

Abstract
Computational assessment of the binding interactions of drugs is an important component of computer-aided drug design paradigms. In this perspective, a set of 30 1-(substituted phenyl)-3-(naphtha[1, 2-d] thiazol-2-yl) urea/thiourea derivatives showing antiparkinsonian activity were docked into inhibitor binding cavity of human adenosine A2A receptor (AA2AR) to understand their mode of binding interactions in silico. Lamarckian genetic algorithm methodology was employed for docking simulations using AutoDock 4.2 program. The results signify that the molecular docking approach is reliable and produces a good correlation coefficient (r2 = 0.483) between docking score and antiparkinsonian activity (in terms of % reduction in catalepsy score). Potent antiparkinsonian agents carried methoxy group in the phenyl ring, exhibited both hydrophilic and lipophilic interactions with lower energy of binding at the AA2AR. These molecular docking analyses should, in our view, contribute for further development of selective AA2AR antagonists for the treatment of Parkinson's disease.
Keywords: Adenosine A2A receptor antagonist; Parkinson's disease; naphtha [1, 2-d] thiazol-2-amine; Urea derivatives
Background
Adenosine A2A receptors (AA2AR) are highly distributed in the central nervous system and are found in abundance in the basal ganglia, a region of the brain associated with motor function [1]. The colocalization of AA2AR and dopamine D2 receptors in the striatopallidal neurons provides the anatomical basis for the existence of a functional antagonistic interaction between these receptors [2]. Stimulation of the dopamine D2 receptors with dopamine or other dopamine D2 receptor agonists enhances motor activity whereas stimulation of AA2ARs reduces this effect [3]. Likewise, AA2AR antagonists may provide a novel therapy for the treatment of Parkinson's disease (PD) with lower risk of dyskinesias. In addition, they may exhibit neuroprotective effects [4]. Furthermore, AA2AR antagonists were found not only to diminish the symptoms of PD but also to potentiate the effect of levodopa [5], so, it may be possible to reduce the dose of the dopaminergic drugs and therefore the occurrence of side effects. AA2AR antagonists are therefore a promising adjunctive to dopamine replacement therapy, and several companies have now advanced selective antagonists of this receptor into clinical development [4]. Significant progress has been made in computer-aided drug design by pharmaceutical companies at different stages of drug discovery such as identifying new hits, enhancing molecule binding affinity in hit-to-lead, and lead optimization [6]. Moreover, in silico approaches are routinely used in modern drug design to help understand drug-receptor interactions. It has been shown in the literature that computational techniques can strongly support and help the design of novel, more potent inhibitors by revealing the mechanism of drug-receptor interactions [7].
Thiazole derivatives have evoked considerable attention in recent years as these are endowed with wide range of biological activities as well as drugs of PD [8–12]. Hoffmann-La Roche discovered benzothiazole derivetives as novel scaffold for AA2AR antagonists [13]. Furthermore, certain thiazoles with a urea moiety [14] have also demonstrated AA2AR antagonistic activities for the development of a suitable approach to the treatment of PD that may be the starting point for the future drug design. Recently, we have discovered urea derivatives of naphtha [1, 2-d]thiazol-2-amine as novel anti-Parkinsonian agents that cause neuroprotection against haloperidol-induced oxidative stress in mice [10]. These findings have motivated us to reveal their interactions with AA2ARs insilico by using AutoDock program and to attest anti-Parkinsonian activity associated with them.
Materials and Methodology
For the present study, crystal structure of human AA2AR with PDB ID: 3EML [15] was downloaded from the protein data bank (www.rcsb.org/pdb). For docking experiments with AutoDock 4.2, antiparkinsonian molecules reported by our group [10] were drawn in ChemBioDraw Ultra 12.0 and converted to their three dimensional structures in ChemBio3D Ultra 12.0, energy minimized by PM3 method using MOPAC Ultra 2009 program (http://OpenMOPAC.net) and saved in pdb format. The prepared ligands were used as input files for AutoDock 4.2 in the next step. Lamarckian genetic algorithm method [16] was employed for docking simulations. The standard docking procedure was used for a rigid protein and a flexible ligand whose torsion angles were identified (for 10 independent runs per ligand). A grid of 60, 60, and 60 points in x, y, and z directions was built with a grid spacing of 0.375 Å and a distancedependent function of the dielectric constant were used for the calculation of the energetic map. The default settings were used for all other parameters. At the end of docking, the best poses were analyzed for hydrogen bonding/π-π interactions and root mean square deviation (RMSD) calculations using Discovery Studio Visualizer 2.5 (Accelrys Software Inc.) and Pymol (The PyMOL Molecular Graphics System) programs. From the estimated free energy of ligand binding (ΔGbinding, kcal/mol), the inhibition constant (Ki) for each ligand was calculated Table 1 (see Table 1).
Results and Discussion
Validation of the docking protocol
To evaluate the accuracy of AutoDock 4.2 as an appropriate docking tool for the present purpose, the co-crystallized ligand (ZM241385) was redocked within the inhibitor binding cavity of human AA2AR, and the docked position was compared to the crystal structure position by calculating RMSD values (0.88 Å, Figure 1). In general, RMSD values smaller than 2.0 Å indicate that the docking protocol is capable of accurately predicting the binding orientation of the co-crystallized ligand [17]. In this study, RMSD values were within 2.0 Å, indicating our docking methods are valid for the given structures and AutoDock 4.2, therefore deemed reliable for docking of naphtha [1, 2- d]thiazolyl urea/thiourea derivatives into the inhibitor binding cavity of AA2AR.
Figure 1.
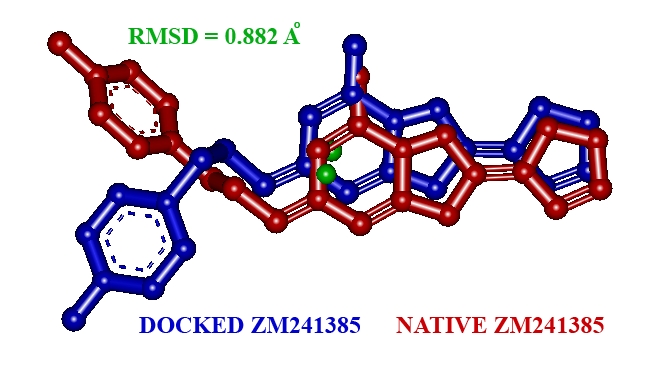
The native co-crystallized ligand ZM241385 docked with the active site of AA2AR.
Correlation between docking scores and antiparkinsonian activity
The results signify that the molecular docking approach is reliable and produces a good correlation coefficient (r2 = 0.483) between docking score and antiparkinsonian activity (in terms of % reduction in catalepsy score). Removal of 4 compounds (compounds 17, 26, 28 and 30) identified as outliers from the docking dataset and yield a better model with correlation coefficient (r2) of 0.646 with 26 compounds (Figure 3). Outliers in the molecular docking studies are anticipated because, docking calculations simulate the interaction between a compound and a protein's active site and the results are comparable to those of biochemical assays/animal experiments; however, it does not take into account, the bioavailability, toxicity, and other factors present in the body.
Figure 3.
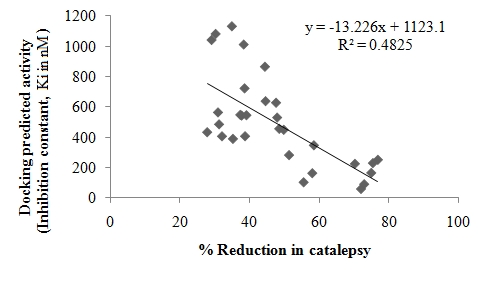
A correlation for docking predicted activity and experimental antiparkinsonian activity.
Binding interactions of 1-(substituted phenyl)-3-(naphtha [1, 2-d] thiazol- 2-yl) urea/thiourea derivatives with human AA2AR
All of the naphtha [1, 2-d] thiazolyl urea/thiourea derivatives were docked into the active site of AA2AR (PDB code: 3EML) to study the possible mode of their interaction. Docking of these compounds into inhibitor binding cavity of AA2AR confirms that these compounds dock in a similar binding modus like native co-crystallized ligand, ZM-241385 (Figure 2). Inhibitor binding cavity of AA2AR is outlined by residues Ile-66, Ala-81, Leu-85, Phe-168, Glu-169, Met-177, Trp-246, Leu-249, His-250, Asn-253, His-264, Leu-267, Met-270, Tyr-271, Ser-277 and His-278. Analysis of the receptor/ligand complex models generated after successful docking of the urea and thiourea derivatives was based on parameters such as: 1) Hydrogen bond interactions; 2) π-π stacking/hydrophobic interactions; 3) binding energy; 4) RMSD of active site residues; 5) Orientation of the docked compound within the active site. The bicyclic triazolotriazine core of ZM241385, native co-crystallized AA2AR antagonist, is anchored by an aromatic stacking interaction with Phe-168 [15], an aliphatic hydrophobic interaction with Ile 274 [18] and a hydrogen bonding interaction with Asn 253 [19]. Likewise, the tricyclic naphthothiazole ring was found to interact with aromatic ring of Phe-168 by π-π stacking interactions and compound 22 participated in hydrogen bonding with Asn 253. Adjacent to Phe- 168, a polar residue Glu-169 donates a hydrogen bond to oxygen atom of methoxy group substituted in phenyl ring of compounds 4, 6, 16, 21 and 22. Similarly, exocyclic amino group (N-15 atom) of ZM241385 also participates in hydrogen bonding with Glu-169 [15, 20]. As a general rule, in most of the potent and fair antiparkinsonian compounds, both hydrogen bond and π-π stacking/hydrophobic interactions between the compound and the active site residues of the receptor have been found to be responsible for mediating the biological activity. In addition to be involved in π-π stacking/hydrophobic interactions with most docked compounds, Phe-168 contributes to H-bond interactions in compounds 5, 6, 9, 10, 18 and 29. Very potent antiparkinsonian agents (compounds 4, 5, 6, 7, 21 and 22) carried methoxy group in the phenyl ring and interacted with Phe-168, Glu-169 and His-278 residues for H-bonding. However, replacement of methoxy group with phenoxy group (compound 8) lost H-bond that resulted in decreased antiparkinsonian activity. Generally, compounds substituted with methyl group or halogen in the aromatic ring of urea or thiourea derivatives (compounds 3, 8, 9, 10, 15, 16, 19, 20, 23 24, 25, 26, 27, 31 and 32) demonstrated poor binding energies as well as both hydrophilic and lipophilic interactions. These compounds were also noted to be poor antiparkinsonian agents.
Figure 2.
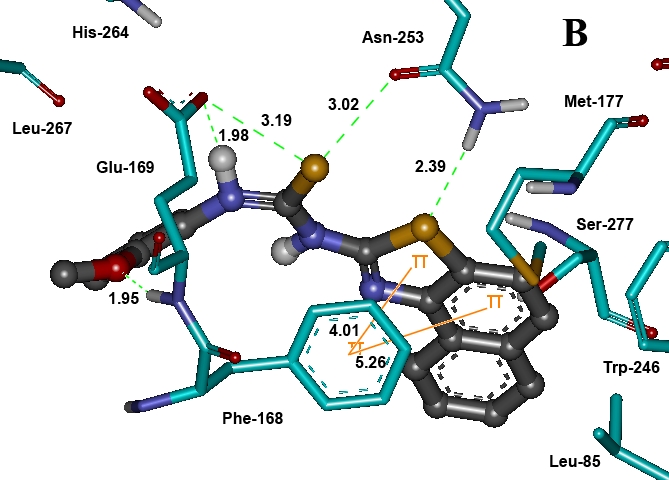
Docked conformation of compound 22 shown as ball and stick in grey color. The residues of binding pocket are shown as stick in cyan color. Dashed lines in green indicate H-bonds and π-π stacking interactions are shown in orange. Bond distances are given in Å.
Conclusion
Molecular docking studies of 1-(substituted phenyl)-3-(naphtha[1,2-d]thiazol- 2-yl)urea/thiourea derivatives with AA2AR exhibited very good binding interactions and warrants further studies to confirm their binding with human AA2AR for the design and development of potent antagonists as novel antiparkinsonian agents. These results clearly indicate that before synthesis and biochemical testing of new analogs, one can use molecular docking studies for assessment of relative binding affinities with AA2AR for most promising drug candidates.
Supplementary material
Footnotes
Citation:Azam et al, Bioinformation 6(9): 330-334 (2011)
References
- 1.P Svenningsson, et al. Prog Neurobiol. 1999;59:355. doi: 10.1016/s0301-0082(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 2.A Pinna, et al. Life Sci. 2005;77:3259. doi: 10.1016/j.lfs.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 3.JD Salamone, et al. Behav Brain Res. 2009;201:216. doi: 10.1016/j.bbr.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.F Azam, et al. Pharmazie. 2009;24:771. [Google Scholar]
- 5.S Rose, et al. Eur J Pharmacol. 2006;546:82. doi: 10.1016/j.ejphar.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 6.H Sun, DO Scott. Chem Biol Drug Des. 2010;75:3. doi: 10.1111/j.1747-0285.2009.00935.x. [DOI] [PubMed] [Google Scholar]
- 7.V Srivastava, et al. Bioinformation. 2008;3:180. [Google Scholar]
- 8.F Azam, et al. Eur J Med Chem. 2009;44:3889. doi: 10.1016/j.ejmech.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 9.F Azam, et al. J Enzyme Inhib Med Chem. 2009;24:808. doi: 10.1080/14756360802399183. [DOI] [PubMed] [Google Scholar]
- 10.F Azam. Med Chem Res. 2009;18:287. [Google Scholar]
- 11.F Azam, et al. J Enzyme Inhib Med Chem. 2010;25:818. doi: 10.3109/14756361003671052. [DOI] [PubMed] [Google Scholar]
- 12.F Azam, et al. Eur J Med Chem. 2009;44:203. [Google Scholar]
- 13.CE Muller, S Ferre. Recent Pat CNS Drug Discov. 2007;2:1. doi: 10.2174/157488907779561772. [DOI] [PubMed] [Google Scholar]
- 14.JE van Muijlwijk-Koezen, et al. J Med Chem. 2001;44:749. [Google Scholar]
- 15.VP Jaakola, et al. Science. 2008;322:1211. [Google Scholar]
- 16.GM Morris, et al. J Comput Chemistry. 1998;19:1639. [Google Scholar]
- 17.J Bostrom, et al. J Mol Graph Model. 2003;21:449. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 18.O Yuzlenko, K Kiec-Kononowicz. J Comput Chem. 2008;30:14. [Google Scholar]
- 19.J Kim, et al. J Biol Chem. 1995;270:13987. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanson, et al. Structure. 2008;16:897. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.