtype OFT cushions (I), in Pax3 nulls the lacZ-positive cells are ectopically located on the surface of the OFT (indicated by * in J). (K,L) Wholemount lacZ staining of RARE-lacZ reporter expression in E13.5 wildtype/RARE-lacZ (K) and Pax3 null/RARE-lacZ (L) hearts. Note two robust streams of RA-responsive CNC within the wildtype OFT (indicated by arrowheads in K) but are absent in the Pax3 null littermate OFT exhibiting PAt (L). Abbreviations: Ao, aorta; rv, right ventricle; lv, left ventricle; ra, right atria; la, left atria; fore, foregut.
Figure 3. Wnt1Cre/R26R lineage mapping of NC cells and their derivatives.

(A,B) Dorsal (A) and left lateral (B) views of E9 wildtype and Pax3Δ5 null (−/−) X-Gal stained embryos, illustrating impaired Pax3Δ5 null NC emigration and migration towards the 2nd PA, when compared to wildtype littermates (embryos on left). However, 1st arch and cranial NC migration appears unaffected. (C) Right lateral views of E9.5 wildtype (+/+), heterozygous (+/ −) and Pax3 null (−/−) littermates showing fewer CNC populate the heterozygous 3/4/6th PAs (middle embryo), and still even less CNC colonize the Pax3 null 3/4/6th arches (right embryo). There is also a significant reduction in NC contribution to the Pax3 null dorsal root ganglia (indicated by * in C). However, cranial NC migration appears unaffected. (D,E) Histology through the OFT region of embryos shown in C, confirmed a lack of Wnt1Cre-marked CNC within the Pax3 null 4th PAs (D) and AP septum (indicated by * in wildtype in E) and OFT cushions. However, the dorsal-ventral boundary of Wnt1-Cre-marked neuroepithelial cell within the NT was similar amongst all three genotypes (D). (F) Lateral view of E12.5 wildtype and Pax3 null, illustrating extensive NC reduction along the anterior-posterior axis of the nulls (right embryo), absent lacZ staining of dorsal root ganglia, as well as spina bifida (Sb) and exencephaly (Ex). (G,H) Wholemount lacZ staining of Wnt1Cre reporter expression in E12.5 wildtype/Wnt1Cre/R26R and Pax3Δ5/Δ5/Wnt1Cre/R26R hearts. Note Pax3 null CNC reduced OFT colonization (−/−), viewed frontally (G) and from the right (), but that the length and width of the mutant OFT is similar to control. (I) Reduced CNC colonization is most evident in E13.5 Pax3 null hearts (right heart), and some lacZ-marked NC are ectopically located on the mutant OFT surface (arrowheads). Stage-matched embryos and isolated hearts are shown photographed at the same magnification.
Pax3Δ5/Δ5 systemic knockout displays specific regression of left 6th PAA
Although the mechanism underlying OFT morphogenesis remains controversial, it is becoming evident that cell lineages within the pharyngeal arches, second heart field and outflow regions are all thought to be required for normal development of the great vessels that exit the heart (Hutson & Kirby, 2007; Conway and Snider, 2007; Snider et al., 2007; Kirby et al., 2008; Li et al., 2010). Prior to completion of mouse cardiac OFT septation at E14.5 (Waldo et al., 1999), the initially symmetrical paired caudal E11.5 PAAs undergo a complex set of sequential asymmetric remodeling steps resulting in the persistence of a left-sided aortic arch (reviewed in Snider et al., 2007). To determine whether systemic loss of Pax3 was involved in remodeling of PAAs, we performed intracardiac India ink injections at different developmental stages. At E10.5, there are no observable differences in PAA patterning (not shown) or length of the OFT (Fig. 3C,E) between Pax3 mutants and control littermates. However, consistent with PAt rather than PTA (Kirby, 2008), by E11.5 the left 6th PAA undergoes abnormal regression in mutant embryos when compared to controls (Fig. 2A,B). Histology revealed that there was no left 6th PAA vessel, which is similar to the normally regressed right 6th PAA. Interestingly, there are no observable differences in left 3rd, 4th PAA remodeling between the two genotypes. Also, the cranially located 1st and 2nd PAA's also undergo normal regression (not shown), suggesting that the caudal 6th PAA is preferentially affected in Pax3Δ5 nulls. The observed Pax3Δ5 null OFT phenotype is consistent with an initial failure of the left 6th to persist, which normally gives rise to the ductus arteriosus. Thus, in its absence, there is only a single outlet via the left 4th (normally gives rise to aorta), resulting in subsequent PAt with attendant brachiocephalic, common carotid and subclavian arteries branching off (Fig. 2D). The pulmonary arteries originate from the back the remaining aorta.
As the majority of NC molecular markers are down-regulated prior to the CNC colonizing the OFT (Snider et al., 2007), we used the Connexin 43 (Cx43) and Retinoic acid response element (RARE) indicator transgenic mice to determine whether CNC could appropriately populate the pharyngeal arches and OFT cushion regions. The Cx43-lacZ transgene provides β-gal expression in migratory mouse NC cells, including CNC cells. It has previously been used to demonstrate differences in Sp NC distribution patterns (Waldo et al., 1999; Epstein et al., 2000). While the RARE-lacZ transgene has been used to identify regions of RA-mediated gene activation in utero (Colbert et al., 1993). Heterozygous Pax3 mutant mice on either Cx43-lacZ or RARE-lacZ backgrounds were intercrossed. The resulting embryos had NC subpopulations labeled with β-galactosidase expression (Fig. 2E–L), and displayed identical OFT phenotypes to those obtained without the transgenes. Significantly, there are reduced numbers of Cx43-lacZ expressing CNC present in the Pax3 null mutant PAs (Fig. 2F) and OFT (Fig. 2J), and they are abnormally distributed on the OFT surface and not within the aorticoplumonary septum or OFT truncal cushions, despite the mutant OFT being comparable in size with control OFTs (Fig. 2J). Thus, systemic loss of Pax3 does not entirely prevent CNC formation and migration. Additionally, the streams of migrating Cx43-lacZ-positive NC appeared disorganized (Fig. 2F). Furthermore, histology revealed that there was a reduction of Cx43-positive indicator expression within the most caudal arches and that the Pax3 null thyroid primordium was still attached to the foregut endoderm in E11.5 mutants (Fig. 2H). This suggests a delay in detachment and dorso-caudal migration of the primordium to its final position behind the trachea. These expression data are in agreement with the findings in Splotch mutants (Epstein et al., 2000), and reinforce the previous data that the Pax3Δ5 null is a phenocopy of Splotch. Although RARE-lacZ indicator patterns demonstrated that the RA levels are unaffected in the E10.5 Pax3 null NT (not shown), there is a complete absence of RA-responsive cells colonizing the E13.5 mutant OFT (Fig. 2K). Given our previous data demonstrating that expression of both Crabp1 and the retinoic acid-responsive Ap2α transcription factor are reduced (Conway et al., 1997a; 2000), these data indicate endogenous levels of retinoic acid are unaffected, but there is either a lack of retinoic acid-signaling within the mutant migrating NC themselves or that the NC are unable to colonize the Pax3 null OFT. As there is a similar reduction in Cx43-positive NC colonizing the 4th and 6th arches and OFT cushions, combined these data imply a CNC deficiency within the OFT rather than abnormal NC junctional communication and lack of retinoic acid signaling.
Migratory NC in Pax3Δ5/Δ5 mutants is significantly reduced
Next we used the R26R lineage-tracing assay to determine whether permanently labeled CNCs (independent of gene expression changes) could appropriately populate the PAs and OFT region. We wanted to test whether the phenotype of the Pax3Δ5 null mouse is the result of a decrease in the absolute numbers of pre-migratory and/or migratory CNC cells rather than due to gene misexpression within the CNC cells themselves. Wnt1-mediated Cre recombination is initially detectable within the E8 dorsal neural folds of the midbrain/hindbrain junction and within migratory NC from E8.5 onwards (Jiang et al. 2000; Stottmann et al., 2004). As a result, we were able to fate-map the CNC lineage from very early ~E8.5 stages onwards, and examine Pax3 mutant pre-migratory NC progenitors and their progeny. Differences in Wnt1-Cre lacZ patterns were observed as early as E8.5–9.0 in mutants, manifested as a reduction in NC emigration from the neural folds along anterio-posterior axis of neural tube (Fig. 3). This reduction in overall numbers led to reduced colonization of mutant NC-derived structures at this stage, including 1st and 2nd branchial arches (Fig. 3B). By E9.5, NC cells appear to be migrating along appropriate migratory pathways in both mutants and controls. However, consistent with the histological OFT defects and diminished number of Cx43 and RARE-lacZ labeled migratory NCs observed, there is a significant reduction in the number of Wnt1-Cre;R26R lacZ-marked NC cells that colonize the 3rd/4th/6th pharyngeal aortic arches. Similarly, there are significant reductions/absences in several other NC-derived structures (i.e. DRGs, Schwann cells, thymus/parathyroid, enteric nervous system) but NC colonization of the frontonasal regions was spatially unaffected (Fig. 3C,F). Notably, NC absences were more pronounced in posterior regions as a few dysmorphic anterior DRGs were present, suggesting a graded NC deficiency along the rostrocaudal axis (Fig. 3C,F). Moreover, this reduction within the arch region was determined to be Pax3-dose dependent, as detectably fewer lacZ-positive cells were observed in Pax3Δ5/+ heterozygote embryos when compared to wildtype littermates (Fig. 3C). Significantly, there was an even more drastic reduction in migratory CNC observed in Pax3Δ5/Δ5 nulls even compared to Pax3Δ5/+ embryos (Fig. 3C). Similarly, serial transverse sectioning of whole mount embryos (E10–E13) and analysis of positively stained cells also revealed that the number of NC colonizing the PAs and subsequently invading the OFT cushions and aorto-pulmonary (AP) septum is reduced in mutants (Fig. 3D,E). The AP septum and OFT cushions in Pax3Δ5/+ embryos are also less populated with NC cells when compared to wildtype, however, this slight reduction is not sufficient to affect normal OFT remodeling as Pax3 heterozygote hearts undergo normal septation. By E13.5, CNC cells have completely colonized the distal region of the wildtype OFT and can be seen by the observed invasion of two streams of lacZ-positive cells (Fig. 3G). However, despite the ability of Pax3-deficent NC to colonize the mutant OFT, they do so in far fewer numbers (Fig. 3G,H) and are sometimes ectopically located on the OFT surface (Fig. 2I). Despite the significant lack of NC colonization of the Pax3 null OFT, the length and orientation of the mutant OFT is comparable with wildtype.
These Wnt1-Cre lineage mapping studies were complemented with an additional reporter, namely Ap2α-IRESCre knock-in mice (Macatee et al. 2003). The Ap2α gene is expressed in many regions of the mouse embryo, including early neural fold, migrating NC and ectoderm (Brewer et al., 2002). This was to ensure reproducibility of our data and to determine the effect of Pax3 deletion in a slightly earlier population of NC. Although Ap2α-IRESCre is expressed within the ectoderm (Macatee et al. 2003), we found that it is also expressed within ~E8 cranial, cardiac and trunk NC progenitor regions of the neural folds (Fig. 6A and Supplemental Fig. 2), and is thus earlier and more extensive than Wnt1-Cre. Using this R26R NC lineage-tracing strategy, both derivatives of the NC progenitors (region known to express Pax3) and ectoderm (region that does not express Pax3) were permanently labeled and fate-mapped in both wildtype and Pax3 null mutant backgrounds (Fig. 4). Analysis of Ap2α-IRESCre;Pax3Δ5/Δ5;R26R(+/+) wholemount and serial section lacZ patterns correlate with those of Wnt1-Cre;Pax3Δ5/Δ5;R26R(+/+). Altogether, they demonstrate that there is an initial temporary delay in NC emigration, a significant reduction in overall numbers of migratory NC within the PAs (Fig. 4B) and drastically reduced colonization of the mutant OFT cushions (Fig. 4F). Since Pax3 is not expressed within the pharyngeal ectoderm, Ap2α-IRESCre lacZ expression in the Pax3Δ5 null ectoderm was intact (Fig. 4D,F), as expected. Furthermore, there is a cranial-to-caudal progressive loss of migratory CNC observed in Pax3 mutants, from the 3rd (Fig. 4D) to the 4th (Fig. 4F) and finally to a complete absence within the 6th arch (Fig. 4H). Thus, correlating with the observed Pax3Δ5 null PAA vessel regression defects, there are reduced lacZ-marked mutant NC cells surrounding the persisting 3rd and 4th PAAs but virtually no lacZ-positive cells within the 6th arch. Combined, the lineage mapping and transgenic indicator data demonstrate that CNC cells in Pax3 null embryos were capable of populating the PAAs, aortic sac and nascent aorticopulmonary septum but at greatly diminished numbers compared to those of controls.
Figure 6. Analysis of Pax3Δ5/Ap2αΔ5 conditional knockouts.
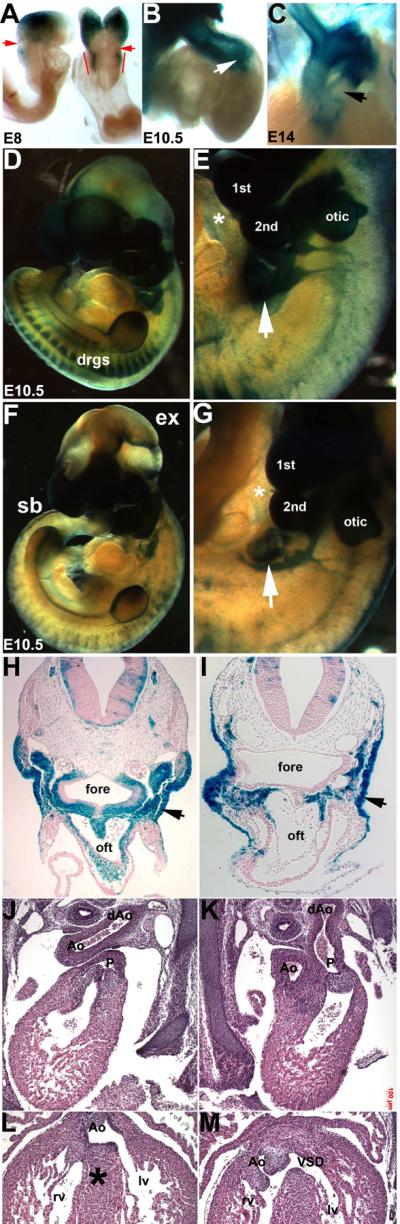
(A–C) Ap2α-IRESCre;R26R lineage mapping in wildtype embryos shows colonization of lacZ-positive cells in E8 neural folds (A) and E10.5 OFT (B,C). Note absence of lacZ cells in pulmonary trunk SHF-derived vascular smooth muscle (arrow in C). (D–G) Examination of lacZ spatiotemporal expression patterns in wildtype (D&E) and conditional Pax3Δ5/Ap2αΔ5 mutant (E&G) embryos at E10.5. Note reduced CNC colonization of E10.5 mutant 4th and 6th PAA's (arrow in G) compared to wildtype (arrow in E). Conditional mutants also exhibit exencephaly (ex) and spina bifida (sb). (H–I) Histological analysis of transverse sections of embryos shown in (D&F). Reduced CNC colonization of OFT region in conditional mutant (I) compared to control (H). (J–M) Conditional E14.5 Pax3Δ5/Ap2αΔ5 mutants display DORV and both the OFT vessels are abnormally aligned over the right ventricle (K) and VSD defects (M) when compared to control littermates (J,L). The aorta abnormally exits the Pax3Δ5/Ap2αΔ5 mutant right ventricle (M) compared to the left ventricle in the control (L), and asterix in L indicates intact interventricular septum. Abbreviations: Ao, aorta; aAo, descending aorta; fore, foregut; lv, left ventricle; otic, otic sulcus (presumptive ear); rv, right ventricle. Scale bar J–M is 100μm.
Figure 4. Ap2α-IRESCre;R26R lineage mapping of NC and their derivatives.

(A,B) Right lateral views of E10.5 wildtype (A) and Pax3Δ5 null (B) littermates illustrating reduced CNC population of Pax3 null 3/4/6th PAs (indicated by red * in B) and significantly diminished DRG (black * in B). However, Ap2α-IRESCre;R26R-marked cranial NC migration appears unaffected despite the presence of exencephaly in the Pax3 null mutant. (C–H) Histology through the 3rd (C,D), 4th (E,F) and 6th (G,H) arch regions of embryos shown in A and B. Sectioning confirmed reduced Ap2α-IRESCre-marked CNC around the Pax3 null 3rd PAA artery (indicated by * in D) and thyroid primordium (D), and around the Pax3Δ5 null 4th (indicated by * in F) and absent NC around the 6th (indicated by * in H) PAA. However, Ap2α-IRESCre ectodermal reporter expression (Macatee et al. 2003) is unaffected in Pax3 nulls (indicated via open arrowhead in F), as Pax3 in not expressed within the ectoderm. Note, only left PAA arteries and left dorsal aorta (dA) are indicated. Abbreviations: t, trachea; fore, foregut.
Wnt1-Cre-restricted deletion of Pax3 is not sufficient to phenocopy Pax3Δ5/Δ5 OFT defects
Given the essential role of CNCs in OFT septation (Kirby et al., 1983; Hutson and Kirby, 2007; Snider et al., 2007) and our systemic Pax3Δ5 null lineage mapping data, the PAt defect observed in Pax3Δ5/Δ5 embryos suggests Pax3 is required for heart development via a direct influence on CNC morphogenesis. To test this assumption and address the ongoing controversy surrounding the NC cell-autonomous versus environmental Pax3 requirement (Li et al., 1999; Conway et al., 2000; Mansouri et al., 2001; Kwang et al., 2002; Chan et al., 2004), we specifically ablated Pax3 within the NC lineage using Wnt1-Cre (Fig. 5). Wnt1-Cre is the current gold standard NC-restricted Cre line that robustly expresses Cre in the dorsal NT and pre-migratory NC progenitors. Thus, it is expressed in emigrating NC progeny (Jiang et al., 2000; Stottmann et al., 2004). Wnt1-Cre activity is detectable in the NC at 5 somites and this coincides with the time at which the first NC cells have been detected by cell labeling studies (Serbedzija and McMahon, 1997), thus we used it to drive recombination of the floxed Pax3 allele within the NC from very early stages of development. Initially, Pax3flox/+;Wnt1-Cre;R26R(+/+) mice were crossed to Pax3flox/flox mice to obtain conditional knockout embryos that lack Pax3 in only dorsal NT and emigrating NC lineages. By including the recombination reporter R26R in our breeding scheme to ablate Pax3 in the NC, such that each resulting embryo carries one copy of R26R, we simultaneously marked both NCs and cells lacking Pax3 (though we cannot be certain that any specific labeled cell necessarily lacks Pax3, as the loxP sites in R26R and Pax3 recombine independently). Genotyping of >100 embryos (E10.5–E19.5) demonstrated that the conditional Pax3Wnt1Δ5/Wnt1Δ5 mutants were present at expected Mendelian ratios, indicating an absence of OFT defects and embryonic lethality. This was also confirmed via histology (not shown).
Figure 5. Wnt1-Cre-medidated conditional deletion of Pax3 within the NC lineage does not affect OFT septation but does perturb NC-derived Schwann cell morphogenesis.

(A–F) Wnt1-Cre;R26R lacZ lineage mapping of wildtype (A–C) and Pax3Δ5/Wnt1Δ5 conditional mutant NC cells (D–F). CNC colonization of E10.5 mutant 4/6th PAAs and OFT is equivalent to wildtype, but E11.5 mutants exhibit disorganized Schwann cell morphogenesis and clump on neurons (arrows in E). Conditional mutants also exhibit exencephaly (D, F), but are otherwise identical to wildtype littermates. (G&H) In situ hybridization with Pax3 full length probe (G) and Pax3 exon5-specific probe (H) on adjacent conditional mutant E10 sections. Note Δ5 transcripts are missing in uppermost dorsal NT (arrow in H). Phase contrast images are inset to verify tissue integrity. Both exon5-specific and full length probes are identically expressed in somites (som). (I&J) Consistent with in situ data, Pax3 protein is absent in mutant uppermost dorsal NT (arrows in J), but in region below Wnt1-Cre expression, Pax3 immunohistochemistry signal intensity is similar to control littermates. (K&L) Gross view of P10 pups showing generalized pigmentation defects in mutants (inset in L). LacZ staining- revealed melanocytes (arrow in K) in control epidermal basal layer (*) that are positive for melanin (brown), a marker for mature melanocytes. Mutants exhibit lacZ-positive melanocytes at similar densities but are all negative for melanin synthesis.
In order to maximize Cre recombination efficiency, Pax3Δ5/+;Wnt1-Cre;R26R(+/+) mice were crossed to Pax3flox/flox mice. Haplo-insufficient conditional Pax3Δ5/Wnt1Δ5 mutants were present at expected Mendelian ratios at birth (n=50). However, 56% (n=28/50) of Pax3Δ5/Wnt1Δ5 exhibited excencephaly (Fig. 5D,F) and die shortly after birth, but are not cyanotic. Consistent with Wnt1-Cre expression initiating ~E8.0 head-to-tail and posterior NT closure occurring prior to onset of Wnt1-Cre expression, the spina bifida evident in Pax3Δ5 nulls was not observed (Fig. 5F). Subsequently, the remaining surviving postnatal conditional mutants (n=20) are readily identified by their pigmentation defects (Fig. 5L). Examination of serial sections of E13.5 and newborn conditional mutant hearts (n= 20; 10 had exencephaly and 10 did not) did not reveal any structural heart defects and were indistinguishable from serially sectioned control littermates (n=6). Furthermore, both DRG and thymus development, which are also dependent on NC cells, were unaffected in Pax3Δ5/Wnt1Δ5 mutants. Consistent with observed normal heart histology, Wnt1-Cre;R26R lacZ staining did not reveal any gross alterations within CNC distribution during emigration, migration to arches or within OFT cushions in conditional mutant embryos (Fig. 5D). However, conditional Pax3Δ5/Wnt1Δ5 mutants NC-derived Schwann cells are aberrantly affected following Wnt1-Cre-mediated loss of Pax3 (Fig. 5E), as fewer fiber-like CNS neuronal connections to DRGs are present and Schwann cell distribution is non-uniform resulting in lacZ-positive clumps. Similarly, newborn conditional mutants displayed pigmentation defects due to anomalous differentiation of NC-derived melanocytes. This was determined based on the similar distribution of NC-derived melanocytes in the basal epidermis of both conditional mutants and control pups (10 day old), but negative for melanin synthesis in mutants (Fig. 5L). In support of these late gestational Pax3-dependent defects, it has been shown that Pax3-expressing adult melanocyte stem cells are committed but undifferentiated until Pax3-repression is relieved by an external βcatenin stimulus (Lang et al., 2005).
To confirm successful Wnt1-Cre-mediated recombination of the Pax3f/Δ5 allele, in situ hybridization was performed at E10 using both an exon5-specific Pax3 probe, which detects only wildtype Pax3 transcript. The exon5-specific probe did not detect signal in the dorsal-most region of the NT corresponding to the known Wnt1-Cre-expression domain (Fig. 5H). The exon5-specific probe, is robustly expressed in remaining non-Wnt1-Cre expressing E10 conditional mutant NT and is normally expressed throughout the somites (as Wnt1-Cre is not expressed in somite; Fig. 5H). In the adjacent conditional mutant NT sections, a N-terminus Pax3 probe (detects both wildtype and mutant Pax3Δ5 transcripts) revealed uniform expression throughout the Pax3 expression domain. In parallel, we measured recombination efficiency in purified NC-derived Schwann cells. Using a PCR strategy that amplifies both floxed and wildtype but not the recombined Δ5 allele, we assayed equal amounts of DNA from Pax3flox/+ cells with or without Wnt1-Cre. As expected given the altered lacZ expression (Fig. 5D,E), we were able to amplify the floxed allele in Wnt1-Cre-negative Schwann cells (Supplemental Fig. 1) but not within Wnt1-Cre-positive Schwann cells (even after 44 cycles). As an internal control, the wildtype Pax3 allele was readily amplified with same intensity in both genotypes, again supporting efficient recombination in Wnt1-Cre-expressing NC cells (Supplemental Fig. 1). Finally, immunohistochemistry analysis was performed using anti-Pax3 antibody at E10 to determine presence/absence of Pax3 protein. The antibody failed to detect any Pax3 protein in the Wnt1-Cre-expressing region of the NT of conditional mutants (Fig. 5J). Together, these results confirmed successful deletion of exon5 within the Wnt1-Cre-expressing dorsal most domain of the NT and in NC progeny.
Targeted early neural fold Pax3 deletion can result in OFT alignment defects
Due to the absence of PAA and/or OFT remodeling defects in the Pax3Δ5/Wnt1Δ5 conditional mutants, an alternative conditional approach was used in an attempt to recapitulate the Pax3Δ5/Δ5 phenotype. As Wnt1-Cre driven Cre recombinase itself is restricted to the dorsal NT (Danielian et al., 1998) and coincides with the onset of Pax3 expression (Goulding et al., 1991), we ablated Pax3 gene function with the targeted Ap2α-IRESCre knock-in driver (Macatee et al., 2003). Lineage analyses of Ap2α-IRESCre;R26R embryos revealed that Cre activity can be seen as early as E8 within the neural folds (Fig. 6A). Importantly, it marks CNC-derived cells that eventually colonize the OFT (Fig. 6B,C). As a result, this alternative conditional approach was used to determine if the lack of phenotype seen in Pax3Δ5/Wnt1Δ5 mutants was either due to an inability of Wnt1-Cre to successfully delete Pax3 early enough within the NC before they had undergone proper formation and specification. As anticipated, conditional Pax3Δ5/Ap2αΔ5 mutants exhibited OFT defects, although not as severe as systemic Pax3Δ5/Δ5 nulls. Significantly, they exhibited double outlet right ventricle (DORV; the aorta and pulmonary trunk both exit from the right ventricle) and VSD (n=14/14 mutants). Moreover, Pax3Δ5/Ap2αΔ5 mutants survived to term but they then rapidly developed cyanosis and died within 10 to 120 min after birth. DORV is an alignment defect thought to occur either when the OFT does not lengthen sufficiently to allow the juxtaposition of the OFT with the septating ventricular chambers (Li et al., 2010), abnormal myocardial rotation (Bajolle et al., 2006) and/or reduced colonization of the OFT by CNC lineage (Kirby et al., 1983; Epstein et al., 2000). Wholemount embryo analysis of lacZ-positive cells revealed decreased CNC migration and colonization of caudal PAA's (3rd/4th/6th) and OFT region (Fig. 6E,G). Histological analysis confirmed reduced distribution of Pax3Δ5/Ap2αΔ5 mutant CNC cells colonizing the arches and invading the OFT (Fig. 6I), but not as severe as the reductions observed in Pax3Δ5 nulls (Fig. 4). However, ectodermal derivatives were unaffected, as Pax3 is not expressed in this lineage (Fig. 6I; Goulding et al., 1991). Similar to Pax3Δ5/Wnt1Δ5 conditional mutants, the Pax3Δ5/Ap2αΔ5 conditional mutants exhibited 100% fully penetrant NT defects (Fig. 6F) and the DRG were hypoplastic. The presence of caudal NT defects supports the R26R results demonstrating earlier more extensive rostal-caudal Cre activity (Fig. 6A). Finally, in order to assess whether sustained Cre expression within migrating NC is required for OFT pathogenesis, we used the P0-Cre line (Yamauchi et al., 1999). P0-Cre activity is absent from the CNC-containing NT itself, but is present in E9 migratory NC and their progeny. As expected, Pax3Δ5/P0Δ5 mutant embryos were unaffected and were recovered postnatally at appropriate Mendelain ratios (n=8 litters). Gross examination revealed normal NT formation, and histology confirmed the absence of any structural cardiovascular abnormalities. P0-Cre;R26R reporter expression demonstrated that the Pax3Δ5/P0Δ5 NC lineage was unaffected (not shown). To conclude, Pax3Δ5/Wnt1Δ5 and Pax3Δ5/P0Δ5 mutants consistently exhibit normal hearts, which differed significantly from the characteristic DORV and PAt phenotypes seen in Pax3Δ5/Ap2aΔ5 and Pax3Δ5/Δ5 mutant embryos. This genetic data demonstrates that Pax3 function is specifically required within the early E8–8.5 neural fold prior to CNC emigration, but is dispensable during subsequent NC migration and colonization of the arches and OFT.
Pax3 function is not required within the cardiomyocytes, endothelial or secondary heart field lineages
Previously, we have shown that Sp2H mutants embryos exhibit compromised Ca2+ handling (Conway et al., 1997b), implying that Pax3 may play an indirect role in heart development. Similarly, an abnormally thinned myocardium is seen in Sp mutants (Li et al., 1999), but this is also thought to be indirect, as NC do not contribute directly to the myocardium (Jiang et al., 2000). Kirby et al. have shown that pre-migratory CNC ablation in chick results in myocardial dysfunction prior to CNC arrival within the OFT (Waldo et al., 1999). There are also reports that Pax3 may play a direct pathological role, as mice lacking the homeobox gene Lbx1, which is expressed in a subpopulation of CNC exhibit ectopic Pax3 upregulation in E12 mutant myocardium (Schafer et al., 2003). Further, Pax3-positive cells have been found in the E16 Pax3-FKHR knock-in fetal heart (Lagutina et al., 2002). Given these reports, we systematically ablated Pax3 in the cardiomyocyte, endothelial and anterior heart field lineages.
Initially, we crossed Pax3flox/Δ5;R26R(+/+) and Pax3+/Δ5; αMHC-Cre mice. αMHC-Cre drives Cre expression in myocardium from ~E9.5 onwards (Oh et al., 2003), to generate mutant embryos (n=6 litters) that lack Pax3 gene in cardiomyocytes. However, these mutants are unaffected in utero or postnatally (Supplemental Fig. 3). As αMHC-Cre may not express early enough, we crossed used the Nkx2.5Cre knockin mice (Moses et al., 2001). Nkx2.5Cre initiates Cre expression in early as ~E7.5 within the cardiac crescent cells. In addition to deletion of Pax3 from cardiomyocytes, Nkx2.5Cre also deleted Pax3 from pharyngeal arches through which the CNCs travel. However, conditional Pax3Δ5/Nkx2.5Δ5 mice (n=6 litters) live to adulthood without overt cardiovascular defects (Supplemental Fig. 4B). As it has been reported that loss of Pax3 and an absence of NC can disrupt the distribution of SHF cells (Bradshaw et al., 2009) and that signaling between the SHF and NC is critical for OFT morphogenesis (Li et al., 2010), we used both the Nkx2.5Cre and Wnt1-Cre drivers in tandem to delete Pax3 from the NC, SHF and myocardium (n=22 litters). Surprisingly, the addition of the Nkx2.5Cre allele did not alter the already observed Pax3Δ5/Wnt1Δ5 phenotypes and Pax3Δ5/Wnt1-Nkx2.5 Δ5 OFT morphogenesis was unperturbed (Supplemental Fig. 4D). Finally, given the cross-talk between the myocardium and endocardium during early heart development, we intercrossed Pax3flox/Δ5;R26R(+/+) female and Pax3+/Δ5:Tie2-Cre male mice. However, resultant conditional Pax3Δ5/Tie2Δ5 mice (n=8 litters) are viable and unaffected (not shown). As Pax3 is not normally thought to be expressed in myocardium or endocardium (Engleka et al., 2005), these data demonstrate that normal Pax3 function is not required during myocardial, SHF or endocardial morphogenesis.
Wnt1-Cre-restricted genetic cell ablation results in OFT defects
As we did not detect the expected OFT phenotypes in NC-restricted conditional Pax3 knockouts, we sought to address whether Pax3 functions earlier within the NT and the Wnt1-Cre expressing lineage is downstream of Pax3. Additionally, it was important to test whether Wnt1-Cre was consistently expressed in sufficient CNC progenitors within the dorsal most NT and whether genetic ablation of Wnt1-Cre-expressing cells is sufficient to always result in PAt and/or PTA. Null Wnt1 mice are viable but Wnt1/Wnt-3a double nulls have a marked deficiency in NC derivatives and a pronounced reduction in dorsolateral neural precursors suggesting Wnt signaling regulates the expansion of dorsal neural precursors (Ikeya et al., 1997). Therefore, we crossed Wnt1-Cre and R26rDTA mice (Ivanova et al. 2005). Cre-mediated activation of diphtheria toxin fragment-A (DTA) kills cells via apoptosis by interfering with the RNA translation machinery in each individual cell. This system is highly cell autonomous as only cells that express the DTA are ablated without any effects on neighboring non-Cre expressing cells (Ivanova et al. 2005; Snider et al., 2009). Ablation of Wnt1-Cre-expressing NC lineage led to fully penetrant PTA defects (Fig. 7; n=24/24 mutants). Histology and immunohistochemistry confirmed the presence of Type-II PTA (Van Praagh & Van Praagh, 1965) as they had a single persisting stunted OFT with a single valve, and concomitant VSDs in 100% of the Wnt1-Cre;R26rDTA ablated hearts (Fig. 7D,F). However, instead of a single OFT vessel with 3 semilunar valve cusps as seen in Pax3Δ5 nulls, the Wnt1-Cre;R26rDTA mutants had a single OFT with multiple variable odd-shaped valve leaflets (usually 4 but sometimes as many as 6 disproportionate cusps). This data is consistent with previous findings using Herpes simplex virus-1 thymidine kinase and Wnt1-Cre to ablated NC (Porras and Brown, 2008). Wnt1-Cre;R26rDTA ablated mutants also displayed craniofacial defects (Fig. 7A) including severe microcephaly consistent with significant loss of cranial NC contribution to the craniofacial region. An absence of NC-derived structures such as thymus, DRG, and Schwann cells was also observed (not shown), as well as a diminutive neural tube (Supplemental Fig. 5). However, consistent with the report that apoptosis is not involved in neurulation (Massa et al., 2009), the Wnt1-Cre;R26rDTA ablated mutants did not exhibit any NT closure defects. Given that NT closure defects are consistently observed in Pax3Δ5/Δ5, Pax3Δ5/Wnt1Δ5 and Pax3Δ5/Ap2αΔ5 mutants and NT closure and NC migration defects have been functionally dissociated in Splotch mice (Franz, 1992; Estiberio et al., 1993), these data demonstrate that development of Pax3-associated NT and NC defects are caused by independent mechanisms. Although it would have been useful, unfortunately Ap2α−IRESCre;R26rDTA and P0-Cre;R26rDTA genetic ablation resulted in very early embryonic lethality prior to OFT septation due to Cre expression within the allantois (Supplemental Fig. 2B) and yolk sac (not shown).
Figure 7. Evaluation of OFT phenotypes following Cre/loxP-mediated genetic NC ablation in Wnt1-Cre;R26-EGFP-DTA embryos.

(A–C) Gross examination of E15 Wnt1-Cre;R26-EGFP-DTA (A), R26-EGFP-DTA only (B) and wildtype non-transgenic sibling controls (C) viewed under UV. Note lack of craniofacial structures (open arrowhead) and internalized eyes following genetic NC ablation (A). Insets show same embryos viewed under brightfield. The R26-EGFP-DTA non-transgenic sibling embryos are normal, indicative of absent non-specific DTA expression in non-appearance of the Wnt1-Cre transgene. (D–G) Subsequent transverse sectioning and counterstaining with either (D,E) αSMA immunohistochemistry (brown DAB staining) or (F,G) Hematoxylyn/Eosin (blue/pink) revealed that the Wnt1-Cre;R26-EGFP-DTA mutants exhibit PTA with stunted OFT and multiple irregular valve leaflets (indicated by * in D) and a large VSD (* in F), when compared to normal wildtype or R26-EGFP-DTA only (G) littermate control hearts.
In order to assess when and where the Wnt1-Cre expressing lineage was ablated, we crossed in the R26R reporter (Soriano, 1999) to assess Cre-mediated recombination activity (Fig. 8A–C). Significantly, R26R reporter expression was largely absent by E8.5 (Fig. 8A) and there were no lacZ marked NC cells detected along the NC migration route, within the arches or colonizing the OFT endocardial cushions (Fig. 8A,B). Therefore, this data suggests that the DTA is working very quickly-within ~6 hours of Cre expression and successive recombination. Additionally, elevated TUNEL staining was detectable within the E8–8.5 Wnt1-Cre;R26rDTA mutant neural folds (not shown). This suggests that Wnt1-Cre mediated DTA ablation of NC progenitors occurs within the NT itself. Finally, as this ablation system completely abrogated NC migration and colonization of the PAs/OFT but Pax3Δ5 nulls continue to have some mutant migratory CNC cells within the arches and OFT, we assessed whether PAA remodeling was similarly affected. Intracardiac ink injections revealed that in contrast to the Pax3Δ5 nulls, PAA remodeling was randomized in Wnt1-Cre;R26rDTA mutants (Fig. 8D–I). Significantly, anterior arch arteries that usually regress now persist, while remodeling of the Wnt1-Cre;R26rDTA 3rd, 4th and 6th PAAs is completely variable. In order to determine if indeed the entire NC population was being ablated, marker analyses on ablated mutant embryos at E12 were performed (Supplemental Fig. 5). Using a series of well-established markers of premigratory NC (Wnt1), NT and emigrating NC (Pax3) and both premigratory and migratory NC (Ap2α), the efficiency of Wnt1-Cre-mediated ablation was confirmed. As expected, both Wnt1 and the dorsal-most Pax3 expression domain were absent, and there was a total lack of Ap2α expressing NC progeny within the ablated mutants. These data demonstrate that Wnt1-Cre is consistently expressed in CNC progenitors and loss of the Wnt1-Cre progeny along a rostrocaudal axis is sufficient to cause OFT defects. However, Wnt1-Cre mediated genetic ablation of the NC does not exactly phenocopy the Pax3Δ5 null mouse heart defects and suggests that the presence of Pax3Δ5 null NC is able to drive normal 3rd and 4th PAA remodeling. Thus, the pathogenesis of congenital cardiovascular defects which are due to a complete absence of CNC cells themselves may not be akin to those in which mutant CNC are present, either in reduced numbers or in normal numbers but exhibiting compromised differentiation ability.
Figure 8. Pathogenesis of Wnt1-Cre;R26-EGFP-DTA-mediated genetic NC ablation cardiovascular defects.

(A) In order to visualize the DTA expressing cells and to determine how quickly the DTA caused NC apoptosis, we utilized the R26R reporter mice that express lacZ in every cell in which Wnt1-Cre is expressed. By E8.5, the lacZ positive Wnt1-Cre-expressing NC are almost completely ablated within the embryo and only a few Wnt1-Cre cells remain in the Wnt1-Cre;R26-EGFP-DTA/R26R triple transgenic cranial region (indicated via open arrowhead). This demonstrates that this system ablated NC within ~6 hours following onset of Wnt1-Cre-mediated recombination. Note that in the Wnt1-Cre;R26R double transgenic non-ablated control littermate, that cranial and CNC are lacZ positive but that the more posterior trunk NC are yet to be labeled (*). (B,C) Similarly, E11Wnt1-Cre;R26-EGFP-DTA;R26R triple transgenic ablated embryos (B) completely lack any CNC within the pharyngeal arches or any colonization of the mutant OFT, but normal Wnt1-Cre;R26R double transgenic non-ablated control littermates (C) exhibit robust CNC colonization. Also note the absence of Wnt1-Cre-marked NC in the mutant DRG, when compared to normal littermates (indicated by * in C). (D–I) Ink was injected into beating hearts of live E11.5 embryos to visualize the vasculature. By E11.5, the PAAs should have undergone asymmetrical remodeling. Consequently in normal embryos, the right 4th and 6th PAAs regress, and the left 4th and 6th persist to become the aortic artery and pulmonary trunk. Three ink injected Wnt1-Cre;R26-EGFP-DTA;R26R triple transgenic ablated mutants illustrating the various PAA remodeling abnormalities both the left and right side of the mutant embryos. (D,E) Mutant one has just a single abnormal persisting PAA on both sides. (F,G) Mutant two has a 3rd and 6th on the right, but persisting 1st, 3rd, 4th PAA on the left. (H,I) Mutant three has persisting 1st, 2nd, 3rd, 4th and 6th on the right, as well as persisting 1st, 2nd, 4th and 6th on the left but no left 3rd PAA.
DISCUSSION
In this work, we sought to determine the requirements for Pax3 in CNC morphogenesis by comparing systemic Pax3Δ5 mutants with those that had Pax3 specifically deleted in the NC domain from early somite stages. Our study reveals a critical requirement for Pax3 in NC progenitor formation within the neuroepithelium, and that this early loss of Pax3 results in subsequent anomalous NC delamination and migration from the NT and defective 6th PAA remodeling and conotruncal PAt defects. Significantly, as the Pax3Δ5 allele cardiac PAt OFT phenotypes are 100% penetrant and do not exhibit variability, this enabled us to confidently examine their in utero pathogenesis. We further demonstrated that systemic mutation of Pax3 does not entirely prevent CNC specification and that the Sp2H and Pax3Δ5 mutations are allelic. Surprisingly, Wnt1-Cre-mediated NC-restricted loss of Pax3 does not affect CNC migration, colonization or differentiation in the cardiovascular system. This is despite the fact that Wnt1-Cre-mediated genetic ablation of Wnt1-Cre expressing NC progenitors results in fully penetrant PTA OFT defects. The NC progenitor function for Pax3 is supported by Ap2α−IRESCre lineage restricted Pax3 deletion, as Pax3Δ5/Ap2αΔ5 embryos lacking early neural fold Pax3 function do exhibit discernable defects in CNC morphogenesis and OFT maldevelopment. Finally, Cre/loxP analysis revealed that the Δ5 Pax3 mutant transcript does not play a dominant-negative role in NC, cardiomyocyte, endothelial or second heart field-derived lineages. Although the deduced Δ5 protein structure was confirmed by Western analysis using an antibody that recognizes the C-terminus of Pax3 (Fig. 1A), lacking an antibody which recognizes the Pax3 N-terminus, we cannot verify the presence of truncated corresponding Δ5 protein in Pax3Δ5/Δ5 samples (despite the presence of Δ5 transcripts; Fig. 5G). Nevertheless, Western analysis confirms that deletion of exon5 leads to elimination of Pax3 wildtype protein and phenocopies Sp2H, suggesting the allele is behaving as designed (Koushik et al., 2002). Thus, these studies confirm that the Wnt1-expressing lineage is essential for CNC morphogenesis and that Pax3 function in the CNC progenitors is necessary prior to Wnt1-Cre expression within the NC lineage. Recent studies involving the use of Wnt1-Cre to restore Pax3 enhancer function in the NC in an already Pax3-deficient environment (Degenhardt et al., 2010), supports our data and suggests that the Wnt1-Cre-expressing population of NC cells is sufficient to regulate OFT remodeling when these cells are properly specified. Moreover, new data suggests that Ap2α itself may play a unique multistep role in early Xenopus NC development, first in positioning of the neural border and directly activating XPax3 in cooperation with XWnt signals, followed by specification of the NC (de Croze et al., 2011). However, the fact that systemic Ap2α null mice exhibit mainly DORV, survive until birth and Pax3 expression is not dependent upon Ap2α (Schorle et al., 1996; Brewer et al., 2002); and that our Pax3Δ5/Ap2αΔ5 conditional mutants only exhibit DORV and also survive until birth, suggest that in mammals Ap2α is unlikely to be upstream of Pax3 within the cardiac NC lineage. As Pax3 is one of the first transcription factors to be expressed within the neuroepithelium coincident with NC induction (Goulding et al., 1991; Bang et al., 1997; Sauka-Spengler & Bronner-Fraser, 2008; Chang et al., 2008), our lineage-restricted Pax3 deletion and genetic NC ablation approaches are consistent with the hypothesis that Pax3 is a key CNC-specific inducing factor within the early NT.
Reduced colonization of the OFT has been proposed to be the primary defect that ultimately leads to a lack of a Pax3-deficent CNC phenotypes (Conway et al., 1997a; Epstein et al., 2000; Conway et al., 2000; Chan et al., 2004). The severity of these defects increases along the rostrocaudal axis, as no NC derivatives are present in the caudal portion of the Pax3-mutant embryos (Serbedzija & McMahon, 1997). Transgenic Pax3 re-expression in Sp nulls under a proximal 1.6kb Pax3 promoter, which is capable of driving Pax3 in the dorsal NT and early migratory NC (Li et al., 1999; Milewski et al., 2004), is sufficient to rescue in utero CNC defects. Similarly, Maxson et al. showed combined deletion of the Msx2 transcription factor, which is co-expressed within the Pax3 expression domain in the NT, can rescue the Sp mutant cardiovascular system (Kwang et al., 2002). Moreover, direct labeling of premigratory NC and orthotopic transplantation has confirmed that Sp2H mutant CNC are capable of migrating along normal pathways, but in significantly reduced numbers (Chan et al., 2004). Collectively these studies support the suggestion that a decrease in the number of migrating CNC is the primary defect that ultimately leads to a lack of CNC-derived cells in the OFT of Sp2H embryos (Conway et al., 2000). While these data indicate Pax3 plays a NC-specific role, there remains controversy surrounding the issue of the cell-autonomy vs. non-cell-autonomy (Chan et al., 2004). Specifically, Sp2H mutant NC derived from the caudal trunk level (Serbedzija & McMahon, 1997) or CNC vagal level (Conway et al., 2000) can follow normal migratory pathways after transplantation to chick embryos, suggesting the environment plays a crucial role in regulating NC migration. Similarly, reciprocal grafting of premigratory NC between wildtype and Sp2H embryos revealed retardation of CNC migration in Sp2H mutant embryos requires the genetic defect in both NC and their migratory environment (Chan et al., 2004). Chimera studies, in which wildtype and Pax3-deficient cells co-exist in the same embryo (Mansouri et al., 2001), have additionally supported a non-cell autonomous mechanism for the Sp NC defect. Extracellular matrix abnormalities are also consistent with a non-permissive migratory environment along the CNC pathways (Henderson et al., 1997). This suggests that migrating NC might also actively participate in shaping the molecular composition of their own migratory routes. Unexpectedly, given this wealth of transgenic data, our lineage restricted Pax3 conditional knockout studies have revealed that Pax3 function is not required in CNC cells once they have initiated emigration from the NT, but is required cell autonomously in the early neural folds within a brief time window in pre-somitic ~E8 embryos.
The ultimate identity of a NC cell is determined in response to both intrinsic and extrinsic influences that operate differently along the neural axis, migratory paths, and homing sites (Raible, 2006; Sauka-Spengler & Bronner-Fraser, 2008). Significantly, streams of migrating Cx43-lacZ-positive NC appeared disorganized in Pax3Δ5 nulls, suggesting either the permissive pathways are inappropriately specified or that Pax3-deficient NC lack guidance signals. Additionally, the Wnt1-Cre;R26R marked CNC within the Pax3Δ5 null OFT appear to have lost retinoic acid responsiveness (Fig. 2). As both the Pax3Δ5 and Wnt1-Cre;R26rDTA mutant embryos exhibited fully penetrant PAA defects and conotruncal defects but Pax3Δ5/Wnt1Δ5 and Pax3Δ5/P0Δ5 mutants failed to exhibit any CNC-associated phenotypes, this suggests that either Pax3 is required in non-NC lineages or that Wnt1-Cre is unable to appropriately conditionally delete Pax3 early enough. Although our data and that of Chang et al. (2008) indicates that the onset of Pax3 precedes Wnt1-Cre expression within the neural folds and that Wnt1-Cre only deletes the dorsal most domain of Pax3 expression, Wnt1-Cre is able to effectively delete Pax3 and disrupt both NC-derived melanocyte and Schwann cell morphogenesis (Fig. 5 and Supplemental Fig. 1). As Wnt1-Cre;R26rDTA mutant embryos exhibit fully penetrant CNC-associated defects, this reveals that the dorsal most domain of Pax3 expression is the source of the CNC lineage and that the remaining NT Pax3-expressing cells are unable to compensate for the loss of the ablated Wnt1-Cre population. Furthermore, the finding that Pax3Δ5/Ap2αΔ5 embryos exhibit DORV and reduced CNC colonization of the PAs and OFT, supports the hypothesis of a cell autonomous function of Pax3 within CNC themselves. Given that the mesodermal Pax3-expressing somites are thought to control the rostrocaudal patterning of the migratory NC (Bronner-Fraser, 1995), that somites can coordinate the timing of NC emigration from the NT (Sela-Donenfeld & Kalcheim, 2000) and that the more caudal CNC are known to migrate through the somites; these results suggest Pax3Δ5 null CNC-associated phenotypes involve abnormal NC patterning and signaling independent of the adjacent Pax3-deficent somites. This is supported via previous NT-restricted transgenic rescue of Sp nulls (Li et al., 1999) and our Wnt1-Cre and P0-Cre results.
The lack of an observable phenotype within the Pax3Δ5/Wnt1Δ5 mutants raises the intriguing possibility that Wnt1-Cre may not be marking the entire CNC lineage. Based on our lineage mapping using the earlier expressing Ap2α-IRESCre mice, we detected lacZ-positive NC cells within the cardiac neural fold region within pre-somitic ~E7.5 embryos (Supplemental Fig. 2). Thus, it is possible that an earlier wave of CNC cells may be sufficient for modulating OFT remodeling in the absence of properly specified Wnt1-Cre-expressing CNC cells. The fact that Pax3Δ5/Ap2αΔ5 conditional mutants and Pax3Δ5/Δ5 systemic mutants exhibit different OFT septation defects, lends some credence to this speculation. In fact, other studies have confirmed the inability of Wnt1-Cre-mediated deletion of genes, which are known to be important for NC induction, to affect conotruncal phenotypes. Wnt1-Cre-mediated conditional knockouts of both Foxd3 and Connexin43 failed to yield fully penetrant OFT defects. However, more severe OFT defects were observed when the earlier and more extensive transgenic Pax3-Cre line was used to ablate these genes (Liu et al., 2006; Nelms et al., 2010). This suggests that while the Wnt1-Cre-expressing CNC cell subpopulation is essential for OFT morphogenesis, its remodeling may not be solely dependent on this cell population. Rather, an earlier non-Wnt1-Cre-expressing NC subpopulation may also play a role in tandem and/or in parallel. Nevertheless, these data demonstrate the need to further explore the contribution, if any, of a non-Wnt1-Cre-expressing cell subpopulation within mammalian OFT morphogenesis.
Abnormal regression of left 6th PAA is the first observable anatomical defect in Pax3Δ5 null cardiovascular system, and is the site at which the consequences of abnormal CNC development are initially most pronounced. Conotruncal defects can either be caused via a defect in OFT septation or a failure of the left 6th artery to persist. Furthermore, defects within OFT septation are not always associated with the failure of this arch artery to persist (Kirby et al., 1997; Ya et al., 1998). Thus, the single OFT Pax3Δ5 PAt phenotype is primarily the result of the left 6th PAA remodeling defect. The specific regression in the left 6th, despite reductions in migratory CNC to both 3rd and 4th Pax3Δ5 null PAAs could suggest a potential left 6th specific role encoded by Pax3 within the CNC. It may also be indicative of a 6th PAA over-sensitivity to loss of CNC when compared to the other more cranial PAA's (i.e., 3rd/4th). Surely, the rostrocaudal progressive loss of migratory CNC observed in Pax3Δ5/Δ5 mutant PAA's (based on both Wnt1-Cre and Ap2αCre lineage mapping data), which is most severe in the 6th compared to 3rd/4th PAAs, lends credence to this theory. Furthermore, the discovery of a single OFT vessel with 3 well-proportioned semilunar valve cusps in Pax3Δ5 nulls as opposed to a single OFT with multiple odd shaped (4–6) cusps in Wnt1-Cre;R26rDTA PTA mutants, supports the idea that loss of the left 6th PAA is key and that the presence of even reduced Pax3Δ5 null CNC numbers within the E11 aortic sac region during OFT septation is not sufficient to initiate division. These data also imply that presence of CNC within the OFT, even mutant CNC, is more likely to result in PAt rather than PTA which is the hallmark of an OFT that is totally devoid of CNC (as seen with chick neural fold surgical ablation and within Wnt1-Cre;R26rDTA genetic ablation mutants). Furthermore, although migratory NC's are not important for the initial formation of PAA's, they are required for their remodeling (Waldo et al., 1996). This has been successfully demonstrated by surgical neural fold ablation studies done in chick embryos, which result in a wide variation of PAA anomalies (Kirby et al., 1983; Waldo et al., 1996; Creazzo et al., 1998). The complete absence of migratory CNC in Wnt1-Cre;R26rDTA mutant PAs and the randomized PAA defects observed, indicate that the abnormal regression of left 6th PAA is most likely due to CNC deficiency rather than any Pax3 encoded function within the CNC.
In patients, haplo-insufficient PAX3 mutations lead to Waardenburg syndrome, an autosomal-dominant disorder that consists of defects in NC-derived tissues and is characterized by pigmentation, hearing, and facioskeletal anomalies (Read & Newton, 1997). Cardiac defects have also been reported in some Waardenburg children (Khaldi et al., 1990; Mathieu et al., 1990). By methodically comparing Pax3Δ5/Δ5 systemic mutants to both Pax3 NC-restricted conditional and Wnt1-Cre;R26rDTA NC-ablated mutants, we have been able to tease out the Pax3-specific cell autonomous role in NC-related congenital heart defects. Interestingly, the phenotype of the Pax3Δ5 null, Pax3Δ5/Ap2aΔ5 and Wnt1-Cre;R26rDTA mutant mice each have similarities to the human disease DiGeorge syndrome (DGS). DGS patients typically present with craniofacial, cardiovascular and thymic anomalies arising from a 3-Mb deletion of chromosome 22q11 (Scambler, 2010). Significantly, at least one of the genes within the critical chromosome 22q11 region called HIRA, is known to interact with Pax3 and is co-expressed with Pax3 in the early neural folds (Scambler, 2010). The results presented here suggest a crucial role for Pax3 in specifying the CNC lineage progenitors and might have implications for the potential contribution of anomalous NC dysregulation in human neurocristopathies.
Supplementary Material
Acknowledgements
We are grateful to Drs. Cecelia Lo (Cx43-lacZ), Melissa Colbert (RARE-lacZ), Henry Sucov (Wnt1-Cre), Weinian Shou (αMHCCre), D. Wade Clapp (P0-Cre), Lak Koni (Tie2-Cre) and Andy Copp (ROSA26-eGFP-DTA) for providing mice lines and to Prof. Margaret Kirby for her many helpful and insightful comments. The Pax3 and Pax7 antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. These studies were supported, in part, by American Heart Association Pre-doctoral Fellowship (to MO), Riley Children's Foundation (HZ), Indiana University Department of Pediatrics (Neonatology) and NIH HL60714 grant (SJC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–79. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajolle F, Zaffran S, Kelly RG, Hadchouel J, Bonnet D, Brown NA, Buckingham ME. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ Res. 2006;98:421–8. doi: 10.1161/01.RES.0000202800.85341.6e. [DOI] [PubMed] [Google Scholar]
- Bang AG, Papalopulu N, Kintner C, Goulding MD. Expression of Pax-3 is initiated in the early neural plate by posteriorizing signals produced by the organizer and by posterior non-axial mesoderm. Development. 1997;124:2075–85. doi: 10.1242/dev.124.10.2075. [DOI] [PubMed] [Google Scholar]
- Bradshaw L, Chaudhry B, Hildreth V, Webb S, Henderson DJ. Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch(2H) J Anat. 2009;214:245–57. doi: 10.1111/j.1469-7580.2008.01028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP-2alpha in cardiac outflow tract morphogenesis. Mech Dev. 2002;110:139–49. doi: 10.1016/s0925-4773(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M. Patterning of the vertebrate neural crest. Perspect Dev Neurobiol. 1995;3:53–62. [PubMed] [Google Scholar]
- Chan WY, Cheung CS, Yung KM, Copp AJ. Cardiac neural crest of the mouse embryo: axial level of origin, migratory pathway and cell autonomy of the splotch (Sp2H) mutant effect. Development. 2004;131:3367–79. doi: 10.1242/dev.01197. [DOI] [PubMed] [Google Scholar]
- Chang CP, Stankunas K, Shang C, Kao SC, Twu KY, Cleary ML. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development. 2008;135:3577–86. doi: 10.1242/dev.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–9. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Colbert MC, Linney E, LaMantia AS. Local sources of retinoic acid coincide with retinoid-mediated transgene activity during embryonic development. Proc Natl Acad Sci U S A. 1993;90:6572–6. doi: 10.1073/pnas.90.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res. 2000;47:314–28. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Godt RE, Hatcher CJ, Leatherbury L, Zolotouchnikov VV, Brotto MA, Copp AJ, Kirby ML, Creazzo TL. Neural crest is involved in development of abnormal myocardial function. J Mol Cell Cardiol. 1997a;29:2675–85. doi: 10.1006/jmcc.1997.0499. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997b;124:505–14. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–86. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–6. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- de Crozé N, Maczkowiak F, Monsoro-Burq AH. Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc Natl Acad Sci U S A. 2011;108:155–60. doi: 10.1073/pnas.1010740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt KR, Milewski RC, Padmanabhan A, Miller M, Singh MK, Lang D, Engleka KA, Wu M, Li J, Zhou D, Antonucci N, Li L, Epstein JA. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol. 2010;339:519–27. doi: 10.1016/j.ydbio.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–78. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Parmacek MS. Recent advances in cardiac development with therapeutic implications for adult cardiovascular disease. Circulation. 2005;112:592–7. doi: 10.1161/CIRCULATIONAHA.104.479857. [DOI] [PubMed] [Google Scholar]
- Estibeiro JP, Brook FA, Copp AJ. Interaction between splotch (Sp) and curly tail (ct) mouse mutants in the embryonic development of neural tube defects. Development. 1993;119:113–21. doi: 10.1242/dev.119.1.113. [DOI] [PubMed] [Google Scholar]
- Franz T. Neural tube defects without neural crest defects in splotch mice. Teratology. 1992;46:599–604. doi: 10.1002/tera.1420460609. [DOI] [PubMed] [Google Scholar]
- Goddeeris MM, Schwartz R, Klingensmith J, Meyers EN. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development. 2007;134:1593–604. doi: 10.1242/dev.02824. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–47. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson DJ, Ybot-Gonzalez P, Copp AJ. Over-expression of the chondroitin sulphate proteoglycan versican is associated with defective neural crest migration in the Pax3 mutant mouse (splotch) Mech Dev. 1997;69:39–51. doi: 10.1016/s0925-4773(97)00151-2. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18:101–10. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S. Wnt signalling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–70. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- Ismat FA, Xu J, Lu MM, Epstein JA. The neurofibromin GAP-related domain rescues endothelial but not neural crest development in Nf1 mice. J Clin Invest. 2006;116:2378–84. doi: 10.1172/JCI28341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis. 2005;43:129–35. doi: 10.1002/gene.20162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Khaldi F, Serbegi M, Mokadem H, Lazzem B, Bennaceur B. Waardenburg syndrome. Report of a familial case. Ann Pediatr (Paris) 1990;37:55–8. [PubMed] [Google Scholar]
- Kirby ML. Pulmonary atresia or persistent truncus arteriosus: is it important to make the distinction and how do we do it? Circ Res. 2008;103:337–9. doi: 10.1161/CIRCRESAHA.108.174862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–61. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Hunt P, Wallis K, Thorogood P. Abnormal patterning of the aortic arch arteries does not evoke cardiac malformations. Dev Dyn. 1997;208:34–47. doi: 10.1002/(SICI)1097-0177(199701)208:1<34::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–54. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushik SV, Chen H, Wang J, Conway SJ. Generation of a conditional loxP allele of the Pax3 transcription factor that enables selective deletion of the homeodomain. Genesis. 2002;32:114–7. doi: 10.1002/gene.10051. [DOI] [PubMed] [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, Maas RL, Maxson RE., Jr. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129:527–38. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- Lagutina I, Conway SJ, Sublett J, Grosveld GC. Pax3-FKHR knock-in mice show developmental aberrations but do not develop tumors. Mol Cell Biol. 2002;22:7204–16. doi: 10.1128/MCB.22.20.7204-7216.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D, Lu MM, Huang L, Engleka KA, Zhang M, Chu EY, Lipner S, Skoultchi A, Millar SE, Epstein JA. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature. 2005;433:884–7. doi: 10.1038/nature03292. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Dev Biol. 1974;41:162–84. doi: 10.1016/0012-1606(74)90291-7. [DOI] [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Li P, Pashmforoush M, Sucov HM. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev Cell. 2010;18:480–5. doi: 10.1016/j.devcel.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley A, Snider P, Zhou H, Rogers R, Wang J, Olaopa M, Kruzynska-Frejtag A, Koushik SV, Lilly B, Burch JB, Firulli AB, Conway SJ. Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Dev Biol. 2007;307:340–55. doi: 10.1016/j.ydbio.2007.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu F, Schneider AE, St Amand T, Epstein JA, Gutstein DE. Distinct cardiac malformations caused by absence of connexin 43 in the neural crest and in the non-crest neural tube. Development. 2006;133:2063–73. doi: 10.1242/dev.02374. [DOI] [PubMed] [Google Scholar]
- Lo CW, Cohen MF, Huang GY, Lazatin BO, Patel N, Sullivan R, Pauken C, Park SM. Cx43 gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev Genet. 1997;20:119–32. doi: 10.1002/(SICI)1520-6408(1997)20:2<119::AID-DVG5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130:6361–74. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A. The role of Pax3 and Pax7 in development and cancer. Crit Rev Oncog. 1998;9:141–9. doi: 10.1615/critrevoncog.v9.i2.40. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Pla P, Larue L, Gruss P. Pax3 acts cell autonomously in the neural tube and somites by controlling cell surface properties. Development. 2001;128:1995–2005. doi: 10.1242/dev.128.11.1995. [DOI] [PubMed] [Google Scholar]
- Massa V, Savery D, Ybot-Gonzalez P, Ferraro E, Rongvaux A, Cecconi F, Flavell R, Greene ND, Copp AJ. Apoptosis is not required for mammalian neural tube closure. Proc Natl Acad Sci U S A. 2009;106:8233–8. doi: 10.1073/pnas.0900333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Bourges E, Caron F, Piussan C. Waardenburg's syndrome and severe cyanotic cardiopathy. Arch Fr Pediatr. 1990;47:657–9. [PubMed] [Google Scholar]
- Meulemans D, Bronner-Fraser M. Gene-regulatory interactions in neural crest evolution and development. Dev Cell. 2004;7:291–9. doi: 10.1016/j.devcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA. Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator of Pax3. Development. 2004;131:829–37. doi: 10.1242/dev.00975. [DOI] [PubMed] [Google Scholar]
- Morgan SC, Relaix F, Sandell LL, Loeken MR. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008;82:453–63. doi: 10.1002/bdra.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2–5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–80. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Nelms BL, Pfaltzgraff ER, Labosky PA. Functional interaction between Foxd3 and Pax3 in cardiac neural crest development. Genesis. 2010 doi: 10.1002/dvg.20686. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A. 2003;100:12313–8. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras D, Brown CB. Temporal-spatial ablation of neural crest in the mouse results in cardiovascular defects. Dev Dyn. 2008;237:153–62. doi: 10.1002/dvdy.21382. [DOI] [PubMed] [Google Scholar]
- Raible DW. Development of the neural crest: achieving specificity in regulatory pathways. Curr Opin Cell Biol. 2006;18:698–703. doi: 10.1016/j.ceb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–65. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist TH, Finnell RH. Another key role for the cardiac neural crest in heart development. Am J Physiol Heart Circ Physiol. 2007;292:H1225–6. doi: 10.1152/ajpheart.01218.2006. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. Evolution of the neural crest viewed from a gene regulatory perspective. Genesis. 2008;46:673–82. doi: 10.1002/dvg.20436. [DOI] [PubMed] [Google Scholar]
- Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol. 2010;31:378–90. doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- Schafer K, Neuhaus P, Kruse J, Braun T. The homeobox gene Lbx1 specifies a subpopulation of cardiac neural crest necessary for normal heart development. Circ Res. 2003;92:73–80. doi: 10.1161/01.res.0000050587.76563.a5. [DOI] [PubMed] [Google Scholar]
- Scholl AM, Kirby ML. Signals controlling neural crest contributions to the heart. Wiley Interdiscip Rev Syst Biol Med. 2009;1:220–7. doi: 10.1002/wsbm.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–8. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- Sela-Donenfeld D, Kalcheim C. Inhibition of noggin expression in the dorsal neural tube by somitogenesis: a mechanism for coordinating the timing of neural crest emigration. Development. 2000;127:4845–54. doi: 10.1242/dev.127.22.4845. [DOI] [PubMed] [Google Scholar]
- Serbedzija GN, McMahon AP. Analysis of neural crest cell migration in Splotch mice using a neural crest-specific LacZ reporter. Dev Biol. 1997;185:139–47. doi: 10.1006/dbio.1997.8551. [DOI] [PubMed] [Google Scholar]
- Snider P, Conway SJ. Developmental biology: the power of blood. Nature. 2007;450:180–1. doi: 10.1038/450180a. [DOI] [PubMed] [Google Scholar]
- Snider P, Fix JL, Rogers R, Peabody-Dowling G, Ingram D, Lilly B, Conway SJ. Generation and characterization of Csrp1 enhancer-driven tissue-restricted Crerecombinase mice. Genesis. 2008a;46:167–76. doi: 10.1002/dvg.20379. [DOI] [PubMed] [Google Scholar]
- Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald R, Conway SJ. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008b;102:752–60. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Olaopa M, Firulli AB, Conway SJ. Cardiovascular development and the colonizing cardiac neural crest lineage. ScientificWorldJournal. 2007;7:1090–113. doi: 10.1100/tsw.2007.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Tang S, Lin G, Wang J, Conway SJ. Generation of Smad7(-Cre) recombinase mice: A useful tool for the study of epithelial-mesenchymal transformation within the embryonic heart. Genesis. 2009;47:469–75. doi: 10.1002/dvg.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stewart RA, Sanda T, Widlund HR, Zhu S, Swanson KD, Hurley AD, Bentires-Alj M, Fisher DE, Kontaridis MI, Look AT, Neel BG. Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev Cell. 2010;18:750–62. doi: 10.1016/j.devcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–15. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–18. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang LS, Wlodarczyk BJ, Santillano DR, Miranda RC, Finnell RH. Developmental consequences of abnormal folate transport during murine heart morphogenesis. Birth Defects Res A Clin Mol Teratol. 2004;70:449–58. doi: 10.1002/bdra.20043. [DOI] [PubMed] [Google Scholar]
- Tang S, Snider P, Firulli AB, Conway SJ. Trigenic neural crest-restricted Smad7 over-expression results in congenital craniofacial and cardiovascular defects. Dev Biol. 2010;344:233–47. doi: 10.1016/j.ydbio.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor P, Krumlauf R. Development. Riding the crest of the Wnt signaling wave. Science. 2002;297:781–3. doi: 10.1126/science.1075454. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Dixon J, Dixon MJ. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet. 2009;17:275–83. doi: 10.1038/ejhg.2008.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Praagh R, Van Praagh S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am J Cardiol. 1965;16:406–25. doi: 10.1016/0002-9149(65)90732-0. [DOI] [PubMed] [Google Scholar]
- Waldo K, Zdanowicz M, Burch J, Kumiski DH, Stadt HA, Godt RE, Creazzo TL, Kirby ML. A novel role for cardiac neural crest in heart development. J Clin Invest. 1999;103:1499–507. doi: 10.1172/JCI6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. 2005;281:66–77. doi: 10.1016/j.ydbio.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev Dyn. 1996;205:281–92. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Ya J, Schilham MW, de Boer PA, Moorman AF, Clevers H, Lamers WH. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ Res. 1998;83:986–94. doi: 10.1161/01.res.83.10.986. [DOI] [PubMed] [Google Scholar]
- Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212:191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, Clegg T, White H, Mead L, Wenning MJ, Williams DA, Kapur R, Atkinson SJ, Clapp DW. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–61. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Shiratori H, Hamada H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature. 2007;450:285–8. doi: 10.1038/nature06254. [DOI] [PubMed] [Google Scholar]
- Zhou HM, Wang J, Rogers R, Conway SJ. Lineage-specific responses to reduced embryonic Pax3 expression levels. Dev Biol. 2008;315:369–82. doi: 10.1016/j.ydbio.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.