

Abstract
Fatty acid analysis is essential to a broad range of applications including those associated with the nascent algal biofuel and algal bioproduct industries. Current fatty acid profiling methods require lengthy, sequential extraction and transesterification steps necessitating significant quantities of analyte. We report the development of a rapid, microscale, single-step, in situ protocol for GC–MS lipid analysis that requires only 250 μg dry mass per sample. We furthermore demonstrate the broad applications of this technique by profiling the fatty acids of several algal species, small aquatic organisms, insects and terrestrial plant material. When combined with fluorescent techniques utilizing the BODIPY dye family and flow cytometry, this micro-assay serves as a powerful tool for analyzing fatty acids in laboratory and field collected samples, for high-throughput screening, and for crop assessment. Additionally, the high sensitivity of the technique allows for population analyses across a wide variety of taxa.
Keywords: Biobased products, Biofuels (Energy), Genetics, Breeding, Biotechnology biocatalysis, Genomics, Fats and oils, Nutraceuticals, Functional foods, Food and feed science, Nutrition and health, Lipid chemistry, Lipid analysis, Chromatography
Introduction
High-resolution fatty acid analysis is essential to many medical, industrial, and experimental applications [1]. The analytical method chosen should be quantitative, rapid, highly reproducible, cost-effective, and applicable across a wide range of sample types and sizes.
Most recommended methods for the assay of fatty acids begin with the extraction of the targeted product from an organic matrix with a non-polar solvent such as hexane [2, 3], followed by a two-step transmethylation that converts the acids to fatty acid methyl esters (FAME). The AOAC and AOCS methods for fatty acid transmethylation recommend treating the extracted samples with a base in methanol, followed by acid catalysis in methanolic boron trifluoride [4, 5]. The FAME generated by this multi-step process are suitable for separation and quantitation by GC–MS.
There has been increased interest in the development of alternative methods that streamline fatty acid analysis [6–10]. The aims have been twofold: (a) to directly transmethylate lipids from an organic product (in situ methods), thus eliminating the need for a preliminary, non-polar extraction step, and (b) to use a single-step derivatization procedure for generating FAME [2]. These technical refinements expedite analysis, reduce required volumes of expensive reagents, and minimize experimental error [6]. Unfortunately, sample size remains an issue. Frequently, large amounts of testable material are difficult to obtain, especially in a laboratory setting.
In this paper we describe the development of a single-step, in situ micro-method for fatty acid analysis that requires a minimum of ~250 μg dry mass per sample. To demonstrate the utility of the Sub-Microscale In Situ (SMIS) method, we have utilized this technique to assay the fatty acids of an algal culture as it progresses through a circadian cycle. We additionally used this technique to demonstrate fatty acid variability among several algal species and to assess fatty acid profiles of terrestrial plant pollen and spores, Daphnia magnus, Brachionus calyciflorus, and Drosophila melanogaster.
Materials and Methods
Algal Culture and Harvest Conditions
All media chemicals were of reagent grade. Wastewater was obtained from the Sequim Sewage Treatment Plant (Sequim, Washington) from the pre-UV treated effluent. An MP-11A distillation apparatus (Corning, Corning, NY, USA) was used to produce distilled water for cultures.
Unless noted, algae were routinely maintained as described in Table 1. These cultures were grown in 250-mL Erlenmeyer flasks containing 110 mL of medium that were stoppered with gas-permeable foam plugs (Bellco Glass, Vineland, NJ, USA) and covered with small autoclave bags. All cultures were maintained on a 12-h light:12-h dark photoperiod using 100 μE s−1 m−2 full-spectrum illumination at 20 °C without agitation. For the cell cycle study, the alga Chrysochromulina sp. was grown in a 36 × 52 × 15 cm translucent HDPE vessel that was completely covered with a sheet of transparent polycarbonate. This culture was grown in 5.5 L of proprietary wastewater medium under the standard conditions listed above.
Table 1.
Culture conditions for algal maintenance
| Name of algae | Algal class | Growth medium | Source | Agitation | Mean cell diameter (μm) | Cell covering |
|---|---|---|---|---|---|---|
| Mallomonas splendens | Synurophyceae | DY-V + Sia | CCMP 1782 | No | 12.2 | Silica scales |
| Nannochloropsis oculata | Eustigmatophyceae | f/2 + Sia | CCMP 525 | No | 1.5a | Cellulose wall |
| Chrysochromulina sp. (strain P4) | Haptophyceae | RAC1 + WWb | UWC 1985A | No | 3.9 | Plasma membrane |
| Emiliania huxleyi | Haptophyceae | L1 + Sia | CCMP 1742 | 60 RPM | 4.5a | Calcium carbonate scales |
| Rhodomonas sp. | Cryptophyceae | Oc3 | CCMP 318 | No | 8.9 | Pellicle |
| Prorocentrum micans | Dinophyceae | f/2a | CCMP 1329 | No | 10.5 | Theca |
aFrom the Provasoli–Guillard National Center for Culture of Marine Phytoplankton website (https://ccmp.bigelow.org/)
bProprietary medium (Cattolico, Hardin, Vo and Barker, University of Washington)
cMcIntosh and Cattolico [20]
Unless indicated, cell counts were determined using a Beckman Z2 Coulter Counter equipped with a 100-μm aperture (Beckman Coulter, Brea, CA, USA). For fatty acid analysis, 10 mL samples were collected in 16 × 125 mm screw-threaded clear glass tubes with phenolic screw caps (Fisher Scientific, Hampton, NH, USA) and centrifuged at 3,950×g for 20 min at 4 °C in a Sorvall RC-5 superspeed refrigerated centrifuge (Fisher Scientific). The recovered cell pellets were immediately flash frozen in liquid nitrogen and stored at −80 °C until lyophilization. Lyophilization was performed with a Labconco Freezone 2.5 (Labconco, Kansas City, MO, USA) to equilibrium dryness at a pressure at or below 1.33 mbar over a 48-h period. Sample tubes were then capped and stored at −20 °C. The tubes were removed from the freezer and allowed to come to room temperature prior to analysis.
Neutral lipid content was measured by placing a 0.5 mL aliquot of cell culture into a well of a 96-well plate (BD Biosciences, San Jose, CA, USA) that contained 5 μL 0.5 mM BODIPY 505/515 solution. The solution was prepared by adding 10 μL of 5 mM BODIPY 505/515 (Invitrogen Molecular Probes, Carlsbad, CA, USA) solubilized in DMSO (≥98%) to 90 μL Chrysochromulina sp. medium. Flow cytometric measurements were made using a Millipore guava easyCyte 8HT flow cytometer (Millipore, Billerica, MA, USA) with at least two clean cycles between each measurement. The easyCyte system supports excitation at 488 nm and emission at 525 nm (±30 nm). Fluorescence of unstained cells was measured concurrently for background subtraction. It was found that background from BODIPY was negligible—more than four orders of magnitude below signal. Separation between the emission ranges of BODIPY and chlorophyll auto-fluorescence was sufficient to eliminate interference.
Daphnia magnus Growth Conditions
Daphnia magnus neonates were grown in a 6-well titer plate (BD Biosciences) in 10 mL L16 media [11] per well. After 18 h, five neonates were taken at random from each well and placed in 250 mL Erlenmeyer flasks containing 100 mL L16 media that were capped with foam plugs. 5–10 mL of Chrysochromulina sp. culture were added to the Daphnia flasks as live food each day. Chrysochromulina sp. feedstock cells were grown in a 250-mL flask as described in Table 1 to a harvest concentration of 1.02 × 106 cells mL−1. Growth of the mixed culture took place at 20 °C under ~100 μE s−1 m−2 full-spectrum illumination. Five adult Daphnia magnus were collected with Pasteur pipettes 30 min after the final feeding and placed in a 16 × 125 mm screw-threaded clear glass tube, flash-frozen in liquid nitrogen, and lyophilized to complete dryness prior to analysis.
Brachionus calyciflorus Growth Conditions
3,000–5,000 Brachionus calyciflorus eggs were grown in 100 mL of Chrysochromulina sp. media in a 250-mL Erlenmeyer flask capped with foam plugs. The rotifers were fed 2.5–5 mL per day of live Chrysochromulina sp. culture as described for Daphnia magnus. The Brachionus calyciflorus were grown at 20 °C under ~100 μE s−1 m−2 full-spectrum illumination. After 7 days, the Brachionus calyciflorus culture was filtered through a 160-μm Nitex nylon mesh and placed in 100 mL of fresh medium. The supernatant was decanted after settling and 8 mL aliquots of the remaining sample were transferred by micropipette to 16 × 125 mm screw-threaded clear glass tubes, flash-frozen in liquid nitrogen, and lyophilized to complete dryness prior to analysis.
Pollen and Lycopodium Spore Collection and Preparation
Centaurea cyanus pollen was collected at the University of Washington greenhouse by scraping mature stamen with a pair of tweezers over a 16 × 125 mm screw-threaded clear glass tube. The pollen was then suspended in 2 mL of Isoton. Then 800 μL of this solution was placed in a second 16 × 125 mm screw-threaded clear glass tube, frozen in liquid nitrogen and lyophilized to complete dryness prior to analysis. Several spore-producing bodies from Lycopodium nummularifolium were cut and placed in a paper envelope. The envelope was then stored in a desiccator for 1 week followed by a second week in a drying oven at ~50 °C. The loose spores were then shaken from the envelope into a 16 × 125 mm screw-threaded clear glass tube. The spores were suspended in 2 mL of Isoton, passed through a 9-inch Pasteur pipette several times to separate the grains from one another, counted using a 10 μL Hy-Lite Ultra plane improved Neubauer hemocytometer (Hausser Scientific, Blue Bell, PA, USA), then prepared for analysis as described for C. cyanus pollen.
Drosophila melanogaster Fly Preparation
Drosophila melanogaster flies were graciously provided by the laboratory of Dr. Barbara Wakimoto, University of Washington, Seattle, WA, USA. The flies were incapacitated by flushing with nitrogen. A single male or female fly was placed into a 16 × 125 mm screw-threaded clear glass tube. Sample tubes were then frozen in liquid nitrogen and lyophilized to complete dryness prior to analysis.
Solvents and Chemicals for Analysis
Water for lipid analysis was purified through a Micropure filtration system (Micropure, Nisku, Canada) to a minimum resistance of 17 MΩ/cm. The solvents isooctane, methylene chloride, and methanolic boron trifluoride (1.3 M) were of analytical grade (Sigma–Aldrich, St. Louis, MO, USA). Sodium chloride was of reagent grade (Fisher Scientific). A 28-component fatty acid methyl ester standard made in methylene chloride (“NLEA FAME mix”; Restek, Bellefonte, PA, USA) was used for GC–MS retention time identification and response curve generation. Surrogate triglycerides C11:0 and C17:0 were of greater than 99% purity (Sigma–Aldrich). A seven-component deuterated aromatic mixture in methylene chloride (“Revised SV Standard”, Restek, Bellefonte, PA, USA) was used as an internal standard for GC–MS quantitation. Nitrogen was of grade 4.8 (Praxair, Danbury, CT, USA).
Standardization and Transesterification
A surrogate mixture of 25.0 μL containing 1.556 g L−1 C11:0 and 1.756 g L−1 C17:0 triglycerides made in methylene chloride was added to each sample tube and the solvent was evaporated under a stream of compressed air. Subsequently, 200 μL methanolic boron trifluoride was added to all tubes which were then flushed with dry nitrogen, capped with PTFE-lined screw caps and vortexed for 10 s. The capped tubes were placed in either a boiling water bath or a heating block that was maintained at 100 °C for 1 h, after which the samples were allowed to cool in air to ~30 °C before being uncapped. Then 100–200 μL isooctane was added, followed by a flush with isooctane-saturated nitrogen. The tubes were immediately recapped and vortexed for 10 s. 500 μL saturated NaCl solution in water was added to all tubes. The tubes were then flushed with isooctane-saturated nitrogen, recapped and vortexed for 10 s, and then centrifuged in a clinical centrifuge at approximately 4,000×g.
Phase Separation
Phase separation was accomplished in two steps. Step 1: all liquid in the sample was slowly withdrawn using a 9-inch Pasteur pipette (Corning) equipped with a blooming rubber bulb (Fisher Scientific). Roughly half the aqueous phase was then expelled back into the 10-mL tube. The remaining liquid in the Pasteur pipette was allowed to settle for 10 s and was then expelled slowly into a 400-μL silanized flat-bottom microvolume insert (Agilent, Santa Clara, CA, USA). At no time was air allowed to bubble into the sample. This step allows for the recovery of the isooctane layer with a micropipetter. Step 2: 50.0 μL of the isooctane phase was withdrawn from the 400-μL microvolume insert with a 50-μL micropipetter (Biohit, Neptune, NJ, USA). This aliquot was then transferred into a 150-μL glass microvolume insert with polymer feet (Agilent) contained in a 2.0-mL screw-cap vial (Agilent). 5.55 μL of 1,000 μg mL−1 Revised SV Standard was added, bringing the concentration of the internal standard to 100 μg mL−1 in the microvolume insert. The 2.0 mL screw-cap vial was then sealed using a screw cap with PTFE/silicon bonded septa (Agilent) and the sample was immediately analyzed via GC–MS.
GC–MS Analysis
GC–MS analysis was performed on an HP 5890 gas chromatograph equipped with an HP 5971A mass spectrometer, an HP 7673A autosampler, and an Agilent HP-5 ms 30 m × 0.25 mm, 0.25 μm phase thickness column (Agilent). The injected volume was 1.0 μL. A splitless injection was used to maximize the amount of analyte on the column. Inlet temperature was 250 °C and detector temperature was 280 °C. Column temperature began at 80 °C and increased to 110 °C at 8.0 °C min−1, immediately followed by a ramp at 14.0 °C min−1 to a final temperature of 310 °C which was held for 3 min before run termination. Helium (Praxair) flowing at 0.5 mL min−1 was used as the carrier gas. Relative quantities of lipid types were determined by comparing single-ion response ratios between each analyte and the nearest eluting internal standard with HP ChemStation software (EnviroQuant module; Agilent), with additional analytical analysis performed in MS Excel. It should be noted that C18:4 and C18:5 coelute on the column used for this assay and have similar fragmentation patterns by EI ionization and therefore are quantitated together as C18:4/5.
Statistical Methods
To assess the precision and reproducibility of the SMIS technique, the small sample statistical method of Dean and Dixon [12] was applied to the fatty acid composition data from the light/dark cycle experiment (n = 542) presented below. The Dean and Dixon method for 2-4 samples was chosen as an alternative to standard ANOVA statistical methods that require large numbers of samples. The principal advantage of the Dean and Dixon method is improved accuracy for the analysis of data sets with a small number of samples.
Results and Discussion
Methods Comparison
The SMIS technique was compared to published fatty acid analytical methods with respect to relative amounts of material, reagents and time required to process a sample (Table 2). Data indicate that the SMIS method uses at least five to twenty-five times less solvent and reagent than four conventional methods, including both AOAC and AOCS recommended techniques. Additionally, the SMIS technique requires much less sample material: eight times less sample is necessary than for the Micro-Direct technique, and about 80 times less sample is necessary than for the AOCS or AOAC standard methods. Another advantage of in situ methods, such as our SMIS protocol, is that samples need not be transferred prior to phase separation.
Table 2.
A comparison of methods used for fatty acid analysis
| Method | Traditional extraction or in situ | Approx. minimum sample mass (g) | Description | Approx. solvent volumes | Number of sample transfers during transesterification | Reference |
|---|---|---|---|---|---|---|
| Sub-microscale in situ (“SMIS”) | in situ | 0.00025 | FAME prepared by exposing total sample to BF3 in MeOH, heated at 100 °C for 1 h, followed by a two-step phase separation | 0.9 mL | 2 | This study |
| Micro-direct | in situ | 0.002 | FAME prepared by exposing total sample to HCl in MeOH, heated at 110 °C for 2 h | 5 mL | ~3 | Viga and Grahl-Nielsen [21]; Theimann et al. [6] |
| Ackman | Traditional extraction | 0.2 | Lipids extracted in non-polar solvent. FAME prepared using BF3 in MeOH, heated at 100 °C for 1 h | 20 mL | 3 | Ackman [2] |
| AOCS | Traditional extraction | 0.2 | Lipids extracted in non-polar solvent. FAME prepared first using NaOH in MeOH, heated at 100 °C for 5 min., then using BF3 in MeOH, heated at 100 °C for 1 h | 25 mL | ~5 | AOCS Method [5]; Barker [3] |
| AOAC | Traditional extraction | 0.2 | Hydrolytic lipid extraction. FAME prepared first using NaOH in MeOH, heated at 100 °C for 5 min, then using BF3 in MeOH, heated at 100 °C for 1 h | 25 mL | ~5 | AOAC Method [4] |
Data adapted from Theimann [6]
The necessity of treating samples with methanolic base during the process of analyzing fatty acids has been questioned [2, 6]. Mazalli and Bragagnolo [13] demonstrated that a saponification reaction longer than 5 min or with excess heat caused free fatty acids to be produced from the esters generated at the initiation of this treatment. Several investigators [14, 15] found that transesterification using a single-step boron trifluoride method produced results comparable to the recommended AOCS or AOAC techniques [4, 5]. Because we felt that the saponification step added non-beneficial complexity to our fatty acid analysis, it has been omitted from the SMIS method. Notably, the SMIS method is an in situ technique wherein fatty acids are transesterified without prior extraction from their organic matrix. In a comparative study, Theimann et al. [6] tested an in situ method against traditional analysis techniques using whale tissue. It was found that the in situ method generated data with less variance. This difference in precision was explained by errors that were generated during the lipid extraction step. Omission of both saponification and matrix extraction in the SMIS method eliminates unnecessary procedural steps, thus saving material cost while generating more reproducible data. This streamlining also reduces technician-time required to process samples, allowing many samples to be run in parallel.
The SMIS method produces GC–MS data with few interfering analytes (Fig. 1). To demonstrate the precision of this method we analyzed the variation among replicate samples for a data set (Fig. 2) that consisted of 104 samples obtained over a 25 h period. Variation among replicate samples was ~5%. Importantly, when results obtained by different technicians using the SMIS technique on duplicate samples were compared, the mean variance was no greater than the variation between replicates when analyzed by the same technician (~4%). These observations validate the accuracy and repeatability of the SMIS technique.
Fig. 1.
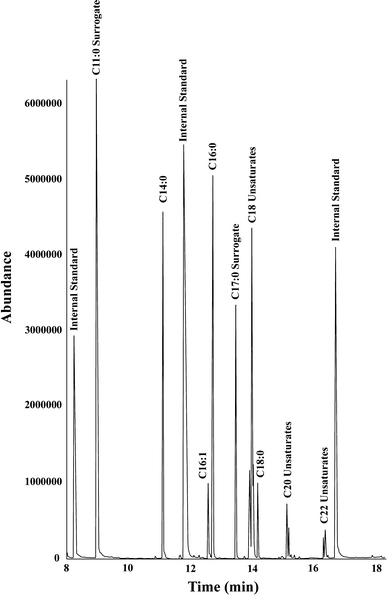
GC–MS total ion chromatogram of a representative sample total ion chromatogram of a sample taken from the Cell Cycle Experiment (D0, replicate 1) showing an absence of interfering compounds. Abundance is electron multiplier counts. The three visible internal standards, in order of elution, are: Acenaphthene d-10, Phenanthrene d-10, and Chrysene d-12
Fig. 2.

A comparison of cell cycle parameters Left axis: filled diamonds culture density (Coulter Counter). Right axis: filled squares neutral lipid concentration in relative fluorescent units (RFU), measured using BODIPY 505/515 dye; filled triangles Fatty acid per cell (picograms); filled circles fatty acid per liter of culture in mg using GC–MS. Error (by 95% confidence interval) for all data points is less than ~5%. Samples were run in quadruplicate
Versatility of Technique
The examples presented here have been chosen to demonstrate the versatility of the SMIS technique. These examples utilize a wide variety of biological materials, and exploit the small sample size required for this assay.
Cell Cycle Analysis
It is well known that circadian rhythms strongly influence biological processes, including lipid biogenesis. Despite the utility of cell cycle lipid research, few quantitative and qualitative results have been published. Previous research has quantitated lipid content using either fluorescent dyes [16] or rarely electron microscopy [17]. A major drawback of these methods is that neither generates information on fatty acid composition.
The highly oleaginous, ~4 μm alga Chrysochromulina sp. was used as a test organism to examine fatty acid production using the SMIS method. The circadian cycle of algal cells within a culture can be synchronized by maintaining the culture on a light:dark photoperiod. A culture maintained on a 12 h light:12 h dark photoperiod was sampled during the logarithmic growth phase over a 25-h period. As seen in Fig. 2, cell number increased linearly from ~L4 (6.43 × 105 cells mL−1) to ~D10 (1.79 × 106 cells mL−1). Concomitantly, total fatty acid per cell, measured using the SMIS method, increased from the onset of light (~3.49 pg cell−1) until ~L6 (~7.51 pg cell−1). The increase in fatty acid content from the onset of light over 12 h is readily visible by fluorescent microscopy using BODIPY 505/515 (Fig. 3). Fatty acid levels plateaued until the onset of dark, then decreased to 2.78 pg cell−1 at the onset of light (L0). The reduction in fatty acid content on day 2 was anticipated. Unlike many algae that increase in fatty acid content per cell as a culture matures, the fatty acid content of Chrysochromulina sp. cells rapidly decreases as they enter the stationary phase of growth (Hardin, Bigelow and Cattolico, unpublished). Total fatty acid per liter of culture increased linearly throughout the light from 2.18 mg L−1 at L0 to a maximum value of 7.57 mg L−1 at D0, and then decreased to 4.86 mg L−1 at L0 on day 2. A positive correlation between cell volume and total fatty acid content per cell was observed (Fig. 4a).
Fig. 3.

Lipid bodies in Chrysochromulina sp. Cells were maintained on a 12 h light:12 h dark photoperiod. D11.5 cells were harvested at 11.5 h into the dark period. L11.5 cells were harvested at 11.5 h into the light. Cells were stained with BODIPY 505/515 dye (green) as described in “Materials and methods”. The chloroplast auto-fluoresces red. The scale bar is 10 μm. The cell volumes (computed from mean diameter) at these points were about 23 μm3 at D11.5 and 44 μm3 at L11.5. Excitation was at 450–490 nm and emission wavelengths were imaged through a 515 nm long-pass filter (color figure online)
Fig. 4.

Fatty acid profiles over the 12 h light:12 h dark photoperiod of Chrysochromulina sp. a The relationship between cell volume (computed from cell diameter) and fatty acid content. b–d Changes in fatty acid composition as measured by GC–MS. Y axis represents the percent of total fatty acids. X axis represents the hour in the light/dark cycle
It should be noted that cell division and fatty acid biogenesis are discrete with respect to their phase (cell division initiates at ~L3; fatty acid biogenesis at the onset of light), amplitude (a 2.78-fold increase occurs in cell number whereas a 2.23-fold increase is seen in fatty acid levels) and period (~18 h for completion of cell division in the culture while lipid biogenesis only continues for ~12 h), suggesting that though the processes are most certainly linked, they may be individually malleable.
Importantly, there also appears to be a circadian-driven shift in lipid identities during the 12 h light:12 h dark Chrysochromulina sp. growth cycle (Fig. 4b–d). Many of the fatty acids can be grouped together by the similarities in their behavior over the tested light/dark photoperiod. Two major groupings appear: fatty acids that increase in relative abundance in the light and then decrease in the dark, and those with the reverse pattern. For clarity, fatty acids are presented as saturated fatty acids (Fig. 4b), unsaturated C18s (Fig. 4c), and the long-chain unsaturates and C16:1 (Fig. 4d). Saturated fatty acids (C14:0, C16:0, C18:0) all increased from a total of 40.3% at the onset of the cell cycle to 47.7% at the end of the light period, then decreased to 35.7% during the dark period (Fig. 4b). Unsaturated C18 fatty acids (C18:1, C18:2, C18:3, C18:4/5) represent 35.0–39.2% of the total fatty acids over the entire cycle. C18:1 increases from 2.3 to 3.2% in the light and decreases to 1.7% in the dark. Similarly, C18:2 has a minimum at hour L2 of 22.1% and a maximum at ~D1 of 27.0%. C18:3 and C18:4/5 both decreased in the light and increased in the dark, with minima and maxima of 1.7 and 2.7% respectively for C18:3; 5.6% and 9.3% for C18:4/5 (Fig. 4c). The long-chain unsaturated fatty acids (C20:4, C20:5, C22:5, C22:6) comprised 13.4% of the total fatty acids at L0. These fatty acids decreased to 10.2% at ~L6, then increased slowly until reaching a final concentration of 21.9% at L0 on day 2. C16:1 followed the same trend, decreasing from the onset of light (10.5%) until the onset of dark (4.0%), then increasing in abundance during the dark period to 6.1%.
BODIPY 505/515 is a fluorescent dye that accumulates in cellular lipid bodies via a diffusion trap mechanism [18]. Algal cells were stained with BODIPY 505/515 at different times during the cell cycle both to determine neutral lipid per cell by flow cytometry and to image lipid bodies in living cells. The flow cytometric data in Fig. 2 show that neutral lipid production began at the onset of light. Neutral lipid quantity increased by 160% by ~L6, and remained at that level until D0. Although at the beginning of the photoperiod the relative changes in neutral lipid signal and total fatty acid are comparable, by ~D3 neutral lipids begins to decrease faster than total fatty acids. In the dark, the total fatty acid per cell decreased by 67% while neutral lipid signal decreased by 92%.
Microscopic examination of Chrysochromulina sp. cells stained with BODIPY 505/515 shows that the cells display a high level of internal organization with respect to lipid packaging. These algae contain only two lipid bodies, each closely associated with one of the two chloroplasts found in the cell. When BODIPY 505/515 stained oil bodies were microscopically monitored over the circadian cycle, a dramatic change in the size of these organelles was noted. As shown in Fig. 3, lipid bodies were large and filled with neutral lipids at the end of the light period. By the end of the dark period, neutral lipid content and lipid body size were minimal. The concomitant increase in saturated fatty acids and neutral lipids during the light suggests that the oil bodies contain a higher proportion of these fatty acids than the rest of the cell. Furthermore, neutral lipids decrease more rapidly in the dark than do total fatty acid levels per cell, indicating that fatty acids in the oil bodies are being metabolized and/or being utilized elsewhere in the cell during this time period.
To our knowledge, this data represents the first report for an alga wherein changes in fatty acid quality and quantity determined by GC–MS are coupled with BODIPY 505/515 flow-cytometric measurement of neutral lipids as well as observation of lipid body size. Understanding the timing of fatty acid synthesis is particularly significant, for it appears that algal species differ with respect to fatty acid production during a circadian cycle. For example, though the eustigmatophyte Nannochloropsis gaditana also synthesizes its fatty acids during the light portion of an imposed photoperiod [16], the raphidophyte Heterosigma akashiwo synthesizes fatty acid in the dark [19]. Such diverse observations suggest that these algae may use different metabolic pathways to generate the bulk of their lipids. Production of fatty acids in the dark may suggest that a direct homotropic pathway to fatty acid synthesis is disallowed, implying either that the lipids are produced by heterotrophic metabolism of nutrients in the medium or by the conversion of compounds that were generated in the light.
Fatty Acid Assays of Oleaginous Algae
Algal fatty acids have been recognized as an attractive source of oil for biofuel and pharmaceuticals (e.g., omega 3 and 6 fatty acids), and as a food source for organisms of higher trophic levels. Algae vary extensively in cell morphology, which can affect efficient lipid recovery. The SMIS technique is effective in assaying fatty acid composition in algae possessing a variety of cell coverings, ranging from the very fragile (e.g., naked cells—those delineated by a plasma membrane or cells embellished with delicate scales such as Chrysochromulina sp. or Mallomonas splendens), to the significantly robust (e.g., cells having a theca or multilayered cell walls such as Prorocentrum micans or Nannochloropsis oculata). In our studies we have assayed over 20 algal taxa, representing 11 algal classes, using this method. A representative data set presented in Table 3 shows that fatty acid composition can vary markedly among organisms. For example, Nannochloropsis oculata, suggested as a source of oil for biofuel, has essentially no C22:6 (DHA), whereas Prorocentrum micans, often cultured for human consumption, is a rich source of this nutritionally important unsaturated fatty acid.
Table 3.
Fatty acid profiles of selected algae
| Algae | Mallomonas splendens | Nannochloropsis oculata | Chrysochromulina sp. | Emiliania huxleyi | Rhodomonas sp. | Prorocentrum micans |
|---|---|---|---|---|---|---|
| %a C14:0 | 13.9 ± 0.3 | 0.9 ± 0.0 | 16.9 ± 0.4 | 4.4 ± 0.1 | 1.1 ± 0.0 | 0.9 ± 0.0 |
| % C16:0 | 7.0 ± 0.2 | 10.4 ± 0.3 | 24.0 ± 0.6 | 4.1 ± 0.1 | 3.5 ± 0.1 | 14.7 ± 0.4 |
| % C16:1 | 6.1 ± 0.3 | 14.1 ± 0.6 | 5.8 ± 0.2 | 1.2 ± 0.1 | 0.5 ± 0.0 | 1.4 ± 0.1 |
| % C18:0 | 1.1 ± 0.1 | 0.4 ± 0.0 | 4.6 ± 0.2 | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.6 ± 0.0 |
| % C18:1 | 9.8 ± 1.8 | 1.7 ± 0.3 | 3.7 ± 0.5 | 6.6 ± 1.2 | 0.7 ± 0.1 | 1.2 ± 0.2 |
| % C18:2 | 12.4 ± 0.4 | 1.7 ± 0.1 | 25.7 ± 0.4 | 1.4 ± 0.0 | 1.3 ± 0.0 | 2.4 ± 0.1 |
| % C18:3 | 12.9 ± 0.9 | 0.5 ± 0.0 | 2.5 ± 0.1 | 2.5 ± 0.2 | 3.6 ± 0.2 | 1.2 ± 0.1 |
| % C18:4/5 | 4.4 ± 0.2 | 0.2 ± 0.0 | 6.6 ± 0.4 | 7.3 ± 0.3 | 20.7 ± 1.0 | 15.0 ± 0.7 |
| % C20:4 | ND | 9.5 ± 0.5 | 2.9 ± 0.1 | 3.8 ± 0.2 | 44.1 ± 2.3 | 3.3 ± 0.2 |
| % C20:5 | ND | 60.7 ± 3.1 | 2.8 ± 0.3 | 2.3 ± 0.1 | 4.0 ± 0.2 | 2.5 ± 0.1 |
| % C22:5 | 14.6 ± 1.0 | ND | 1.6 ± 0.1 | 53.4 ± 3.5 | 14.5 ± 0.1 | 41.9 ± 2.8 |
| % C22:6 | 17.8 ± 0.8 | ND | 2.9 ± 0.1 | 12.7 ± 0.6 | 5.7 ± 0.3 | 14.8 ± 0.7 |
| FA/Cell (pg) | 66.5 ± 4.0 | 1.15 ± 0.07 | 7.51 ± 0.45 | 3.17 ± 0.19 | 128 ± 8 | 51.5 ± 3.1 |
| Harvest density (×105 mL−1) | 1.1 | 164.0 | 6.52 | 48.8 | 1.9 | 1.9 |
ND not detected
aPercent of total fatty acid
The pursuit of algal oil for fuel has generated renewed interest in the cultivation of algae for various products. The SMIS method of lipid profiling and quantitation offers advantages in this expanding field. The technique can rapidly identify algal taxa that contain oil compounds of commercial interest. It also greatly simplifies experiments on: the cell cycle, physiological responses to environmental stimuli (e.g. light, temperature, and water chemistry changes), and life history events (e.g. logarithmic to stationary growth phases). Such monitoring facilitates the optimization of algal growth conditions and enables timed harvesting of algal cultures, thus maximizing commercial potential.
Fatty Acid Assays of Zooplankton, Insects, and Plant Material
In addition to algae, the SMIS method has been used to analyze fatty acid composition in a broad array of taxa that are of interest to both the research community and industry (Table 4). The sensitivity of the SMIS technique is evidenced by the fact that a single Drosophila melanogaster sufficed to determine the fatty acid profile of this fly. When Brachionus calyciflorus was fed Chrysochromulina sp. (Table 3), the levels of unsaturated fatty acids in this rotifer increased significantly when compared to control organisms fed an alga (Scenedesmus sp.) that contained low levels of these products. Plant pollen and spores are often difficult materials to assay, given the dense coverings of these reproductive cells. The fatty acid profiles of Centaurea cyanus (corn flower) pollen and Lycopodium nummularifolium (club moss) spores (Table 4) appear different from most algae, in that they lack long chain unsaturated fatty acids. The technique was also used to successfully obtain profiles from exudate of Nepenthes khasiana (pitcher plant) before and after prey capture, proving that its small sample size requirements permit the analysis of materials which are difficult to obtain in large quantities.
Table 4.
Fatty acid profiles of other organisms
| Organism | Daphnia magnus | Drosophila melanogaster male | Drosophila melanogaster female | Brachionus calyciflorus fed Chrysochromulina sp. | Centaurea cyanus pollen | Lycopodium nummularifolium spores |
|---|---|---|---|---|---|---|
| Phylum | Arthropoda | Arthropoda | Arthropoda | Rotifera | Magnoliophyta | Lycopodiophyta |
| %a C8:0 | ND | ND | ND | ND | ND | trace |
| % C9:0 | ND | ND | ND | ND | ND | trace |
| % C10:0 | ND | ND | ND | ND | ND | trace |
| % C12:0 | ND | ND | ND | ND | ND | trace |
| % C14:0 | 8.5 ± 0.2 | 8.3 ± 0.2 | 12.1 ± 0.3 | 4.2 ± 0.1 | ND | 0.9 ± 0.0 |
| % C15:0 | ND | ND | ND | ND | ND | Trace |
| % C16:0 | 17.1 ± 0.4 | 17.2 ± 0.4 | 19.2 ± 0.5 | 10.4 ± 0.3 | 72.5 ± 1.8 | 15.1 ± 0.4 |
| % C16:1 | 4.2 ± 0.2 | 21.2 ± 0.9 | 25.7 ± 1.1 | 9.8 ± 0.4 | Trace | 21.4 ± 0.9 |
| % C18:0 | 5.6 ± 0.4 | 2.9 ± 0.2 | 3.4 ± 0.2 | 4.5 ± 0.3 | 1.3 ± 0.1 | 2.2 ± 0.2 |
| % C18:1 | 4.8 ± 0.9 | 15.8 ± 2.9 | 27.0 ± 5.0 | 54.2 ± 10.1 | 14.8 ± 2.8 | 42.9 ± 8.0 |
| % C18:2 | 14.7 ± 0.5 | 34.6 ± 1.2 | 12.6 ± 0.5 | 6.4 ± 0.2 | 11.4 ± 0.4 | 8.5 ± 0.3 |
| % C18:3 | 5.9 ± 0.4 | ND | ND | 3.8 ± 0.3 | ND | ND |
| % C18:4/5 | 10.0 ± 0.5 | ND | ND | 0.6 ± 0.0 | ND | 0.1 ± 0.0 |
| % C20:0 | ND | ND | ND | ND | Trace | Trace |
| % C20:4 | 12.7 ± 0.7 | ND | ND | 1.0 ± 0.1 | ND | 0.2 ± 0.0 |
| % C20:5 | 16.4 ± 0.9 | ND | ND | 1.9 ± 0.1 | ND | 1.0 ± 0.1 |
| % C22:0 | ND | ND | ND | ND | ND | Trace |
| % C22:5 | ND | ND | ND | 1.1 ± 0.1 | ND | 7.6 ± 0.5 |
| % C22:6 | ND | ND | ND | 2.0 ± 0.1 | ND | ND |
| % C24:0 | ND | ND | ND | ND | ND | Trace |
| Total fatty acid per organism or cell (g) | (5.68 ± 0.34) × 10−7 | (4.78 ± 0.29) × 10−5 | (9.13 ± 0.55) × 10−5 | NA | NA | (2.00 ± 0.12) × 10−9 |
| Organisms or cells used in Assay | 5 | 1 | 1 | Not counted | Not counted | 2.26 × 106 |
ND not detected, NA not applicable
aPercent of total fatty acid
Conclusion
The broad application of the SMIS technique allows for the assessment of fatty acid levels in spores, pollen, algae, and other organisms where sample size may vary significantly. Critically, the technique can be used when probing small samples or difficult to obtain material. Because the SMIS method is a rapid technique, a researcher can process as many as 100 samples per day. Its high sensitivity (a single Drosophila can be assayed) enables population analysis of many small organisms at a refined level.
Minimal sample size requirements and high potential throughput make this technique an ideal candidate for large-scale algal strain screening projects. It may also be applied to screening for successful expression in genetically modified organisms. Lastly, this technique has broad applications in aquaculture.
Acknowledgments
We would like to thank William Yost for assistance with cell cycle experiments, Chloe Deodato for providing algal cultures, Matthew Stephens for providing Daphnia magnus and Brachionus calyciflorus, Barbara Wakimoto for providing Drosophila melanogaster, and Doug Ewing for his help in obtaining plant pollen and spores. We would also like to thank Megan Black for waste-water acquisition, Lewis Johnson for help in manuscript review, and Loren Kruse and Martin Sadilek for analytical assistance. We would like to acknowledge funding of this work by the US Department of Energy under contract DE-EE0003046 awarded to the National Alliance for Advanced Biofuels and Bioproducts.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- HDPE
High density polyethylene
- PTFE
Polytetrafluoroethylene
- AOAC
American Association of Analytical Chemists
- AOCS
American Oil Chemists Society
- SMIS Method
Sub-microscale in situ method
- CCMP
Provasoli–Guillard National Center for Culture of Marine Phytoplankton
- UWC
University of Washington Culture Collection
References
- 1.Chisti Y. Biodiesel from microalgae beats bioethanol. Trends Biotechnol. 2007;26(3):126–131. doi: 10.1016/j.tibtech.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Ackman RG. Remarks on official methods employing boron trifluoride in the preparation of methyl esters from the fatty acids of fish oils. J Am Oil Chem Soc. 1998;75:541–545. doi: 10.1007/s11746-998-0263-9. [DOI] [Google Scholar]
- 3.Barker JP, Mescher A, Kramlich J (2009) Fatty acid compositions of solvent extracted lipids from two microalgae. In: 2009 SAE Aerotech conference
- 4.AOAC . Fatty acid methyl esters. Washington, DC: Association of Official Analytical Chemists; 2005. p. 22. [Google Scholar]
- 5.AOCS (2007) AOCS methods for biodiesel feedstock quality. Official methods and recommended practices of the AOCS, 5th edn, AOCS, USA
- 6.Thiemann GW, Budge SM, Iverson SJ. Determining blubber fatty acid composition: a comparison of in situ direct and traditional methods. Mar Mammal Sci. 2004;20(2):284–295. doi: 10.1111/j.1748-7692.2004.tb01157.x. [DOI] [Google Scholar]
- 7.Georgogianni KG, Kontominas MG, Pomonis PJ, Avlonitis D, Gergis V. Alkaline conventional and in situ transesterification of cottonseed oil for the production of biodiesel. Energy Fuels. 2008;22:2110–2115. doi: 10.1021/ef700784j. [DOI] [Google Scholar]
- 8.Wyatt VT, Haas MJ. Production of fatty acid methyl esters via the in situ transesterification of soybean oil in carbon dioxide-expanded methanol. J Am Oil Chem Soc. 2009;86:1009–1016. doi: 10.1007/s11746-009-1438-8. [DOI] [Google Scholar]
- 9.Zeng J, Wang X, Zhao B, Sun J, Wang Y. Rapid in situ transesterification of sunflower oil. Ind Eng Chem Res. 2009;48:850–856. doi: 10.1021/ie8008956. [DOI] [Google Scholar]
- 10.Lepage G, Roy CC. Improved recovery of fatty acid through direct transesterification without prior extraction or purification. J Lipid Res. 1984;25:1391–1396. [PubMed] [Google Scholar]
- 11.Ravet JL, Brett MT. Phytoplankton essential fatty acid and phosphorus content constraints on Daphnia somatic growth and reproduction. Limnol Oceanogr. 2006;51(5):2438–2452. doi: 10.4319/lo.2006.51.5.2438. [DOI] [Google Scholar]
- 12.Dean RB, Dixon WJ. Simplified statistics for small numbers of observations. Anal Chem. 1950;23(4):636–638. doi: 10.1021/ac60052a025. [DOI] [Google Scholar]
- 13.Mazalli MR, Bragagnolo N. Validation of two methods for fatty acid analysis in eggs. Lipids. 2007;42:483–490. doi: 10.1007/s11745-007-3046-4. [DOI] [PubMed] [Google Scholar]
- 14.Carvalho AP, Malcata XF. Preparation of fatty acid methyl esters for gas-chromatographic analysis of marine lipids: insight studies. J Agr Food Chem. 2005;53:5049–5059. doi: 10.1021/jf048788i. [DOI] [PubMed] [Google Scholar]
- 15.Vicente G, Martines M, Aracil J. Integrated biodiesel production: a comparison of different homogeneous catalyst systems. Bioresour Technol. 2004;92:297–305. doi: 10.1016/j.biortech.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Fabregas J, Maseda A, Dominguez A, Ferreira M, Otero A. Changes in the cell composition of the marine microalga, Nannochloropsis gaditana, during a light:dark cycle. Biotechnol Lett. 2002;24:1699–1703. doi: 10.1023/A:1020661719272. [DOI] [Google Scholar]
- 17.Dodge JD. The fine structure of algal cells. New York: Academic Press; 1973. [Google Scholar]
- 18.Cooper MS, Hardin WR, Petersen TW, Cattolico RA. Visualizing “green oil” in live algal cells. J Biosci Bioeng. 2009;109(2):198–201. doi: 10.1016/j.jbiosc.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Wada M, Hara Y, Kato M, Yamada M, Fujii T. Diurnal appearance, fine structure, and chemical composition of fatty particles in Heterosigma akashiwo (Raphidophyceae) Protoplasma. 1987;137:134–139. doi: 10.1007/BF01281148. [DOI] [Google Scholar]
- 20.McIntosh L, Cattolico RA. Preservation of algal and higher plant ribosomal RNA integrity during extraction and electrophoretic quantitation. Anal Biochem. 1978;91:600–612. doi: 10.1016/0003-2697(78)90546-8. [DOI] [PubMed] [Google Scholar]
- 21.Viga A, Grahl-Nielsen O. Genotypic and phenotypic fatty acid composition in the tissues of salmon, Salmo salar. Comp Biochem Phys B. 1990;96(4):721–727. doi: 10.1016/0305-0491(90)90220-N. [DOI] [Google Scholar]