

Abstract
The GEIRA (Gene-Environment and Gene–Gene Interaction Research Application) algorithm and subsequent program is dedicated to genome-wide gene-environment and gene–gene interaction analysis. It implements concepts of both additive and multiplicative interaction as well as calculations based on dominant, recessive and co-dominant genetic models, respectively. Estimates of interactions are incorporated in a single table to make the output easily read. The algorithm is coded in both SAS and R. GEIRA is freely available to non-commercial users at http://www.epinet.se. Additional information, including user’s manual and example datasets is available online at http://www.epinet.se.
Keywords: GWAS, Interaction, Program
Introduction
The most common human diseases have a complex aetiology involving both genetic and environmental factors. Genome-wide association studies (GWAS) applying a selected set of single nucleotide polymorphism (SNP) covering the entire genome have recently become popular and quite successful methods in search for genetic determinants of common diseases. However, the genetic variants discovered so far account for only a small portion of common disease heritability, indicating that there still are many genes or loci with small effects to be discovered. The possibility exists that a gene’s effect is more easily detected within a framework that accommodates gene-environment and gene–gene interactions. For example, a gene’s overall effect might be too small to detect with any reasonable sample size, or a genetic effect is entirely dependent on an environmental exposure and may show a substantial effect when a specific environmental factor or another genetic variant is present. Results from many association studies have not been replicated, possibly due in part to the omission of incorporating interactions among disease associated loci in the analysis where allele frequencies for interacting genes or environmental exposures may have differed in the investigated populations [1]. In fact, gene-environment interactions and gene–gene interactions are increasingly recognized phenomena in the field of human genetics [1–4]. This pinpoints the needs for the development of a computation tool for detecting gene-environment and gene–gene interactions in a genome-wide fashion, i.e. a tool that allows the analysis of interaction between a specific environmental or genetic factor and a large number of SNPs.
In statistics, the term “interaction” is used to refer to departure from the underlying form of a statistical model. Because there are various statistical models, interaction does not have a consistent meaning [5]. Interaction is often assessed by entering a product term into the linear or multiplicative regression model. For example, in a logistic regression model a product term is estimated as departure from multiplicativity of odds ratios. Interaction in epidemiology commonly refers to the situation where the joint effect of two risk factors on disease risk differs from the sum of their independent effects. This concept of interaction has a consistent meaning and is particularly applicable in the context of the sufficient cause model as described by Rothman [5, 6]. A sufficient cause is defined as minimal sets of actions, events, or states of nature that together initiated a process that inevitably resulted in the outcome. For a particular outcome there would likely be many different sufficient causal mechanisms. In the sufficient cause context, interaction is defined as two causes being component causes of the same sufficient cause. It can be shown that in the absence of interaction in this sense, disease rates are additive [5, 7, 8]. It can also be shown that when the joint effect of two contributory causes of a disease exceeds the sum of their independent effects (on the additative scale) then sufficient cause interaction is present (but not vice versa). Rothman has argued that interaction as departure from additivity better reflects biologic interaction [5, 7]. He presents three measures of biologic interaction: RERI, the relative excess risk due to interaction; AP, the attributable proportion due to interaction; and S, the synergy index [5–7, 9–12]. These measures are defined as follows:
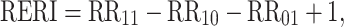 |
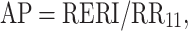 |
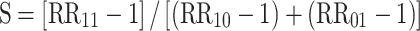 |
where RR11 is the relative risk in the exposure category when both risk factors are present, RR10 is the relative risk for the first risk factor in the absence of the second and RR01 is the relative risk for the second risk factor in the absence of the first. Those who are unexposed to both the first and the second risk factor are used for reference category, i.e., RR00 = 1. Thus, there are three relative risks to be estimated. If there is no interaction, RERI and AP equals 0 and S equals 1. For the calculation of confidence interval there are a few options [13, 14]. In the current version of GEIRA program, we use Hosmer and Lemeshow’s delta method, which is a straight forward Taylor expansion of the variances and covariances based on Wald-type statistics using approximate variance estimators. The calculation will be faster in terms of computation time compared with the re-sampling based bootstrap method [14] in the context of genome-wide scale. The limited number of SNPs that come out from the screening can then easily be re-calculated with confidence limits based on e.g. Assmann’s method [14].
In the literature on gene-environment and gene–gene interactions, there is no definite agreement on which computational methods for calculations of interactions are most appropriate in which contexts [15–17]. Irrespective of what choice of method that is preferred, there is a definite need for better computational methods for quantification of interactions. The primary goal of present article is to provide tools for large scale interaction calculation for several of the most utilized methods for detection of interactions. We have chosen to use the term “additive interaction” referring to Rothman’s “biologic interaction” and “multiplicative interaction” referring to the product term in a multiplicative model. In the program, multiplicative interaction is estimated by inclusion of an interaction term in a logistic regression model while the estimation of additive interaction is based on the three measures, i.e., RERI, AP and S as described above.
Methods
SAS version 9.2 for windows and R version 2.6.2 were used to develop the GEIRA program.
Features
The program analyzes genome-wide SNP data or any list of bi-allelic genetic markers or dichotomous environmental factors in a case–control study setting using toward the single risk factor of choice (“major” risk factor). It includes simple data processing steps, which makes it easy and user-friendly and enables users to modify and extend the program. After user defined single factor of choice and other variables are determined, the program will run automatically. All relevant measures for both additive interaction and multiplicative interaction will be included in the output in format of a table. Users can easily generate a ranked top list of SNPs based on certain user defined criteria.
GEIRA consists of six steps which are described as follows (Fig. 1):
Fig. 1.

A pictorial representation of the GEIRA algorithm
Step 1 (Data importing)
Here we assume that some other software package has previously been used to quality control genome-wide genotype data, including dataset filtering on the basis of SNP genotype call rate, minor allele frequency, Hardy–Weinberg equilibrium, individual missing genotype, outliers, etc. The program will read in transposed PLINK format data files, i.e., TPED (containing SNP and genotype information where one row is a SNP) and TFAM (containing individual and family information where one row is an individual) (see PLINK documentation for details. http://pngu.mgh.harvard.edu/~purcell/plink/pdf.shtml). In addition to the TPED and TFAM files, a covariate file containing covariate information (including an environmental variable or a major genetic variable of choice) is needed.
Step 2 (Risk allele assigning)
A minor allele is determined using all subjects (both cases and controls). A risk allele is determined by comparing the minor allele frequency (MAF) in cases and controls. If the MAF in cases is greater than or equal to that in controls, the minor allele is assigned to the risk allele. If the MAF in cases is less than that in controls, the major allele is assigned to the risk allele (Fig. 2 and user’s manual for details).
Fig. 2.

Detailed algorithm for step 2 (risk allele assignment). Example 1 illustrates a situation where a major allele is assigned to a risk allele. Example 2 illustrates a situation where a minor allele is assigned to a risk allele. c_gen1_f genotype count of wide type homozygote (homozygote 1) in controls, c_het_f genotype count of heterozygote in controls, c_gen2_f genotype count of homozygote 2 in controls, d_gen1_f genotype count of wide type homozygote (homozygote 1) in cases, c_het_f genotype count of heterozygote in cases, c_gen2_f genotype count of homozygote 2 in cases. c_p:control minor allele frequency; d_p:case minor allele frequency; Wid wide type, Het heterozygote, Hom homozygote, MA minor allele, MJ major allele
Step 3 (Data converting)
The raw genotype dataset will then be converted into a dataset based on a dominant, recessive or co-dominant model, according to the following coding:
Assuming C is the minor allele and also the risk allele
Dominant model coding: A_A → 0, A_C → 1, C_C → 1.
Recessive model coding: A_A → 0, A_C → 0, C_C → 1
Co-dominant model coding: A_A → 0, A_C → 1, C_C → 2
Step 4 (Interaction calculation)
Users can choose one of these models, i.e., a dominant, recessive or a co-dominant model. Calculate estimates for both additive and multiplicative interactions, incorporating all estimates into one output table.
Step 5 (Supervising module)
Steps 1–4 should be executed in proper order. We therefore created a supervising module that will pass the correct parameters to each step in order.
Step 6 (Adjustment for multiple testing)
P-value adjustments using Bonferroni, Sidak (A technique slightly less conservative than Bonferroni) and False Discovery Rate (FDR) are corrected for total tests performed and are calculated using simple functions of the raw p-values.
Application to real genome-wide data
Rheumatoid arthritis (RA) is a complex autoimmune disorder with both genetic and environmental influences on the disease pathogenesis [18]. Family aggregation and twin studies have estimated a genetic component of approximately 50% [19, 20]. Smoking is an established risk factor for RA [18, 21]. We applied GEIRA to the Swedish Epidemiologic Investigation of Rheumatoid Arthritis (EIRA) GWAS data (Illumina 300 K). EIRA is an ongoing population-based case–control study aiming at studying gene-environment interactions in rheumatoid arthritis. The study design and population description were detailed previously [22, 23]. A total of 1,147 autoantibody to citrullinated protein antigens (ACPA)-positive rheumatoid arthritis cases and 1,079 age, gender and residence area matched controls were used for calculation.
In order to demonstrate the use of the GEIRA program, we chose to investigate interactions between a large number of genetic variants available in cases (ACPA-positive RA) and controls as well as data on the environmental exposure smoking in the same cases and controls. We used already published GWAS data on these cases and controls as our basic genetic information [22, 24]. We conducted an extra quality control procedure in PLINK [25], thereafter 301,238 autosomal SNPs remained for analysis and were used as an input in the GEIRA program. The environmental factor, smoking status, was coded as 1 for ever smoker and 0 for never smoker. From the analysis, a total of 15 SNPs were found to be statistically significantly interacting with smoking on the additive scale. Of these 15 SNPs, two are from HLA region (HLA-DQA2 and HLA-DRA). The genome-wide significance defined as a P for AP of 1.66 × 10−7 was used for claiming a statistical significance. This application took about 7 h to complete on a desktop with two Intel Xeon processors running at 3.2 GHz and a RAM of 32G. The operating system was Windows Vista Business (64-bit). The further results of these experiments together with biological interpretation of the findings are submitted separately for publication (submitted).
Discussion
The GEIRA program aims at detecting gene-environment as well as gene–gene interactions, and in this first report, we describe both the generic algorithms, and their application in a specific case of gene-environment interaction. The main effect of each SNP can be estimated using other whole-genome association toolsets, such as PLINK [25]. The results from the GEIRA program can be considered as a first screening step for selecting potential candidate gene-environment (or gene–gene) interactions across the genome. SNPs on the top of the list can be furthered examined by follow-up studies, such as replication in another independent population, dense mapping, and so on. Adjustment for confounding factors can be easily done by assigning the macro variables, covar_cat or covar_cont, to the confounding variables (see user’s manual for details). For adjustment of multiple testing, an additional step (step 6) for correction for multiple testing was included in the program. Notwithstanding the fact that the step 2 is developed to ensure that the odds ratio (OR) for the risk allele is greater than 1, the OR associated with the genetic factor might still be less than 1 among subject unexposed to the environmental factor. In this case, the interpretation for interaction should be cautious. AP is an estimate of the proportion of disease, which is due to interaction among persons with both exposures. If RR11 < [(RR10 + RR01)−1], AP will be negative. In this case the joint effect from the simultaneous presence of the two factors is less than what is expected by summing their independent effects. The factors then partly balance each others effects when simultaneously present. In this case, the factors in question may have antagonistic effects. We advise users to read the user’s manual of the program for more details. Displays in the output window are suppressed and contents in the log window are saved in an external directory so that users can trace the program running process if there are some unexpected results in the outputs. To analyze only certain chromosomes, users only need assign a chromosome number of interest to the macro variable chr and run the program. It takes only 7–35 min, depending on which chromosome selected, to complete the analyses for a single chromosome. This program can be easily modified to estimate genome-wide gene–gene interactions. In this case, the only parameter needed to be changed is the environmental variable (envir). The environmental variable should be replaced by a dichotomous genetic variable (SNP, CNV, haplotype etc.) coded as 0 (unexposed) and 1 (exposed) with the remaining parameters unchanged. When applying to the very large GWAS data, parallel computing on a multiple CPUs/clusters server is strongly recommended.
In future versions of GEIRA, we will extend the interaction estimation to one continuous and one dichotomous variable, and further to two continuous variables. The algorithm and the program GEIRA presented in this article offers a powerful, user-friendly tool for performing interaction analyses with whole-genome data.
Acknowledgments:
This work has been financially supported by the Swedish Medical Research Council [to L.A.], the Swedish King Gustaf V’s 80-year foundation [to B.D.], the Swedish Ulla and Gustaf af Uggla Foundation Research Grant [to B.D.], the Swedish COMBINE program [to L.K.], Swedish council for working life and social science COFAS [to H.K.].
Conflict of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Cardon LR, Bell JI. Association study designs for complex diseases. Nat Rev Genet. 2001;2:91–99. doi: 10.1038/35052543. [DOI] [PubMed] [Google Scholar]
- 2.Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003;56:73–82. doi: 10.1159/000073735. [DOI] [PubMed] [Google Scholar]
- 3.Sing CF, Stengard JH, Kardia SL. Dynamic relationships between the genome and exposures to environments as causes of common human diseases. World Rev Nutr Diet. 2004;93:77–91. doi: 10.1159/000081252. [DOI] [PubMed] [Google Scholar]
- 4.Thornton-Wells TA, Moore JH, Haines JL. Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004;20:640–647. doi: 10.1016/j.tig.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Rothman KJ. An introduction: epidemiology. New York: Oxford University Press; 2002. [Google Scholar]
- 6.Rothman KJ. Causes. Am J Epidemiol. 1976;104:587–592. doi: 10.1093/oxfordjournals.aje.a112335. [DOI] [PubMed] [Google Scholar]
- 7.Rothman KJ, Greenland S, Lash TL. Modern epidemiology. Philadelphia: Lippincott Williams and Wilkins; 2008. [Google Scholar]
- 8.VanderWeele TJ. Sufficient cause interactions and statistical interactions. Epidemiology. 2009;20:6–13. doi: 10.1097/EDE.0b013e31818f69e7. [DOI] [PubMed] [Google Scholar]
- 9.Rothman KJ. Synergy and antagonism in cause-effect relationships. Am J Epidemiol. 1974;99:385–388. doi: 10.1093/oxfordjournals.aje.a121626. [DOI] [PubMed] [Google Scholar]
- 10.Rothman KJ. The estimation of synergy or antagonism. Am J Epidemiol. 1976;103:506–511. doi: 10.1093/oxfordjournals.aje.a112252. [DOI] [PubMed] [Google Scholar]
- 11.Hallqvist J, Ahlbom A, Diderichsen F, Reuterwall C. How to evaluate interaction between causes: a review of practices in cardiovascular epidemiology. J Intern Med. 1996;239:377–382. doi: 10.1046/j.1365-2796.1996.431782000.x. [DOI] [PubMed] [Google Scholar]
- 12.Andersson T, Alfredsson L, Kallberg H, et al. Calculating measures of biological interaction. Eur J Epidemiol. 2005;20:575–579. doi: 10.1007/s10654-005-7835-x. [DOI] [PubMed] [Google Scholar]
- 13.Hosmer DW, Lemeshow S. Confidence interval estimation of interaction. Epidemiology. 1992;3:452–456. doi: 10.1097/00001648-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Assmann SF, Hosmer DW, Lemeshow S, Mundt KA. Confidence intervals for measures of interaction. Epidemiology. 1996;7:286–290. doi: 10.1097/00001648-199605000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Gorwood P, Wohl M, Le Strat Y, Rouillon F. Gene-environment interactions in addictive disorders: epidemiological and methodological aspects. C R Biol. 2007;330:329–338. doi: 10.1016/j.crvi.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Weinberg CR. Less is more, except when less is less: Studying joint effects. Genomics. 2009;93:10–12. doi: 10.1016/j.ygeno.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahdi H, Fisher BA, Kallberg H, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009;41:1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 18.Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373:659–672. doi: 10.1016/S0140-6736(09)60008-8. [DOI] [PubMed] [Google Scholar]
- 19.Cornelis F, Faure S, Martinez M, et al. New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc Natl Acad Sci USA. 1998;95:10746–10750. doi: 10.1073/pnas.95.18.10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacGregor AJ, Snieder H, Rigby AS, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 21.Klareskog L, Padyukov L, Lorentzen J, Alfredsson L. Mechanisms of disease: genetic susceptibility and environmental triggers in the development of rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:425–433. doi: 10.1038/ncprheum0249. [DOI] [PubMed] [Google Scholar]
- 22.Plenge RM, Seielstad M, Padyukov L, et al. TRAF1–C5 as a risk locus for rheumatoid arthritis–a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stolt P, Bengtsson C, Nordmark B, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. 2003;62:835–841. doi: 10.1136/ard.62.9.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding B, Padyukov L, Lundstrom E, et al. Different patterns of associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in the extended major histocompatibility complex region. Arthritis Rheum. 2009;60:30–38. doi: 10.1002/art.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]