

Abstract
BACKGROUND
The androgen receptor (AR) is a ligand-dependent transcription factor that mediates androgenic hormone action in cells. We recently demonstrated the involvement of phosphoinositide 3-OH kinase (PI3K) p110beta in AR transactivation and gene expression. In this study, we determined the upstream signals that lead to PI3K/p110beta activation and AR transactivation after androgen stimulation.
METHODS
Human prostate cancer LAPC-4 and 22Rv1 cell lines were used for the experiments. AR transactivation was assessed using an androgen responsive element-driven luciferase (ARE-LUC) assay. Cell proliferation was examined using BrdU incorporation and MTT assays. Target genes were silenced using small interfering RNA (siRNA) approach. Gene expression was evaluated at the mRNA level (real-time RT-PCR) and protein level (Western blot). PI3K kinase activities were measured using immunoprecipitation-based in vitro kinase assay. The AR-DNA binding activity was determined using Chromatin-immunoprecipitation (ChIP) assay.
RESULTS
First, at the cellular plasma membrane, disrupting the integrity of caveolae microdomain with methyl-β- cyclodextrin (M-β-CD) abolished androgen-induced AR transactivation and gene expression. Then, knocking down caveolae structural proteins caveolin-1 or -2 with the gene-specific siRNAs significantly reduced androgen-induced AR transactivation. Next, silencing Gαs and Gα12 genes but not other G-proteins blocked androgen-induced AR transactivation and cell proliferation. Consistently, overexpression of Gαs or Gα12 active mutants enhanced androgen-induced AR transactivation, of which Gαs active mutant sensitized the AR to castration-level of androgen (R1881). Most interestingly, knocking down Gαs but not Gα12 subunit significantly suppressed androgen-stimulated PI3K p110beta activation. However, chromatin-immunoprecipitation (ChIP) analysis revealed that both Gαs or Gα12 subunits are involved in androgen-induced AR interaction with the AR target gene PSA promoter region.
CONCLUSION
These data suggest that caveolae-associated G-protein alpha subunits are involved in AR transactivation by modulating the activities of different PI3K isoforms.
Keywords: prostate cancer, caveolae, androgen receptor, G-protein
INTRODUCTION
Androgens play a critical role not only in the physiological development of male sexual phenotype but also in the genesis of prostate cancer (reviewed in ref. 1–5). Upon its entry into target cells, the androgenic hormone transmits its regulatory signal to the nucleus through the cognate androgen receptor (AR), which is a ligand-activated transcription factor and a member of the nuclear receptor super-family.
The conventional activation model of nuclear receptor family describes that before hormone binding the AR is sequestered in the cytoplasm with heat shock proteins and immunophilins (2). After hormone binding, the AR is released from the multi-protein complex and undergoes a conformational change, homo-dimerization, and then is translocated into the nucleus. There, the transcriptionally active AR accumulates within a sub-nuclear compartment where the receptor binds to the specific hormone responsive DNA sequence in the target gene. Thereafter, the AR recruits a number of co-regulatory activating proteins (co-activators/co-repressors) to form a mega-protein complex, which is then poised to interact with the basal transcriptional machinery to modulate target gene transcription (reviewed in ref. 2).
In addition to the conventional working model for hormonal nuclear receptors (2), recent studies have added more details in AR modulations. For example, after androgen stimulation the AR itself and its cofactors are modified by post-transcriptional mechanisms (i.e. phosphorylation, acetylation, etc), indicating that cellular signaling pathways are involved in androgen-stimulated AR transactivation (4). Many groups have described various androgen-initiated intracellular signaling cascades under different experimental conditions and in different cell types, however; only few of them were shown to participate directly in androgen-stimulated genomic effect, AR transactivation (reviewed in ref. 4).
G-protein family consists of three non-identical subunits, α, β and γ. There are four Gα subfamilies; Gαs, Gαi/o, Gαq/11, and Gα12/13. Each of them has multiple members with different expression specificity. Gβγ complex are formed from 5 β-subunits and 13 γ-subunits, and once formed, the dimers will not separate from each other. In an inactive form, a Gα subunit is GDP-bound and interacts with a Gβγ dimer. Once activated, Gα binds to GTP, dissociates from Gβγ dimer, and then both subunits of Gα and Gβγ are free to modulate downstream effectors. Because of their modular architecture, specific expression patterns and numerous diversity of subtypes, G-protein-regulated signaling pathways are very complicated. Recently, heterotrimeric and small G-proteins, as well as their modulators have been shown to enhance AR transactivation by trace-level androgens or cytokines (reviewed in ref. 5).
In attempt to understand the mechanisms involved in androgen-induced AR transactivation, we recently demonstrated the involvement of phosphoinositide 3-OH kinase (PI3K) p110β isoform, the major membrane-associated signaling molecule, in androgen-mediated AR transactivation (6–7). In this study, we examined the signal cascades upstream of PI3K p110β, which are required for androgen-induced genomic effect, AR transactivation and gene expression. We identified membrane caveolae-associated G-protein αs and α12 as essential signaling components for androgen-stimulated AR transactivation. We also demonstrated that G-protein αs or α12 uses different PI3K isoforms to modulate AR transactivation.
MATERIALS AND METHODS
Cell Culture and Reagents
The human prostate cancer cell lines LAPC-4 and 22Rv1 was described previously (6–7). R1881 was obtained from ICN (Aurora, OH). Methyl-β-cyclodextrin (M-β-CD) was purchased from Sigma (St. Louis, MO). Antibodies for caveolins, G-protein subunits, PI3K isoforms and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-AR antibodies were obtained from Upstate (Charlottesville, VA). Charcoal-stripped FBS (cFBS, steroid-free condition) was obtained from Atlanta Biologicals (Norcross, GA). The expressing constructs for active mutants of G α subunits were obtained from Dr Min Pi, as described previously (8).
Immunoprecipitation and Western blot analysis
Protein G immunoprecipitation kit (Sigma, St. Louis) was used according to the manufacturer’s recommendation. Briefly, cell lysates were prepared from 100 mm dishes, and lysed with the cell lysate buffer containing protease inhibitors from the kit. A total of 100 µg proteins and 2.0 µg purified antibody were mixed for 4 hours at 4°C with rotation followed by incubation with protein G beads overnight at 4°C, mixing by inversion. Immuno-complexes were washed three times and collected by centrifugation.
For Western blot analysis, cells were washed in PBS and lysed in a radio-immunoprecipitation assay (RIPA) buffer supplied with protease inhibitors (CytoSignal, Irvine, CA). Equal amount of protein was separated on an 8–15% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel and blotted onto a polyvinyl difluoride membrane (PVDF, Bio-Rad Laboratories, Hercules, CA). Membranes were blocked in a Tris-buffered saline solution with 5% nonfat dry milk containing 0.1% TWEEN-20 and incubated with antibodies overnight at 4°C. Immunoreactive signals were detected by incubation with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) followed by chemi-luminescent detection using SuperSignal substrate kit (Pierce Chemical Co., Rockford, IL).
Small interference RNA transfection
The control siRNAs with scrambled sequences were purchased from Ambion (Austin, TX) and the predefined siRNAs for G-proteins and PI3K isoforms were purchased from Santa Cruz (Santa Cruz, CA). Transfection was carried out with OligoFectamine agent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. For reporter gene assay, three days after transfection with the siRNAs, cells were transfected with reporter genes as described below. To determine the efficiency of the siRNAs, preliminary experiments were conducted by transfection of the siRNAs at various concentrations (1–100 nM), and a dramatic reduction was observed in 100 nM concentration. A scrambled negative siRNA duplex was used as the control.
Luciferase and SEAP reporter assay
A luciferase reporter construct controlled by synthetic androgen response elements was obtained from Dr. Allen Gao, as described previously (9). The reporter construct pCMV-SEAP, expressing secreted alkaline phosphatase (SEAP) under the control of the cytomegalovirus (CMV) promoter, was described previously in our publication (6–7). It was used as an internal reference control for the luciferase assays. For G-protein overexpression experiments, cells were plated in 6-well tissue culture plates and co-transfected on the following day with 1.0 µg pARE-LUC, 0.5 µg pCMV-SEAP and the constructs for G-protein overexpression (as indicated in the figure legend) using the LipoFectamine reagent (Invitrogen, Carlsbed, CA). After 24 h, cells were serum-starved for another 24 h and then treated with the solvent (ethanol) or R1881 in 2% cFBS. After 24 h, culture supernatants were collected and assayed for SEAP activity, as described previously (6–7). Cells were lysed with a lysis buffer supplied by a luciferase assay system (Promega Corp., Madison, WI). Protein concentration in the cell lysates was measured by a protein assay kit (Bio-Rad Laboratories, Hercules, CA). Equal amount of protein from each cell lysate was assayed in triplicate for luciferase enzyme activity by using the Luciferase assay system (Promega Corp., Madison, WI) and Lumat LB9501 reader (Berthold, Oak Ridge, TN). The luciferase activity of each sample was normalized against the corresponding SEAP activity before the fold induction value relative to control cells was calculated.
Real-Time Reverse Transcription (RT)-PCR
Total RNA was prepared using Trizol reagent (Invitrogen). The real-time PCR was performed using Applied Biosystems 7500 PCR systems under following conditions; reaction mixture consisted of diluted sample cDNA solution (5 µl for PSA, 1 µl for β-actin), 10 µl of Taqman Universal PCR Master mix (catalog # 4324018), 1 µl of Taqman Gene expression Assays for FAM conjugated primer mixtures (catalog # 4331182 for PSA, catalog # 433762T for β-actin) and add water to make 20 µl. Denaturation at 95°C for 15 sec, and annealing and extension at 60°C for 1 min was then performed. The denaturation/anealing cycle was repeated 40 cycles. The amplification value of PSA gene was normalized against β-actin genes before relative ratios were calculated. The values for control samples were set as 1.
Cell Proliferation Assay
Cells were seeded in 96-well microtiter plate overnight. Thereafter, cells were transfected with pooled siRNAs and then incubated for additional 24 and 48 h in the presence or absence of 1.0 nM R1881. The proliferation rates of the cell lines were analyzed by measuring 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay using the Cell Proliferation kit I (Roche Molecular Biochemicals, Indianapolis, IN). Formazan crystals were solubilized overnight, and the product was quantified spectrophotometrically by measuring absorbance at 570 nm.
For BrdU incorporation assay, cells were incubated with BrdUrd (BrdUrd labeling and detection kit III; Roche Molecular Biochemicals) for the last 18 h of the experiments. Cells were subsequently fixed with 0.5 M ethanol/HCl and were then incubated with nucleases to partially digest DNA. Monoclonal anti-BrdUrd antibodies conjugated to peroxidase were subsequently added and detected by using 2, 2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) substrate. Quantization was performed colorimetrically at 405–490 nm according to the manufacutrer’s protocol.
PI3K in vitro Kinase Assay
The radio-immunoprecipitation-based in vitro kinase assay was performed as described previously (10). Briefly, cells were scraped into an ice-cold solubilization buffer. After removal of insoluble material by centrifugation, proteins were immunoprecipitated from aliquots of the supernatants with anti-p85 α. Immunoprecipitates were washed extensively. The reaction mixtures were incubated with agitation at room temperature for 10 min before being stopped with 8 M HCl and a 1:1 mix of CHCl3:MeOH. The products were separated by silica gel thin-layer chromatography that used a developing solution of CHCl3:CH3OH:H2O:NH4Cl (60:47:11.6:2). Results were visualized by autoradiography, and the band density of the products co-migrating with a phosphatidylinositol 3-phosphate standard was quantitatively measured with a Gel Logic 100 system (Kodak, New Haven, CT).
The ELISA-based p110 β in vitro kinase assay was carried out using a pre-assembled kit from Echelon Bioscience Inc (Salt Lake City, UT). LAPC-4 cells were transfected with Gα subunit siRNAs as indicated in the figure for 3 days. After serum starvation for 24 h, cells were treated with R1881 (1.0 nM) for 15 min. Cells were then harvested, and equal amount of protein extracts were subjected to the in vitro kinase assay following the protocol provided by the manufacturer.
Chromatin immunoprecipitation (ChIP) assay
Cells were maintained in 10-cm dishes in medium without serum for at least 16 h and treated with or without 1.0 nM R1881 for 12 h. The ChIP assay was performed using a ChIP assay kit and the polyclonal antibody against AR were obtained from Upstate according to the manual (Charlottesville, VA). Normal rabbit serum was used as a negative control and an anti-RNA polymerase II antibody (Santa Cruz Biotechnology) was used in the pilot experiment for kit testing. The primers for PSA in the PCR reactions were listed as follow: 5’-tctgcctttgtcccctagat-3’ and 5’-aaccttcattccccaggact-3’, which amplify a 210-bp fragment corresponding to human PSA gene promoter sequence (–250/–39 from the transcription start site). The GAPDH primers were obtained from the ChIP assay kit. The PCR products were run on 1% agarose gel and stained with ethidium bromide for visualization.
Statistical analysis
All experiments were repeated two or three times. Western blot results are presented from a representative experiment. The mean and standard error of the mean from three experiments for reporter gene assay and cell proliferation assay are shown. The significance of the differences between treatment and control groups was analyzed using the SPSS computer software (SPSS, Inc., Chicago, IL).
RESULTS
Caveolae integrity is essential for androgen-induced AR transactivation
Caveolae is one of plasma membrane microdomains located at or near the cell surface. It exists in most cell types and is involved in regulation of various cell functions, especially the fine-tuning of signaling machinery components on the cell surface including G-proteins and membrane-associated kinases (reviewed in ref. 11). To determine if membrane caveolae is involved in androgen-induced AR transactivation, we tested the effect of a caveolae disruptor, methyl-β-cyclodextrin (M-β-CD, 12), on androgen-induced AR transactivation in a reporter gene assay with a synthetic androgen responsive elements (ARE)-driven luciferase reporter (ARE-LUC). Following serum starvation, LAPC-4 cells were treated with different doses of M-β-CD with or without the synthetic androgen R1881. As shown in Fig 1A, pre-treatment with M-β-CD significantly inhibited R1881-induced ARE-LUC reporter activity. Then, we examined if AR-mediated expression of endogenous AR target gene is suppressed by this disruptor. Real-time PCR analysis revealed that R1881-stimulated PSA expression (the primary AR target gene) was dramatically reduced by M-β-CD pre-treatment (Fig 1B).
Fig. 1. Caveolae integrity is essential for androgen-induced AR transactivation.

(A) LAPC-4 cells were cotransfected with ARE-LUC and pCMV-SEAP reporter constructs overnight. Following serum starvation for 24 h, cells were pretreated with M-β-CD at indicated doses for 45 min followed by the solvent or R1881 (1.0 nM) addition for 16 h.
(B) After serum starvation, LAPC-4 cells were pretreated with M-β-CD at indicated doses for 45 min followed by the solvent or R1881 addition for 16 hrs. Total RNA was extracted for RT-PCR with the primers as indicated on the right side.
(C) LAPC-4 cells were transfected with the control siRNA or siRNAs for Cav-1 or -2 at the final concentration of 100 nM for 3 days and cells were harvested and equal amount of proteins were used for Western blot with antibodies against Cav-1 or -2. Anti-actin blot served as protein loading control.
(D) After transfection with the siRNAs as indicated for 2 days, LAPC-4 cells were transfected again with the reporter constructs (ARE-LUC and CMV-SEAP) overnight. Following serum starvation, cells were treated with the solvent or R1881 (1.0 nM) for 24 h, the media were collected for SEAP assay and cell lysates were subjected for luciferase assay. Average fold induction against mock transfection was calculated after normalized against the internal control SEAP activity and protein concentration, as described in our previous publications (19, 22).
All quantitative data were shown as mean and error bars represent standard error (SEM) from three independent experiments. The asterisk indicates a significant different compared to the control (ANOVA analysis, p < 0.05).
Caveolins (Cav-1, -2 and -3) are the major structural and regulatory components of membrane caveolae and were shown to promote prostate cancer progression (13–14). Cav-1 expression is decreased after castration in the prostate (15), and Cav-2 has been shown to form the caveolae together with Cav-1 (16). Cav-3 is a muscle specific isoform (11). Recently, Cav-1 has been shown to interact with the AR in an androgen-dependent manner and reducing Cav-1 expression by an antisense-based approach suppressed AR transactivation in LNCaP cells (17). To further examine the role of caveolins in AR transactivation, we utilized the small interference RNA (siRNA) approach to knock down the expression of Cav-1 and -2 genes and then tested if caveolins are required for AR transactivation. As shown in Fig 1C, the siRNA-mediated knocking down of the protein levels for Cav-1 and -2 was successfully achieved. Then, these siRNAs were used in ARE-LUC reporter assay. Knocking down either Cav-1 or -2 proteins abolished androgen-induced AR transactivation (Fig 1D). Altogether, these data clearly indicate that the integrity of membrane caveolae is essential for androgen-induced AR transactivation and gene expression, which is supported by a previous report (17).
Gαs and Gα12 subunits are required for AR transactivation
G-proteins are the major signal molecules located on membrane caveolae and have been shown to modulate AR transactivation (18). To understand the specificity of G-protein subunits in AR transactivation, we silenced the representative Gα subunits from each of the four subtypes (s, i/o, q/11 and 12/13). The efficiency of individual siRNAs was confirmed in LAPC-4 cells (Fig 2A). LAPC-4 cells were transfected with the control siRNA or the targeting siRNAs for individual Gα subunits. Two days later, cells were transfected with the ARE-LUC and pCMV-SEAP reporters as described in our recent publication (6–7). As shown in Fig 2B, siRNAs against Gαs and Gα12 but not Gαq and Gαi2 abolished R1881-induced ARE-LUC reporter activity. These data indicate that Gαs and Gα12 subunits are required for androgen-induced AR transactivation.
Fig. 2. Gαs and Gα12 subunits are involved in androgen-induced AR transactivation.
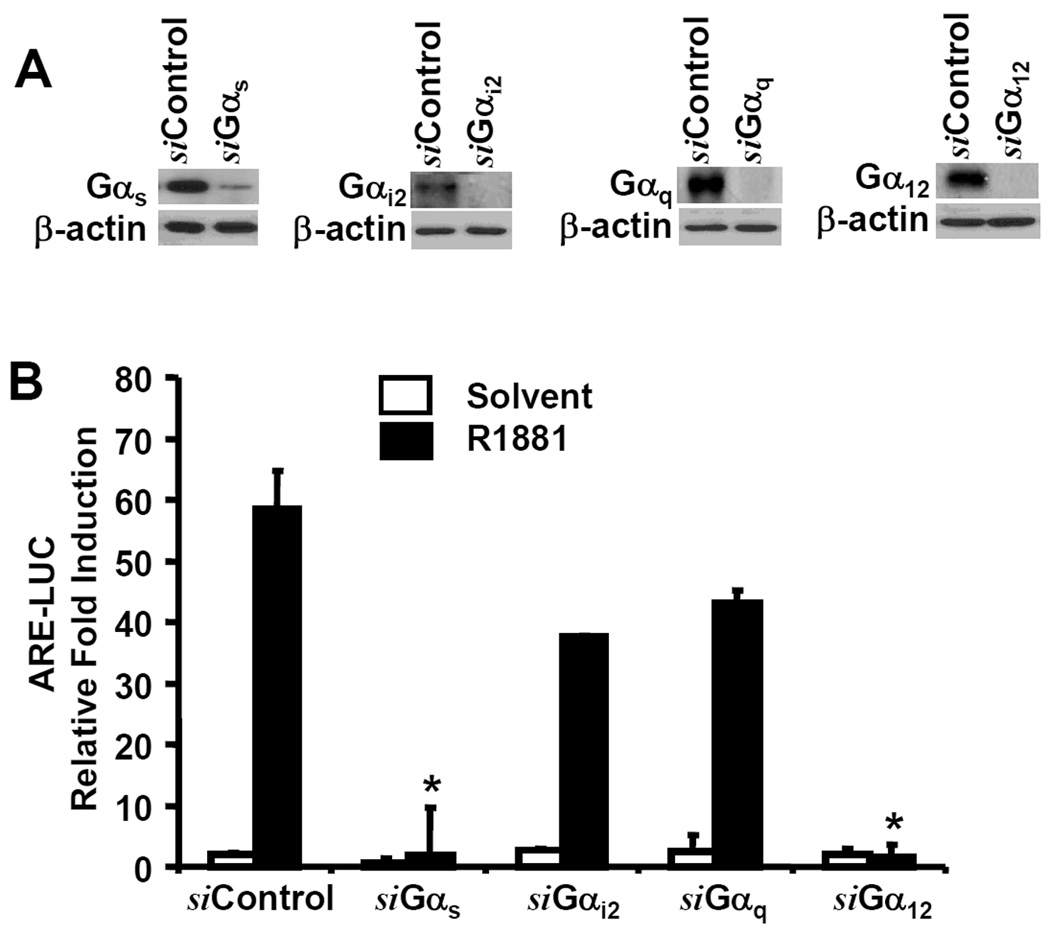
(A) LAPC-4 cells were transfected with the siRNAs against G α subunit genes for 3 days as indicated. Cells were harvested and the target gene proteins were evaluated described in figure 1 legend.
(B) After transfected with the siRNAs as indicated, LAPC-4 cells were co-transfected with ARE-LUC and CMV-SEAP reporter constructs. Cells were treated with the solvent or R1881 (1.0 nM) for overnight. SEAP and luciferase assays were carried out and data were presented as described earlier in figure 1 legend.
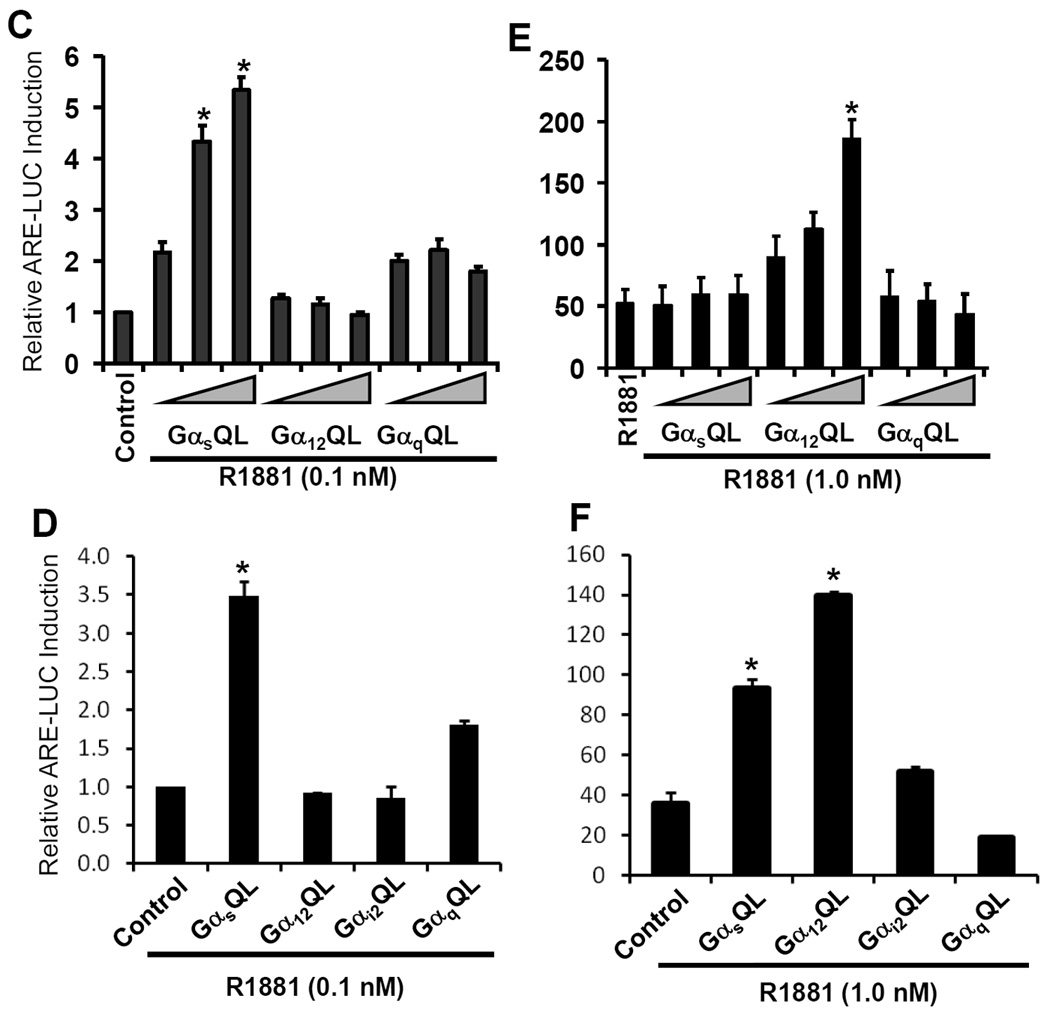
(C-F) Cells, LAPC-4 (penal C & E) or 22Rv1 (penal D & F), were co-transfected with ARE-LUC and CMV-SEAP reporters, as well as different Gα active mutants, which were used with increasing amount of plasmid DNA (0.5, 1.0, 1.5 µg) for LAPC-4 cell or a single dose of 1.5 µg DNA for 22Rv1 cells, as indicated. Control cells (the first column in each panel) received empty vector (pcDNA3.1, 1.5 µg) as the control. After serum starvation, cells were treated with the solvent (C & E) or R1881 (D & F) for 24 h. Luciferase and SEAP activities were assessed and data were presented as described earlier.
To further confirm the involvement of G-proteins in androgen action, we determined if Gα activation leads to AR transactivation in the presence of various levels of androgens. We used the constant active mutants of Gα subunits in the ARE-LUC reporter assay. As shown in Fig 2C & 2D, with a trace level of the synthetic androgen R1881 (0.1 nM), overexpression of active Gαs mutant but not other subunits significantly enhanced R1881-induced ARE-LUC reporter activation in both LAPC-4 and 22Rv1 cells . Interestingly, in the presence of the physiological level of androgen (R1881, 1.0 nM), only Gα12 active mutant but not Gαs mutant dramatically increased R1881-induced ARE-LUC activity (Fig 2E) in LAPC-4 cells, while both Gα12 and Gαs active mutants significantly enhanced R1881-stimulated ARE-LUC activities in 22Rv1 cells (Fig 2F), reflecting a cell specific difference due to distinct androgen responsiveness.
Next, we tested the effect of Gα subunit siRNAs on androgen-induced cell proliferation in LAPC-4 cells. After transfection with the siRNAs, cells were stimulated with androgen for 24–48 h, and cell proliferation was assessed by MTT and BrdU incorporation assays. Consistent with the ARE-LUC reporter assay, the siRNAs against Gαs and Gα12 but not Gαq abolished androgen-induced cell proliferation (Fig 3 A & B).
Fig. 3. Gαs and Gα12 subunits are involved in androgen-induced cell proliferation.

(A) After transfection with the siRNAs as indicated for 2 days, LAPC-4 cells were serum-starved overnight and then treated with the solvent or R1881 for up to 24–48 h. Cell proliferation rate was determined everyday with BrdU incorporation (A) and MTT (B) assays as described in the text. Data represent the average percentage of day 2 and day 3 over day 1 from two independent experiments. Day 1 indicates the starting date of R1881 treatment and the value was set as 100%. Error bars indicate SEM. and the asterisk represents a significant difference (p < 0.05, ANOVA analysis) compared to the control.
In collection, these results indicate that both Gαs and Gα12 are required for androgen-induced AR transactivation and gene expression; however, Gαs activation sensitizes AR response to low level of androgens. Therefore, Gαs and Gα12 may mediate distinct downstream signaling events that act in concert to facilitate AR activation.
Gαs subunit modulates androgen-stimulated PI3K/p110β activation
PI3K is activated through interaction with membrane-associated receptors that are modulated by G-proteins (19–20). Since we found both G-proteins (Fig 2) and PI3K p85α /p110β (7) are required for AR transactivation, we next determined if PI3K p85α/p110β are working downstream of G-proteins following androgen stimulation. Using the siRNA-based screening approach, we found that knocking down the protein levels of Gα12 dramatically reduced p85α-based PI3K activity stimulated by R1881 (Fig 4A). These data indicate that p85α is a downstream component of Gα12-mediated signal cascade.
Fig. 4. Gαs but not Gα12 subunit is involved in androgen-stimulated PI3K p110β activation.
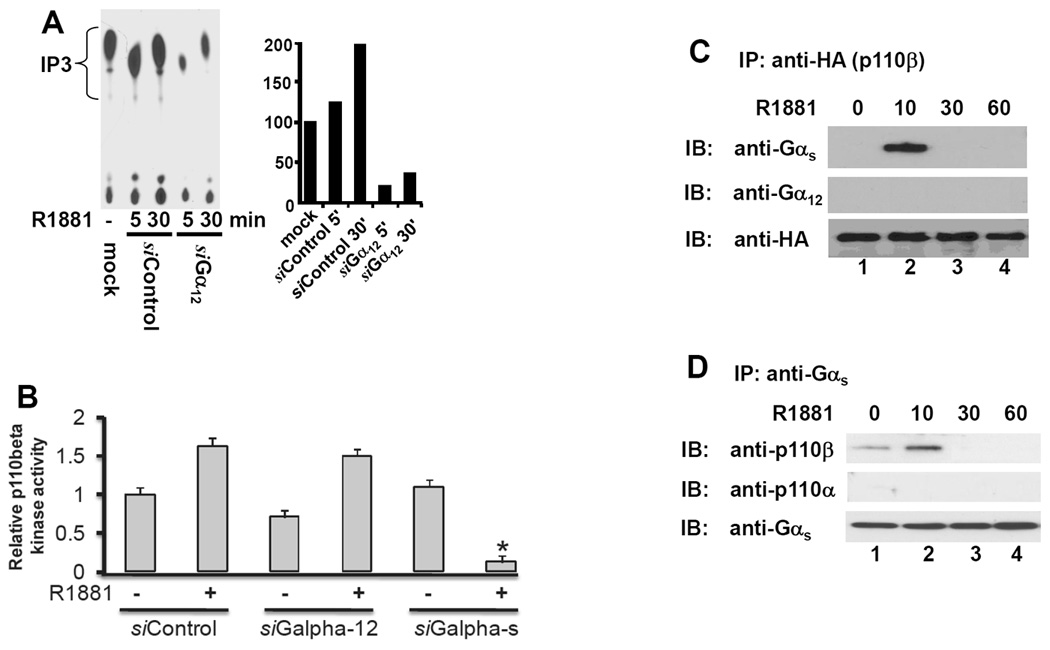
(A) LAPC-4 cells were transfected with the siRNAs as indicated for 3 days, and mock transfection was included as negative control. After serum starvation, cells were left untreated or treated with R1881 (1.0 nM) for 15 min. Cells were harvested and cellular protein extracts were used for anti-p85α immunoprecipitation followed by in vitro kinase assay as described in the text. The left panel shows a representative autoradiography and the right panel represents the relative band density after quantification. Data represents two separate experiments.
(B) LAPC-4 cells were transfected with the siRNA as described above. After serum starvation, cells were treated with R1881 (1.0 nM) for 15 min and then harvested for p110 β kinase assay activity with an immunoprecipitation (IP)-ELISA-based in vitro lipid kinase assay with a pre-assembled kit from Echelon Bioscience Inc (Salt Lake City, UT). Data shown are the average relative values of the ELISA readings compared to the negative control (control siRNA without R1881 treatment) from two independent experiments. Error bars indicate SEM. and the asterisk represents a significant difference (p < 0.05, ANOVA analysis) compared to the control.
(C & D) 22Rv1 cells were transiently transfected with the HA-tagged p110β expression vector (penal C), as described (7), or with the active Gas mutant (panel D), as described (8), for 24 h. After serum starvation, cells were treated with the solvent or R1881 (1.0 nM) for 10–60 min before harvested for protein extraction. A co-immunoprecipitation assay with either anti-HA (p110β) antibodies (panel C) or anti-Gas antibodies (panel D) were carried out to detect their association with G-proteins (C) or PI3K proteins (B). Data shown are representative results from two independent experiments.
Next, we examined the effect of Gα siRNAs on type IB p110β activation in an IP-ELISA-based in vitro lipid kinase assay. As shown in Fig 4B, as expected, R1881 treatment significantly increased p110β activity in LAPC-4 cells. Gαs but not Gα12 siRNA transfection remarkably suppressed R1881-induced p110β activity. These data strongly suggest that Gαs is involved in androgen-stimulated p110β activation.
Lastly, we examined if G-protein alpha subunits were associated with PI3K p110b in response to androgen stimulation using co-immunoprecipitation assay. 22Rv1 cells were transiently transfected with either HA-tagged p110β expression vector or the Gαs active mutant GαsQL for 24 h. After serum starvation for 24 h, cells were stimulated with R1881 for 10–60 before harvested for protein extraction. As shown in Fig 4C, after R1881 stimulation, HA-tagged p110β was transiently associated only with Gαs but not with Gαs12. Consistently, overexpressed GαsQL mutants were transiently associated with p110β but not p110α after R1881 stimulation (Fig 4D), which is supported by our previous report that only p110β is involved in androgen-induced AR transactivation. Taken together, our data indicate that G-proteins modulate PI3K activities in an isoform-specific manner in response to androgen stimulation.
G-protein signal pathway plays a key role in AR-DNA interaction
We previous demonstrated that inhibition of PI3K/p110β activity suppressed androgen-induced AR-DNA interaction (7), suggesting that PI3K signaling is involved in the process of AR-DNA interaction or assembly of the AR-mediated transcription complex. In this study, therefore, we examined if G-proteins are involved in AR-DNA interaction using an in vivo AR-DNA interaction assay (also known as ChIP assay). We first tested the commercial ChIP assay kit with normal rabbit IgG and anti-RNA polymerase II antibodies. A primer pair for GAPDH gene promoter was used in the PCR reaction. As shown in Fig 5A, normal IgG did not pull down any chromatin DNA while anti-RNA polymerase II antibody precipitated GAPDH promoter. The input DNA was used as chromatin loading control. These control experiments indicated that the kit can produce clean results.
Fig. 5. Gαs and Gα12 subunit are essential for androgen-stimulated AR-DNA binding.
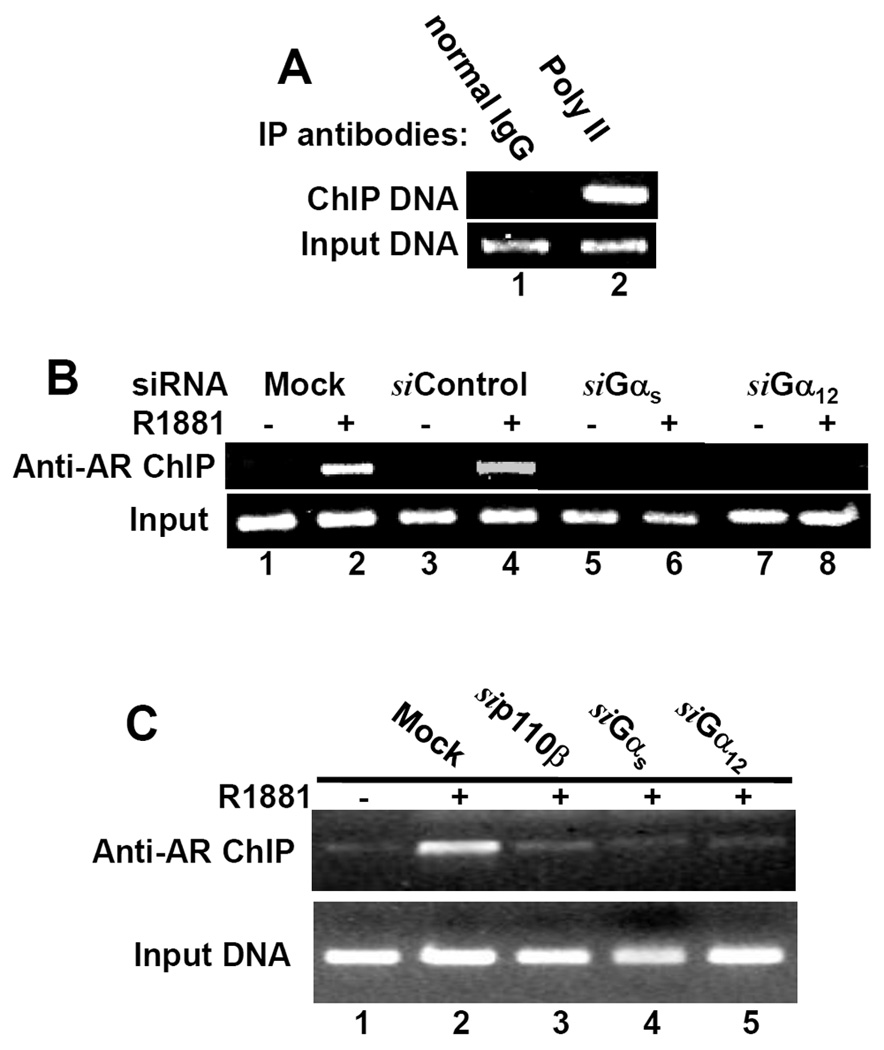
(A) Exponential grown LAPC-4 cells were fixed and harvested for a pilot ChIP assay with normal IgG and anti-RNA polymerase II antibodies as described in the text. The PCR reaction was performed with a primer pair for GAPDH gene promoter.
(B & C) Cells, LAPC-4 (panel B) or 22Rv1 (panel C), were transfected with the siRNAs as indicated for 3 days and mock transfection was included as a control. After R1881 treatment as indicated for 12 h, cells were fixed and harvested for anti-AR ChIP assay with primer pairs for PSA gene promoter region (PSA primer) as described in the text. For all the panels, 1% of the input chromatin was used as PCR templates (positive control of the primers used). Data represent two independent experiments.
Then, we conducted the anti-AR ChIP assay with a primer pair for PSA promoter sequences. As shown in Fig 5B & 5C, either Gαs or Gα12 siRNAs but not the control siRNA abolished androgen-induced AR interaction with PSA promoter region in both LAPC-4 and 22Rv1 cells, indicating that G-protein-derived signaling is needed for androgen-induced AR-DNA binding.
DISCUSSION
Membrane caveolae integrates multiple signal components including G-proteins and receptor tyrosine kinases (11). It interacts with steroid nuclear receptors such as the AR and estrogen receptor (ER) after hormone stimulation (17, 21). Previous studies have shown the involvement of plasma membrane-derived signaling events in androgen-induced genomic and non-genomic effects in various cell types or under different experimental conditions. However, only few of these signaling events were shown to participate directly in androgen-induced AR-mediated gene expression (22, reviewed in ref. 4, 23–24). As such, early studies showed that androgens activated protein kinase A (PKA) via a heterokimeric GTP-binding protein (G-protein)-dependent mechanism in prostate cancer cells (25–27), and activation of Gαs-PKA pathway led to AR transactivation (18, 28–30). In addition, after androgen stimulation, the AR was transiently translocated onto membrane caveolae, which is the site for initiating membrane-associated signaling events, such as the engagement of G-proteins and activation of various tyrosine kinases (17, 31). In this report, we demonstrated that the integrity of caveolae structure is essential for androgen-induced AR transactivation and gene expression, which is supported by a previous report (17). It is plausible that the interaction of AR and Cav-1 following androgen stimulation might be an indirect association through cytoskeleton molecules since both the AR and Cav-1 have been shown to interact with cellular skeleton molecules (32–33).
In this study, we provided the first evidence that Gαs and Gα12 subunits are required for androgen-induced PI3K (p85α and p110β) activation and AR transactivation. Usually, type IA PI3K is activated by tyrosine kinase-based mechanisms through p85 regulatory isoforms, while type IB PI3K p110γ is mainly activated by Gβγ subunits. However, Gα subunits were also reported to activate p110β (20). As discussed earlier, we recently demonstrated that PI3K/p110β activity is required for androgen-induced AR transactivation and gene expression in prostate cancer cells (6–7). However, it is not clear how androgen stimulates PI3K/p110β activation that subsequently regulates AR-DNA interaction and transactivation. Recent report showed that androgen induces PI3K activation via AR interaction with p85α and Src kinase in prostate cancer cells (34), and Src kinase-mediated AR tyrosine phosphorylation after growth factor stimulation is involved in regulation of AR activity (35). However, our study (6) and another report (36) showed that Src kinase inhibitor did not suppress androgen-induced gene expression, indicating that additional cellular signal pathways are involved in androgen-induced genomic effect, AR transactivation. In this study, we determined that androgen-induced PI3K activation is through G-protein α subunit engagement, and different G-protein subunits modulate the activities of distinct PI3K isoforms in response to androgen-stimulation. Most interestingly, our data indicates that aberrant activation of Gαs-p110β pathway might sensitize the AR to castration levels of androgens.
There are two major steps for hormone-bound AR to exert its genomic effect as a transcription factor; nuclear translocation and DNA (promoter region) binding. Currently, it is not clear how precisely androgen induces AR nuclear translocation, although F-actin-binding protein filamin has been found to be required for AR translocation (32) and both p21-activated kinase PAK6 and a Chinese herb extract Emodin inhibit this process (37–38). Once the AR is in the nucleus, it will assemble a mega-protein transcription complex, which is a dynamic process including transient recruitment and then dissociation of co-factors, chromatin remodeling molecules, proteasome subunits and general transcription machinery (24). We recently showed that PI3K inhibitors do not block AR nuclear translocation but inhibit AR-DNA binding and subsequent gene expression (6–7). Indeed, in this study, knocking down the protein levels of Gαs and Gα12 also suppressed AR-DNA binding. A similar effect was also reported by another group using Her2 inhibitor (36). These data indicate that G-protein/PI3K signaling is involved in regulation of AR-DNA interaction or assembly of AR transcription complex.
Because of their important roles in cell signaling, elevated activities of PI3K pathway due to PTEN inactive mutation has long been considered as a key player in cancer pathogenesis (5, 39). Recent emerging evidences indicate that PI3K isoforms themselves (i.e. p110α and p85α) have oncogenic potential through gain-of-function mutations or gene amplification (39). Similarly, Gαs also showed an oncogenic potential to induce cellular transformation and tumor development in some endocrine-related tissue types, including pituitary adenomas, thyroid tumors and testes Leydig cell tumors (reviewed in ref. 40). Currently, no information is available about genetic phenotype and expression pattern of G-protein subunits in human prostate cancers. Interestingly, in this study, we found that overexpression of an active Gαs mutant sensitized AR activation in response to very low level of androgens, suggesting that over-expressed or over-activated G-protein-PI3K (p110β) pathway might be a key factor in castration-resistant progression of prostate cancers.
In conclusion, we identified the signaling components that are essential for androgen-induced AR transactivation. These signaling molecules include membrane caveolins, Gαs and Gα12 subunits, as well as PI3K p85α/p110β. Based on this study and previous reports (6–7, 17–18, 31), we hypothesize that androgen stimulation triggers a signaling cascade at membrane caveolae where the AR interacts with G-proteins and other yet-to-define signaling molecules (likely Her2 kinases, 36). G-protein engagement subsequently activates type IA PI3Ks and other signaling molecules. It is plausible that these G-protein/PI3K isoforms are working towards different downstream effectors, which ultimately regulate AR-DNA interaction or assembly of AR-based transcriptional complex on the promoter region of target genes, as illustrated in Fig 6. Further investigation is desirable for dissecting this complicated process in detail.
Fig. 6. A scheme for G-protein/PI3K regulation of androgen-induced AR transactivation.
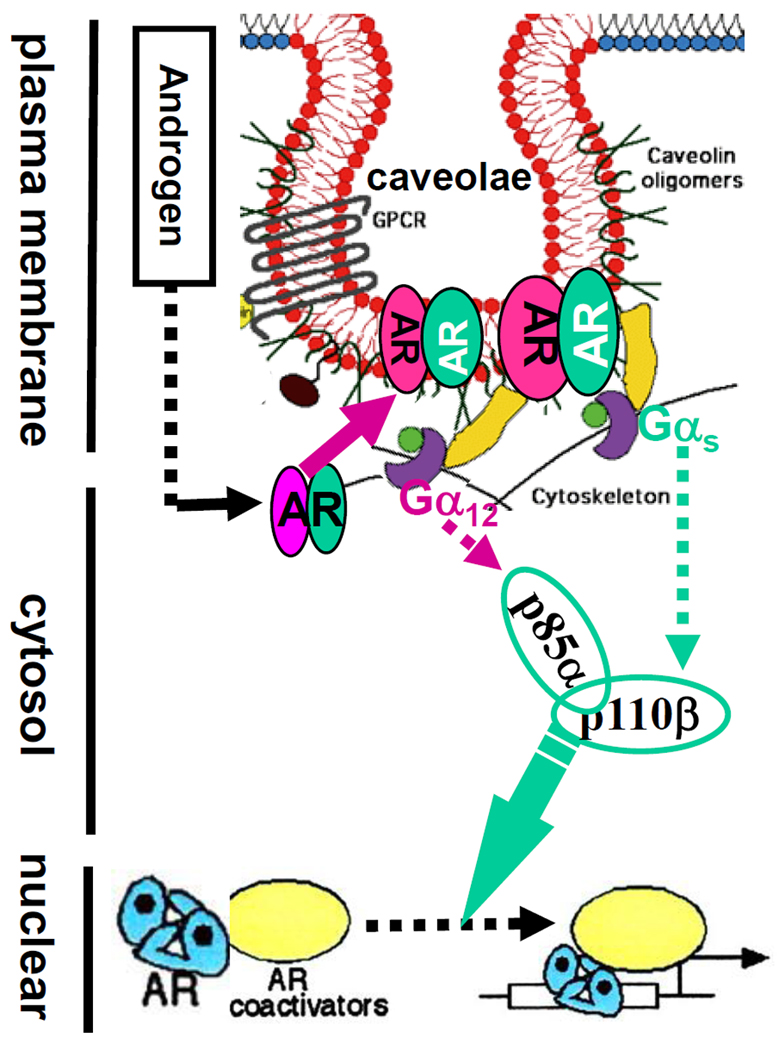
Before androgen stimulation, the AR is sequenced inside cytosol and is associated with cytoskeleton proteins. After androgen binding, the AR interacts with G-proteins and other signal molecules on membrane caveolae through caveolin-dependent mechanism. Engagement of different Gαs and Gα12 subunits induces activation of PI3K isoforms that transduce signals to regulate AR-DNA binding or assembly of AR-dependent transcription complex.
ACKNOWLEDGEMENTS
This study was supported by KU William L. Valk Endowment, Kansas Masonic Foundation and KUMC Lied Foundation. This work was also partially supported by a grant of 1P20RR15563 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), Department of Defense New Investigator Award DAMD17-03-1-0121 and Ideal Development Award W81XWH-04-1-0214 to Dr Benyi Li. This study was also supported partially by grants from the Guangdong Natural Scientific Foundation of China (# 2008-B030301020 and 2006-263041).
Abbreviations
- AR
androgen receptor
- ARE
androgen responsive element
- BrdU
5-bromo-2′-deoxyuridine
- ChIP
chromatin- immunoprecipitation
- CMV
cytomegalovirus
- FBS
fetal bovine serum
- LUC
luciferase
- M-β-CD
methyl-β-cyclodextrin
- PBS
phosphate- buffered saline
- PCR
polymerase chain reaction
- PI3K
phosphoinositide 3-OH kinase
- RIPA
radio-immunoprecipitation assay
- RT
reverse transcription
- SEAP
secreted alkaline phosphatase
- SEM
standard error of mean
- siRNA
small interferencing RNA
REFERENCES
- 1.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 2.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 3.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 4.Edwards J, Bartlett JM. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 1: Modifications to the androgen receptor. BJU Int. 2005;95:1320–1326. doi: 10.1111/j.1464-410X.2005.05526.x. [DOI] [PubMed] [Google Scholar]
- 5.Daaka Y. G proteins in cancer: the prostate cancer paradigm. Sci STKE. 2004;216:re2. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 6.Liao X, Thrasher JB, Holzbeierlein J, Stanley S, Li B. Glycogen synthase kinase-3beta activity is required for androgen-stimulated gene expression in prostate cancer. Endocrinology. 2004;145:2941–2949. doi: 10.1210/en.2003-1519. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Q, Youn H, Tang J, Tawfik O, Dennis K, Terranova PF, Du J, Raynal P, Thrasher JB, Li B. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene. 2008;27:4569–4579. doi: 10.1038/onc.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pi M, Spurney RF, Tu Q, Hinson T, Quarles LD. Calcium-sensing receptor activation of rho involves filamin and rho-guanine nucleotide exchange factor. Endocrinology. 2002;143:3830–3838. doi: 10.1210/en.2002-220240. [DOI] [PubMed] [Google Scholar]
- 9.Lee SO, Lou W, Hou M, Onate SA, Gao AC. Interleukin-4 enhances prostate-specific antigen expression by activation of the androgen receptor and Akt pathway. Oncogene. 2003;22:7981–7988. doi: 10.1038/sj.onc.1206735. [DOI] [PubMed] [Google Scholar]
- 10.Franch HA, Wang X, Sooparb S, Brown NS, Du J. Phosphatidylinositol 3-kinase activity is required for epidermal growth factor to suppress proteolysis. J Am Soc Nephrol. 2002;13:903–909. doi: 10.1681/ASN.V134903. [DOI] [PubMed] [Google Scholar]
- 11.Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol. 2004;32:325–338. doi: 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- 12.Calaghan S, White E. Caveolae modulate excitation-contraction coupling and beta2-adrenergic signaling in adult rat ventricular myocytes. Cardiovasc Res. 2006;69:816–824. doi: 10.1016/j.cardiores.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Nasu Y, Timme TL, Yang G, Bangma CH, Li L, Ren C, Park SH, DeLeon M, Wang J, Thompson TC. Suppression of caveolin expression induces androgen sensitivity in metastatic androgen-insensitive mouse prostate cancer cells. Nat Med. 1998;4:1062–1064. doi: 10.1038/2048. [DOI] [PubMed] [Google Scholar]
- 14.Williams TM, Hassan GS, Li J, Cohen AW, Medina F, Frank PG, Pestell RG, Di Vizio D, Loda M, Lisanti MP. Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. J Biol Chem. 2005;280:25134–25145. doi: 10.1074/jbc.M501186200. [DOI] [PubMed] [Google Scholar]
- 15.Pflug BR, Reiter RE, Nelson JB. Caveolin expression is decreased following androgen deprivation in human prostate cancer cell lines. Prostate. 1999;40:269–273. doi: 10.1002/(sici)1097-0045(19990901)40:4<269::aid-pros9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Sowa G, Pypaert M, Fulton D, Sessa WC. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc Natl Acad Sci U S A. 2003;100:6511–6516. doi: 10.1073/pnas.1031672100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu ML, Schneider MC, Zheng Y, Zhang X, Richie JP. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J Biol Chem. 2001;276:13442–13451. doi: 10.1074/jbc.M006598200. [DOI] [PubMed] [Google Scholar]
- 18.Kasbohm EA, Guo R, Yowell CW, Bagchi G, Kelly P, Arora P, Casey PJ, Daaka Y. Androgen receptor activation by G(s) signaling in prostate cancer cells. J. Biol Chem. 2005;280:11583–11589. doi: 10.1074/jbc.M414423200. [DOI] [PubMed] [Google Scholar]
- 19.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 20.Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem. 1998;273:19080–19085. doi: 10.1074/jbc.273.30.19080. [DOI] [PubMed] [Google Scholar]
- 21.Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–363. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, De Falco A, Varricchio L, Bottero D, Nanayakkara M, Migliaccio A, Auricchio F. Rapid signaling pathway activation by androgens in epithelial and stromal cells. Steroids. 2004;69:517–522. doi: 10.1016/j.steroids.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Heinlein CA, Chang C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol Endocrinol. 2002;16:2181–2187. doi: 10.1210/me.2002-0070. [DOI] [PubMed] [Google Scholar]
- 24.Gioeli D. Signal transduction in prostate cancer progression. Clin Sci (Lond) 2005;108:293–308. doi: 10.1042/CS20040329. [DOI] [PubMed] [Google Scholar]
- 25.Culig Z. Androgen receptor cross-talk with cell signaling pathways. Growth Factors. 2004;22:179–184. doi: 10.1080/08977190412331279908. [DOI] [PubMed] [Google Scholar]
- 26.Nakhla AM, Leonard J, Hryb DJ, Rosner W. Sex hormone-binding globulin receptor signal transduction proceeds via a G protein. Steroids. 1999;64:213–216. doi: 10.1016/s0039-128x(98)00084-1. [DOI] [PubMed] [Google Scholar]
- 27.Rosner W, Hryb DJ, Khan MS, Nakhla AM, Romas NA. Sex hormone-binding globulin mediates steroid hormone signal transduction at the plasma membrane. J Steroid Biochem Mol Biol. 1999;69:481–485. doi: 10.1016/s0960-0760(99)00070-9. [DOI] [PubMed] [Google Scholar]
- 28.Rosner W, Hryb DJ, Khan MS, Nakhla AM, Romas NA. Androgen and estrogen signaling at the cell membrane via G-proteins and cyclic adenosine monophosphate. Steroids. 1999;64:100–106. doi: 10.1016/s0039-128x(98)00108-1. [DOI] [PubMed] [Google Scholar]
- 29.Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 30.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999;274:7777–7783. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem. 1996;271:29182–29190. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000;14:1618–1626. doi: 10.1210/mend.14.10.0541. [DOI] [PubMed] [Google Scholar]
- 33.Stahlhut M, van Deurs B. Identification of filamin as a novel ligand for caveolin-1: evidence for the organization of caveolin-1-associated membrane domains by the actin cytoskeleton. Mol Biol Cell. 2000;11:325–337. doi: 10.1091/mbc.11.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, Nicosia SV, Cheng JQ. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–43000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- 35.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, Qiu Y. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 36.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–527. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 37.Schrantz N, da Silva Correia J, Fowler B, Ge Q, Sun Z, Bokoch GM. Mechanism of p21-activated kinase 6-mediated inhibition of androgen receptor signaling. J Biol Chem. 2004;279:1922–1931. doi: 10.1074/jbc.M311145200. [DOI] [PubMed] [Google Scholar]
- 38.Cha TL, Qiu L, Chen CT, Wen Y, Hung MC. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Res. 2005;65:2287–2295. doi: 10.1158/0008-5472.CAN-04-3250. [DOI] [PubMed] [Google Scholar]
- 39.Stephens L, Williams R, Hawkins P. Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol. 2005;5:357–365. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]