

Abstract
Influenza virus A (IVA) infection is responsible for recent death worldwide. Hence, there is a need to develop therapeutic agents against the virus. We describe the prediction of short interfering RNA (siRNA) as potential therapeutic molecules for the HA (Haemagglutinin) and NA (Neuraminidase) genes. We screened 90,522 siRNA candidates for HA and 13,576 for NA and selected 1006 and 1307 candidates for HA and NA, respectively based on the proportion of viral sequences that are targeted by the corresponding siRNA, with complete matches. Further short listing to select siRNA with no off-target hits, fulfilling all the guidelines mentioned in approach, provided us 13 siRNAs for haemagglutinin and 13 siRNAs for neuraminidase. The approach of finding siRNA using multiple sequence alignments of amino acid sequences has led to the identification of five conserved amino acid sequences, three in hemagglutinin i.e. RGLFGAIAGFIE, YNAELLV and AIAGFIE and two in neuraminidase i.e. RTQSEC and EECSYP which on reveres translation provided siRNA sequences as potential therapeutic candidates. The approaches used during this study have enabled us to identify potentially therapeutic siRNAs against divergent IVA strains.
Keywords: Influenza virus A, Sequence analysis, siRNA, Hemagglutinin, Neuraminidase
Background
Influenza viruses, a cause of significant morbidity and mortality in human population, possess a segmented genome of negative sense RNA encapsulated by a virally specified nucleoprotein. There are three types of influenza viruses that cause illness in humans: type A, B and C, classified according to the serological reactivity of internal proteins. The most common infectious agent of upper respiratory tract, Influenza A virus (IVA) is able to persistently reinfect human populations by continually evading host immunity through the rapid evolution of surface antigens i.e. antigenic drift [1, 2, 3]. Influenza A virus can be further sub-typed according to the serological reactivity of its surface antigens (attachment protein of virus), HA (Haemagglutinin) and NA (Neuraminidase), proteins that are encoded on separate genome segments. To date, 15 HA subtypes and nine NA subtypes have been identified [4, 5]. IVA evolves rapidly because of the low fidelity of the RNA-dependent RNA polymerase. RNA-dependent RNA polymerases generally produce replication errors in 1/104 bases per replication cycle, whereas DNA polymerases produce replication errors in 1/109 bases per replication cycle. Due to this phenomenon individual point mutations lead to amino acid substitutions in the entire influenza genome, and particularly the ones that occur in the surface glycoproteins enabling influenza viruses to evade the host immune system and cause infection and re-infection despite the presence of pre-existing antibody due to ‘antigenic drift’ [3]. Every new antigenic drift variant of influenza A appears to have at least four amino acid substitutions located in two or more antigenic sites of the HA protein [3]. Furthermore, the segmented nature of the influenza genome facilitates the exchange of genetic information between different influenza viruses. This allows the virus to undergo drastic evolutionary changes resulting in ‘antigenic shift’ and hence emergence of antigenically novel influenza viruses. The presence of a large reservoir of influenza A viruses in birds increases the number of viral subtypes available for reassortment, when two different viruses from two different host species coinfect a single individual animal, that have the potential to cause major global pandemics [3]. Due to such processes, human influenza is considered by some as a non eradicable disease, since the range of antigenically diverse subtypes of influenza A viruses are present in waterfowl and can be introduced into humans and may lead to millions of deaths [6–9]. Existing drugs and vaccines against Influenza A virus have limited value and the threat of pandemic persists. Broad spectrum anti-virals have been developed in the past that target an enzyme found in all viruses that have RNA genomes (except retroviruses) i.e. viral RNA-dependent RNA polymerases [10, 11].
Apart from viral proteases and polymerases, some other viral proteins can act as potential drug targets. For instance, Inhibitors of virus entry is another new promising class of drugs that show clinical efficacy. In this regard, inhibitors that block virus entry into cells, by targeting the virus envelope glycoproteins have yielded a clinically approved compound for HIV [12] as well as a promising lead to treat respiratory syncytial virus infections [13]. Similarly, inhibitors of the neuraminidase (glycoprotein) of influenza A and B viruses, two of which are zanamivir [14] and oseltamivir [15], are now approved for therapeutic use in humans. By inhibiting specific cell proteins that are required for virus replication it should be possible to interrupt the virus life cycle [16]. However, possible cross reactivity with host proteins can cause adverse sideeffects inhibiting essential host cell functions [17]. On contrary to agents typically inhibiting the function of crucial viral proteins, RNAi is the relatively newly described natural biological phenomenon that achieves the identical goal by targeting the viral mRNA instead of the proteins they encode. RNA interference (RNAi) is now being widely used to knockdown gene expression, in a sequence specific manner, for therapeutic purposes [18, 19]. The efforts to develop anti-therapeutic agents directly from viral genome sequences has led to short (21-26 nucleotides) interfering RNA (siRNA), double-stranded RNA duplexes that mediate sequence-selective inhibition. In this regard, many experiments have been carried out in the past to clarify possible sequence requirements of functional siRNAs [19, 20]. The guidelines explained in three major previous studies [18– 20] for selecting functional siRNAs are useful in selecting siRNA molecules for therapeutic purpose. However, designing functional siRNAs that target viral sequences is problematic because of their extraordinarily high genetic diversity. This problem suggests a strong need to select highly conserved target sites for designing siRNAs from the pool of various epidemic strains reported to date. Several types of modifications have been incorporated into the ends of the siRNA duplex to study their influence on the silencing ability and duration of their effect in the past [21]. Special care is suggested while designing siRNA as mismatches particularly in the centre of the siRNA duplex prevents target RNA cleavage [22]. The HA and NA have a central role in adaptation of virus, because of their interaction with host cell surfaces and proteases. In this study, we used web-based bioinformatics approach to predict siRNA neutralizing wider range of influenza virus A (IVA) variants by analyzing haemagglutinin (HA) and neuraminidase (NA) genes A (IVA). Moreover, reverse translational approach, using highly conserved amino acid sequences, was used to identify potentially important antiviral siRNAs.
Methodology
Selection & retrieval of sequences
To design siRNA, all the sequences of HA (segment 4) & NA (segment 6) genes of Influenza Virus A already present in the database [http://siVirus.rnai.jp ] were initially selected, and then those sequences were short listed which were derived from viral isolates of a specific geographic region of Asia i.e. Hong Kong, China, Japan and Russia, for this study. Sequence data for gene segments 4 (Haemagglutinin) and 6 (Neuraminidase) of Influenza Virus A was collected for the past 39 years i.e. 1960-1999. The sequences from corresponding countries as mentioned above were downloaded, for multiple alignments, from NCBI Influenza Virus Sequence Database [ http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html]. Sequences for siRNA were retrieved from siVirus based on their degree of conservation, defined as the proportion of viral sequences that are targeted by the corresponding siRNA, with complete matches (i.e. 21/21 matches).
siRNA analysis
siVirus interface was used to determine anti-siRNA sequence results. This interface especially focuses on anti-siRNA design and provides (a) highly conserved target sites for designing anti-siRNA that would resist viral mutational escapes (b) selection of effective siRNAs based on guidelines of Ui- Tei et al., Reynolds et al., Amarzguioui et al. [18–20] (c) Off-target minimized siRNAs within two mismatches against human genes. Off-target hits are those which have dissimilarity to the target sequence. It is desirable to select siRNA that has less off-target hits.
Sequence analysis
TCOFFEE [ http://www.ebi.ac.uk/t-coffee/.html] was used for multiple alignments to observe conservation and functional sites in the HA and NA datasets at amino acid level. The consensus in at least 7 amino acids corresponding to 21 nucleotides was noted. BLAST program was used for similar sequence searches.
Results and Discussion
To design siRNA against influenza virus A, we analyzed all the sequences for gene segment 4 (haemagglutinin) and 6 (neuraminidase) of Influenza Virus A from some of the Asian countries (Tables 3 & 4 see Tables 3 & 4) The selection of effective siRNAs sequences were based on published guidelines [18–20]. Those guidelines were: (a) A/U at the 5' end of the antisense strand, (b) G/C at the 5' end of the sense strand, (c) At least 5 A/U residues at the 5' terminal, one-third of the antisense strand (d) The absence of any GC stretch of more than 9 nucleotides in length. In case any of the above mentioned guideline is not fulfilled, then no gene silencing by a particular siRNA is expected. Furthermore, consensus in amino acid sequences in the HA (H1, H3, H5 and H9) and NA (N1-N2) of different strains was noted and only those strings of at least 7 amino acids were retained which were present in all types studied.
siRNA prediction using siVirus
Figure 1 shows a typical output from siVirus for designing anti-influenza Virus A siRNAs. The sequences from Influenza virus A subtypes A and B, which are the most prevalent genotypes circulating in Asia, were selected. The results were sorted by the degree of conservation and filtered to display siRNAs that satisfy at least one efficacy guideline mentioned above. Out of 90522 siRNA candidates screened, 1006 sequences targeting HA segment, whereas out of 13576 siRNA candidates screened, 1307 sequences targeting NA segment of influenza virus A genome were short listed respectively. Analysis of these siRNA candidates revealed that no conserved siRNAs were obtained when using 90% conservation stringency parameter. However, on decreasing the stringency parameter close to 80%, 100% conservation of siRNAs pertaining to gene segment 4 (haemagglutinin) (Figure 1 & Figure 2) and segment 6 (neuraminidase) (Data Not shown) was noted. Further short listing to select siRNA with no off-target hits, fulfilling all the guidelines mentioned in approach, provided us 13 siRNAs for haemagglutinin and 13 siRNAs for neuraminidase (Tables 1 & 2 see Tables 1 & 2)
Figure 1.

siRNA results for Haemagglutinins (A = siRNA sequence; B = siRNA position; C = siRNA efficacy predictions; D = Off-target search results. The number of human off-targets within two mismatches; E = Degrees of conservation; F = Conservation in the user-selected sequences)
Figure 2.

Graph showing percentage probability of predicted siRNA for Haemagglutinins. White Box represents the small fractions of siRNA filtered through all possible approaches (6.65%)
siRNA prediction using conserved stretches of amino acids
Multiple sequence alignments of amino acids retrieved from NCBI [ http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html] (Tables 3 & 4, see Tables 3 & 4) resulted in obtaining some conserved strings of amino acids i.e., RGLFGAIAGFIE, YNAELLV and AIAGFIE in hemagglutinin (H) among different subtypes of segment 4 (H1, H2, H3, H5, H9) and RTQSEC and EECSYP in neuraminidase (N) among different subtypes of segment 6 (N1 and N2) for different Asian strains (Results not shown). Based on their degree of conservation, we suggest that these conserved amino acid segments could potentially act as useful targets for designing therapeutic siRNAs for different subtypes. These conserved amino acid sequences were reverse translated using a webserver [ http://www.bioinformatics.org/sms2/rev_trans.html] and we obtained the RNA sequence “gcgauugcgggcuuuauugaa” for AIAGFIE, “cgcggcctguuuggcgcgauugcgggcuuuauugaa” for RGLFGAIAGFIE, “uauaacgcggaacugcuggug” for YNAELLV, “cgcacccaggaaagcgaaugc” for RTQESEC and “gaagaaugcagcugcuauccg” for EECSCYP. A number of studies demonstrated inhibition of replication of RNA viruses by RNAi [25, 26], including HIV, Polio virus, Hepatitis C virus, West Nile virus and Influenza virus [27, 28]. The approach of identifying siRNAs by reverse translation of the consensus motifs/conserved amino acid string from different subtypes of haemagglutinin and neuraminidase can be a useful strategy. This needs to be incorporated in any such software tool to obtain more effective translation inhibitor siRNA molecules covering broader range of influenza subtypes. The approach will also help designing antiviral siRNAs that would resist viral mutational escapes.
Conclusion
To identify new potential leads for developing antiviral therapies against IVA we screened an siRNA library targeting haemagglutinin and neuraminidase. For this it was desirable to select an siRNA that had less off target hits and hence to be more specific. The current in silico study resulted in finding various targets for siRNA in haemagglutinin and neuraminidase and the results may have implications in developing prophylactic as well as therapeutic treatment against influenza infection. Another interesting finding of the current study was obtaining few but completely different siRNA targets in HA and NA genes compared to our conventional siRNA identification approach using web based tools, when we reverse translated conserved amino acids back to RNA. We believe that the approach of developing siRNA based on combination of both approaches i.e. Direct search for siRNA against appropriate targets and finding the consensus of amino acids of majority of the circulating virus variants would provide a broad range protection molecule to control the infection.
Supplementary material
Figure 3.
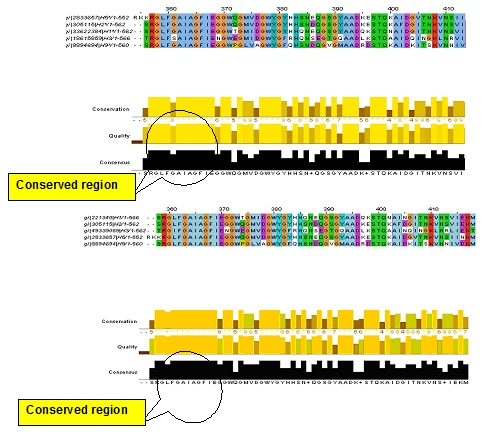
Result showing conserved regions in haemagglutinin using multiple sequence alignment
Acknowledgments
We thank Higher Education Commission of Pakistan and COMSATS Institute of Information Technology, Islamabad for all the support and facilities provided to carry out this work.
Footnotes
Citation:Raza et al, Bioinformation 6(9): 340-343 (2011)
References
- 1.H Hatice, et al. J Med Microbiol. 2009;58:408. doi: 10.1099/jmm.0.006098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WM Fitch, et al. Proc Natl Acad Sci U S A. 1997;94:7712. [Google Scholar]
- 3.MC Zambon. Rev Med Virol. 2001;11:227. doi: 10.1002/rmv.319. [DOI] [PubMed] [Google Scholar]
- 4.C Rohm, et al. Virology. 1996;217:508. doi: 10.1006/viro.1996.0145. [DOI] [PubMed] [Google Scholar]
- 5.RG Webster, et al. Microbiol Rev. 1992;56:152. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.K Subbarao, MW Shaw. Rev Med Virol. 2000;10:337. doi: 10.1002/1099-1654(200009/10)10:5<337::aid-rmv292>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.KD Paterson, GF Pyle. Bull Hist Med. 1991;65:4. [PubMed] [Google Scholar]
- 8.JSP Malik, et al. Clin Microbiol Rev. 2007;20:243. doi: 10.1128/CMR.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.JM Silva, et al. Trend Mol Med. 2002;8:5050. [Google Scholar]
- 10.Q Ge, et al. Proc Natl Acad Sci U S A. 2003;100:2718. [Google Scholar]
- 11.KG Nicholson, et al. Lancet. 2000;355:1845. [Google Scholar]
- 12.RY Chen, et al. Expert Opin Investig Drugs. 2002;11:1837. doi: 10.1517/13543784.11.12.1837. [DOI] [PubMed] [Google Scholar]
- 13.C Cianci, et al. J Antimicrob Chemother. 2005;55:289. doi: 10.1093/jac/dkh558. [DOI] [PubMed] [Google Scholar]
- 14.MV Itzstein, et al. Nature. 1993;363:418. [Google Scholar]
- 15.CU Kim, et al. J Am Chem Soc. 1997;119:681. [Google Scholar]
- 16.D Schurmann, et al. AIDS. 2007;21:1293. [Google Scholar]
- 17.S Esser, et al. J Dtsch Dermatol Ges. 2007;5:245. [Google Scholar]
- 18.A Fire, et al. Nature. 1998;391:806. [Google Scholar]
- 19.CC Mello, D Conte. Nature. 2004;431:338. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- 20.K Ui-Tei, et al. Nucleic Acid Res. 2004;32:936. [Google Scholar]
- 21.A Reynolds, et al. Nat Biotechnol. 2004;22:326. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 22.M Amarzguioui, H Prydz. Biochem Biophys Res Commun. 2004;316:1050. doi: 10.1016/j.bbrc.2004.02.157. [DOI] [PubMed] [Google Scholar]
- 23.RS Dave, RJ Pomerantz. Rev Med Virol. 2003;13:373. doi: 10.1002/rmv.407. [DOI] [PubMed] [Google Scholar]
- 24.M Hamada, et al. Antisense Nucleic Acid Drug Dev. 2002;12:301. doi: 10.1089/108729002761381285. [DOI] [PubMed] [Google Scholar]
- 25.JM Jaqque, et al. Nature. 2002;418:435. [Google Scholar]
- 26.MT Lee, et al. J Virol. 2003;77:11964. doi: 10.1128/JVI.77.22.11964-11972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.G Randall, et al. Proc Natl Acad Sci U S A. 2003;100:235. [Google Scholar]
- 28.M McCown, et al. Virology. 2003;313:514. doi: 10.1016/s0042-6822(03)00341-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.