

Abstract
Recent studies suggest a crucial role for plasminogen activator inhibitor-1 (PAI-1) in mediating stress-induced hypercoagulability and thrombosis. However, the mechanisms by which PAI-1 is released by stress are not well-delineated. Here, we examined catecholaminergic neurosecretory cells for expression, trafficking, and release of PAI-1. PAI-1 was prominently expressed in PC12 pheochromocytoma cells and bovine adrenomedullary chromaffin cells as detected by Northern blotting, Western blotting, and specific PAI-1 ELISA. Sucrose gradient fractionation studies and immunoelectron microscopy demonstrated localization of PAI-1 to catecholamine storage vesicles. Secretogogue stimulation resulted in corelease of PAI-1 with catecholamines. Parallel increases in plasma PAI-1 and catecholamines were observed in response to acute sympathoadrenal activation by restraint stress in mice in vivo. Reverse fibrin zymography demonstrated free PAI-1 in cellular releasates. Detection of high molecular weight complexes by Western blotting, consistent with PAI-1 complexed with t-PA, as well as bands consistent with cleaved PAI-1, suggested that active PAI-1 was present. Modulation of PAI-1 levels by incubating PC12 cells with anti–PAI-1 IgG caused a marked decrease in nicotine-mediated catecholamine release. In summary, PAI-1 is expressed in chromaffin cells, sorted into the regulated pathway of secretion (into catecholamine storage vesicles), and coreleased, by exocytosis, with catecholamines in response to secretogogues.
Introduction
Plasminogen activator inhibitor type 1 (PAI-1) is a member of the serine protease inhibitor (SERPIN) superfamily. Its major recognized function is regulation of the fibrinolytic system by rapidly inhibiting the plasminogen activators tissue plasminogen activator (tPA) and urokinase (uPA), thus preventing activation of plasminogen to plasmin.1 Elevated plasma concentrations of PAI-1 have been observed in a variety of thrombotic disorders, including deep vein thrombosis,2 myocardial infarction,3–5 and disseminated intravascular coagulation.6 Recent studies have suggested an important association and role for the stress response in promoting thrombotic events. Mental and physical stress substantially decrease fibrinolytic activity.7 Acute stress and chronic stress are associated with elevated plasma concentrations of PAI-1 antigen in humans and in rodent models.8,9 Chronic stress is associated with elevated plasma PAI-1 antigen concentrations in middle-aged men.8 Studies using a restraint stress paradigm in mice demonstrated a striking increase in plasma PAI-1 antigen concentrations in response to acute stress.9 In addition, the PAI-1 response correlated with tissue thrombosis, particularly in older stressed mice.9 Moreover, much less tissue thrombosis was induced by restraint stress in both young and aged PAI-1 deficient mice compared with age-matched wild-type mice.9 These results demonstrate that increased expression and release of PAI-1 in response to physiologic and pathologic stress increase the prothrombotic potential, thus promoting thrombotic complications under stressful conditions. Hence, these results suggest a crucial role for PAI-1 in mediating stress-induced hypercoagulability and thrombosis. However, the mechanisms by which PAI-1 plasma concentrations are increased by stress are not well delineated. Of note, the stress response is characterized by, and in large part mediated by, activation of the sympathoadrenal system causing exocytotic release of amines and proteins from catecholamine storages vesicles of the adrenal medulla and sympathetic neurons.10 Interestingly, in the above mentioned murine studies, restraint stress was associated with a pronounced increase in PAI-1 mRNA expression in the adrenal medulla.9 However, the intracellular trafficking of PAI-1 and its release from these cells (ie, via regulated versus constitutive secretory pathways11,12) are fundamental issues that have not been elucidated.
Therefore, we tested the hypothesis that PAI-1 is targeted to and released directly by exocytosis from adrenomedullary catecholamine storage vesicles, by investigating PAI-1 expression, subcellular localization, and secretagogue-mediated PAI-1 release from chromaffin cells. These included rat PC12 cells, a well-established neurosecretory chromaffin cell line with abundant catecholamine storage vesicles,13 as well as primary bovine adrenomedullary chromaffin cells. We also examined plasma PAI-1 and plasma catecholamine responses to acute sympathoadrenal activation in mice in vivo. Our results demonstrate that PAI-1 is prominently expressed in chromaffin cells, and is targeted into catecholamine storage vesicles. In addition, PAI-1 is coreleased by exocytosis with catecholamines in a regulated fashion (via the regulated secretory pathway) in response to chromaffin cell stimulation. In addition, modulation of local PAI-1 levels resulted in substantial alteration of agonist-mediated catecholamine secretion, suggesting that PAI-1 plays a key role in modulating chromaffin cell neurosecretory function.
Methods
Cells and tissues
PC12 cells derived from rat pheochromocytoma13 (at passage number 8) were cultured as described.14–18 Bovine chromaffin cells and bovine chromaffin granules were isolated from fresh bovine adrenal medullae as described.15,16,18,19
mRNA isolation and Northern blot analysis
Total RNA from cells and tissues was prepared using the RNAzol B method according to the manufacturer's instructions (Tel-Test Inc). Northern blot analyses were performed as in our previous studies15 using either a [32P]-labeled 1085-bp murine PAI-1 cDNA EcoR I–Sph I fragment (derived from the PAI-1 cDNA plasmid pGEM-3Z; kindly provided by Dr David J. Loskutoff, The Scripps Research Institute) or control rat cyclophilin cDNA at a concentration of 2 × 106 cpm/mL as probes. PAI-1 mRNA was evaluated in PC12 cells cultured in the absence and presence of the neurotrophin, nerve growth factor (NGF; Gibco-BRL) at 100 ng/mL for 24 hours to induce differentiation of the cells to acquire a sympathetic neuronal phenotype, as described.20
Western blotting
PC12 cells were lysed in 1% Triton X-100 lysis buffer (10mM Tris Cl, pH7.4, 150mM NaCl, 1% Triton X-100, 5mM EDTA, pH 8.0) with the addition of protease inhibitor cocktail (Boehringer Mannheim). Proteins from PC12 cell lysates, or from soluble fractions of isolated bovine adrenal chromaffin granules were electrophoresed on 10% SDS polyacrylamide gels under reducing conditions, transferred to nitrocellulose membranes, and incubated with rabbit anti–rat PAI-1 IgG (American Diagnostica Inc). The membranes were incubated with donkey anti–rabbit IgG-HRP conjugate (GE Healthcare), developed using an ECL substrate (GE Healthcare), and exposed to Kodak X-OMAT AR film (Eastman Kodak).
Sucrose gradient fractionation
Sucrose gradient fractionation studies were performed as in our previous studies.14,15 Briefly, PC12 cells were incubated for 2 hours with 1 μCi/mL [3H]norepinephrine (GE Healthcare). The cells were resuspended in sucrose buffer (10mM Tris HCl pH 7.6, 0.32M sucrose, 1mM EDTA), homogenized, layered over a 30-mL continuous sucrose density gradient (0.32-2.2M), and centrifuged at 100 000g for 1.5 hours at 4°C. Fractions were collected and assayed for [3H]norepinephrine by liquid scintillation counting, sucrose concentration by refractometry, and PAI-1 using a specific ELISA kit (American Diagnostica Inc).
Secretagogue-mediated release of PAI-1
PC12 cells were labeled for 2 hours with [3H]norepinephrine at 1 μCi/mL in cell culture medium, washed 4 times in release buffer (10mM Hepes, pH 7.4, 150mM NaCl, 5mM KCl, 2mM CaCl2), and incubated at 37°C for 30 minutes in release buffer in the presence or absence of secretagogues as described.14–16,18 After collecting the release buffer, the cells were lysed in release buffer containing 0.1% Triton X-100. Release buffer and cell lysates were assayed for [3H]norepinephrine by liquid scintillation counting and PAI-1 using the ELISA for PAI-1.
In vivo sympathoadrenal activation studies
C57Bl/6J mice at 16-18 weeks of age were obtained from the in-house rodent breeding colony. Animal procedures and protocols in these studies were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. Mice were subjected to restraint stress, a potent inducer of sympathoadrenal activation,21,22 using a modification of a previously described method.9 Briefly, mice were restrained in a restraining tube (plastic 50 mL conical centrifuge tube [Corning] with multiple punctures to allow ventilation). After 30 minutes of restraint, animals were immediately anesthetized with 3% to 4% isoflurane in 1L/minutes O2. Cardiac puncture was performed and blood was collected into a syringe containing sodium citrate. Plasma was then obtained by centrifugation of the blood samples at 3000g for 5 minutes and stored at −70°C. Plasma samples from control (unstressed) mice were obtained in the same fashion. PAI-1 total antigen concentration was determined using an ELISA kit (Molecular Innovations), and plasma catecholamines (epinephrine and norepinephrine) were determined by ELISA (CatCombi ELISA; GenWay), according to the manufacturers' instructions. Plasma VWF antigen was determined by ELISA. Briefly, 96-well microtiter plates were coated with rabbit anti–human VWF polyclonal antibody (Dako) diluted in 50mM Na2CO3, pH 9.6. After blocking with 3% BSA diluted in 25mM Tris, pH 7.5, 150mM NaCl (TBS), and washing with TBS containing 1% BSA (TBS/1% BSA), 100 μL of plasma samples, diluted 1/200 in TBS/1% BSA, were incubated in the wells for 18 hours at 4°C. The plates were washed 5 times between incubation steps with TBS containing 0.05% Tween 20. The wells were then incubated with 100μl of HRP-conjugated rabbit anti–human VWF polyclonal antibodies (Dako) diluted 1/2000 in TBS/1% BSA for 18 hours at 4°C, and the color was developed using a TMB peroxidase substrate (Bio-Rad Laboratories) according to the manufacturer's instruction and read at 655 nm using a Versa Max microplate reader (Molecular Devices Inc). Absorbances were converted to percent normal reference mouse citrated plasma (NMP) VWF from a standard curve made from a serial dilution of NMP (1/25 to 1/1600) in TBS/1% BSA analyzed on the same plate.
Reverse fibrin zymography
Reverse fibrin zymography was performed as described.23 Briefly, samples were electrophoresed on 10% SDS-polyacrylamide gels under nonreducing conditions. After removal and neutralization of the SDS by soaking in 2.5% Triton X-100, the gels were incubated on fibrin-agarose indicator films containing 2 mg/mL plasminogen-depleted human fibrinogen (Calbiochem), 0.5 units/mL human thrombin (Calbiochem), 0.05 units/mL human urokinase (Sigma-Aldrich), and 25 μg/mL human plasminogen purified as described.24
Immunoelectron microscopy of PAI-1 in PC12 cells and chromaffin granules
PC12 cells or bovine chromaffin granules were washed in 0.1M cacodylate buffer, pH 7.4, and fixed with 3% paraformaldehyde in cacodylate buffer at 4°C for 24 hours. Samples were dehydrated in ethanol and embedded with LR White resin (medium grade; Ted Pella Inc) according to the manufacturer's instructions.
After embedding, immunolabeling was carried out on thin sections mounted on parlodion coated nickel grids. Sections were blocked in blocking buffer (50mM Tris Cl, pH 7.5, 150mM NaCl, 5% normal goat serum, 0.8% BSA, 0.1% cold water fish skin gelatin), and immunolabeled using rabbit anti–rat PAI-1 IgG (American Diagnostica Inc), followed by detection with goat anti–rabbit IgG conjugated to 10 nm of colloidal gold (Ted Pella Inc).
After immunolabeling, sections were washed, fixed with 1% glutaraldehyde in cacodylate buffer for 10 minutes at room temperature, contrasted with 2% uranyl acetate followed by bismuth subnitrate, and examined with a Zeiss EM10 CR electron microscope.
In quantitative evaluation of immunogold results, the density of gold particles was determined in random electron micrographs, with the gold particles counted and expressed as the number of gold particles per μm2 of secretory vesicle area (or nonvesicular subcellular area) evaluated as described.25 Immunogold particles were considered to be associated with secretory vesicles when they were observed within 50nM of the surface of the vesicle.25
Statistics
Results are reported as mean ± SEM. For secretion studies, statistical significance was determined by Student t test or by ANOVA followed by Student-Newman-Keuls posthoc tests for multiple comparisons. Univariate correlations were assessed by linear least-squares regression analysis. Results from immunoelectron microscopy studies were analyzed by the nonparametric Wilcoxon signed-rank test.
Results
Expression of PAI-1 by chromaffin cells
PAI-1 expression was investigated in rat pheochromocytoma PC12 cells using Northern blot analysis. Hybridization was performed with a 32P-labeled 1085-bp mouse PAI-1 cDNA EcoRI-SphI fragment as probe. A prominent band at 3.0-kb typical of PAI-1 was observed. The PAI-1 signal was increased 2.5-fold after treatment of the cells with neurotrophin (NGF; Figure 1 left panel).
Figure 1.

Expression of PAI-1 in catecholaminergic cells. Left Panel: Northern blot analysis of PAI-1 mRNA expression in rat PC12 pheochromocytoma cells. Twenty-five μg of total RNA from either untreated or NGF-treated PC12 cells were loaded and separated on a 1% formaldehyde agarose gel. [α-32P]-labeled PAI-1 cDNA and rat cyclophilin cDNA were used as specific and control probes, respectively. The positions of 28 S and 18 S ribosomal RNA are shown. Right Panel: Expression of PAI-1 antigen in PC12 cells (A) and bovine adrenal medulla (B). Expression of PAI-1 antigen was investigated by Western blot analyses using specific anti–PAI-1 antibodies, or control normal rabbit IgG.
Next, PAI-1 antigen expression was investigated in both PC12 cells and bovine adrenal chromaffin vesicle lysates in Western blot analysis using an IgG fraction of a rabbit anti–rat PAI-1 antibody. Two major bands were specifically detected in PC12 cell lysates and bovine chromaffin vesicles purified from bovine adrenal glands (Figure 1 right panel), one with an apparent molecular weight (Mrapp) of 50 000, consistent with that of free PAI-1 and the second with an Mrapp of 110,000 consistent with PAI-1 in complex with its target protease, t-PA. The Mrapp 110,000 band was also detected in blots probed with an anti–t-PA antibody, consistent with PAI-1 in a complex with t-PA (see supplemental Figure 1, available on Blood Web site; see the Supplemental Materials link at the top of the online article). The detection of the band at Mrapp 110 000 with anti–PAI-1 antibody is also consistent with the presence of active PAI-1 (suggested by the ability to form a complex with its target protease, t-PA).
PAI-1 antigen was also detected by ELISA. The PAI-1 cell content was 7.4 ± 0.6 ng/106 cells for PC12 cells, and 3.3 ± 0.2 ng/106 cells for bovine chromaffin cells. In controls, cell lysates of bovine chromaffin cells were negative for expression of VWF by Western blotting, indicating minimal contamination of the chromaffin cell preparations with endothelial cells.
Subcellular localization of PAI-1 in chromaffin granules
To explore the subcellular localization of PAI-1 expressed in chromaffin cells, sucrose gradient fractionation of PC12 cell homogenates was performed. PC12 cells were labeled with [3H]norepinephrine, homogenized, and layered over a continuous sucrose density gradient (0.32-2.2M sucrose). After centrifugation, fractions were collected and assayed for PAI-1 antigen by ELISA, [3H]norepinephrine by liquid scintillation counting, and sucrose concentration by refractometry. PAI-1 and [3H]norepinephrine were colocalized to the same subcellular fraction with a major peak at 1.4M sucrose (Figure 2A). The 1.4M sucrose peak is consistent with the buoyant density that we and others have determined previously for catecholamine storage vesicles isolated from PC12 cells.14,15,26,27 In addition, some PAI-1 and [3H]norepinephrine were present in the top fractions of the gradient (fractions 23-25, 0.32-0.6M sucrose). This additional peak at the top of the gradient is consistent with release of granular components from vesicles lysed during the homogenization step (before application of the sample to the gradient) as we and others have shown in studies investigating other secretory vesicle proteins.15,27,28 The fractions at the 1.4M sucrose peak (10 to 19) with the highest [3H]norepinephrine cpm were analyzed further by Western blotting (Figure 2B). PAI-1 antigen migrating with apparent molecular weights of 50 000 and 110 000 was highly enriched in these fractions, suggesting that both free PAI-1 and PAI-1 in a complex with t-PA were present.
Figure 2.

Colocalization of PAI-1 and norepinephrine in sucrose density gradient fractions. PC12 cells were labeled with [3H]norepinephrine and fractionated on a continuous sucrose density gradient as described in “Sucrose gradient fractionation.” Fractions were collected and assayed for [3H]norepinephrine by liquid scintillation counting (squares), PAI-1 by ELISA (circles), and sucrose concentration by refractometry (triangles; A). Fractions 10 to 19 were further analyzed by Western blotting with anti–PAI-1 antibody as described in “Western blotting” (B).
Secretagogue-stimulated corelease of PAI-1 and catecholamines from chromaffin cells
We also investigated the subcellular localization of PAI-1 using functional secretagogue release studies. PC12 cells were loaded with [3H]norepinephrine and stimulated with well established secretagogues that act with varied potency and through different mechanisms to cause regulated exocytotic release from these cells: 60μM nicotine (acting through activation of nicotinic cholinergic receptors); 55mM KCl (resulting in membrane depolarization); and 2mM BaCl2 (acting as a calcium agonist).14,15 Both releasates and whole cell lysates were assayed for PAI-1 antigen and [3H]norepinephrine. Marked increases in PAI-1 secretion were observed in response to each of these secretagogues: 3- to 4-fold for nicotine, 4- to 5-fold for KCl, and 7- to 8-fold for BaCl2 (Figure 3A). Furthermore, secretion of PAI-1 was in parallel with that of [3H]norepinephrine, consistent with release from the same subcellular compartment, the catecholamine storage vesicle.
Figure 3.

Release of PAI-1 in response to catecholaminergic stimulation. (A) Corelease of PAI-1 and norepinephrine from catecholaminergic cells. PC12 cells were labeled with [3H]norepinephrine and incubated at 37°C for 30 minutes in release buffer with or without the presence of secretagogue (60μM nicotine, 55mM KCl, or 2mM BaCl2). The release of PAI-1 antigen (open bars) and [3H]norepinephrine (filled bars) were determined as described in “Secretagogue-mediated release of PAI-1.” Percent release was calculated as the amount in release buffer/total (amount in release buffer + amount in cell lysate), and the results were expressed as fold stimulation compared with basal (unstimulated) values. Values are represented as the mean ± SEM of 6 independent determinations for each group. Release of PAI-1 and [3H]norepinephrine were significantly (P < .001) greater for each of the 3 secretagogues compared with corresponding values for basal secretion. (B) Regulated secretion of PAI-1 from bovine adrenal chromaffin cells in response to secretagogues. Primary bovine adrenal chromaffin cells were incubated at 37°C for 30 minutes in release buffer without (lanes 1 and 2) or with secretagogue (55mM KCl, lanes 3 and 4, or 60μM nicotine, lanes 5 and 6). The releasates from stimulated and unstimulated cells were examined by Western blotting. (C-D) Parallel increases in plasma PAI-1 and plasma catecholamine concentrations in response to acute sympathoadrenal activation in vivo. (C) Groups of aged-matched male C57Bl6/J mice were either unstressed or exposed to restraint stress as described in “In vivo sympathoadrenal activation studies.” Data are represented as the mean ± SEM for n = 6 in each group. Significant (P < .001) increases in plasma PAI-1 concentrations were observed after sympathoadrenal activation, compared with plasma concentrations in unstressed mice. In addition, the increases in plasma PAI-1 were in parallel to those of plasma catecholamines (epinephrine plus norepinephrine), approximately 2-fold increase for both (1.94 ± 0.11-fold increase for PAI-1, and 2.01 ± 0.38-fold increase for catecholamines). (D) Linear regression of plasma PAI-1 and plasma catecholamines in control (unstressed) mice (open circles) and in response to acute sympathoadrenal activation by restraint stress (closed circles; r = 0.825, n = 12, P = .001).
Similar functional secretagogue release studies were performed using primary cultures of bovine adrenal chromaffin cells. PAI-1 was examined using Western blot analysis in cell releasates obtained in either the presence or absence of chromaffin cell secretagogues. Exposure of bovine chromaffin cells to secretagogues, 55mM KCl or 60μM nicotine, resulted in a dramatic increase of PAI-1 in cell releasates (Figure 3B lanes 3, 4, 5, and 6), compared with the trace amount of PAI-1 present in the conditioned media of unstimulated cells (Figure 3B lanes 1 and 2).
Parallel increases in plasma PAI-1 and catecholamine concentrations in response to sympathoadrenal activation in vivo
We also examined plasma PAI-1 and plasma catecholamine concentrations in vivo in response to acute sympathoadrenal activation by restraint stress in C57Bl/6J mice. Significant increases in both plasma PAI-1 and plasma catecholamines were observed after sympathoadrenal activation, compared with plasma concentrations in control (unstressed) mice (for PAI-1, 5.22 ± 0.29 ng/mL [n = 6], vs 2.68 ± 0.35 ng/mL [n = 6], P < .001; for catecholamines, 18.81 ± 3.59 ng/mL [n = 6], vs 9.34 ± 1.74 ng/mL [n = 6], P < .05; Figure 3C). In addition, the increase in plasma PAI-1 concentration was in parallel with that of plasma catecholamines, approximately 2-fold increase for both (1.94 ± 0.11-fold increase for PAI-1, and 2.01 ± 0.38-fold increase for catecholamines). Furthermore, plasma PAI-1 concentrations were highly correlated with plasma catecholamines (r = 0.825, n = 12, P = .001; Figure 3D). In controls, plasma VWF concentrations did not differ between control (unstressed) mice and animals subjected to sympathoadrenal stress (100.9 ± 13.1% [n = 6] vs 110.6 ± 15.2% [n = 6], for control and stressed mice, respectively, P = .638).
Reverse fibrin zymography of chromaffin cell releasates
We also examined neurosecretory cell releasates using reverse fibrin zymography. Using this technique, the presence of the plasminogen activator, uPA, in the indicator gel results in lysis of the entire gel except for zones (which remain opaque) in which PAI-1 activity is present.23 An opaque zone indicating plasminogen activator inhibitory activity was detected in secretagogue-mediated (BaCl2-stimulated) cell releasates (Figure 4A). Only trace amounts of plasminogen activator inhibitory activity were detected in the conditioned media of cells incubated with buffer alone (Figure 4A). This plasminogen activator inhibitory activity migrated with an Mrapp of 50 000, consistent with the molecular weight of the free active form of PAI-1 (and/or latent PAI-1 that is activated during zymography). The same samples were analyzed by Western blotting using a specific anti–rat PAI-1 antibody (Figure 4B), and PAI-1 antigen was detected migrating with the same electrophoretic mobility as plasminogen activator inhibitory activity. Thus, these results suggest that PAI-1 activity is present in the secretogogue-responsive pool.
Figure 4.
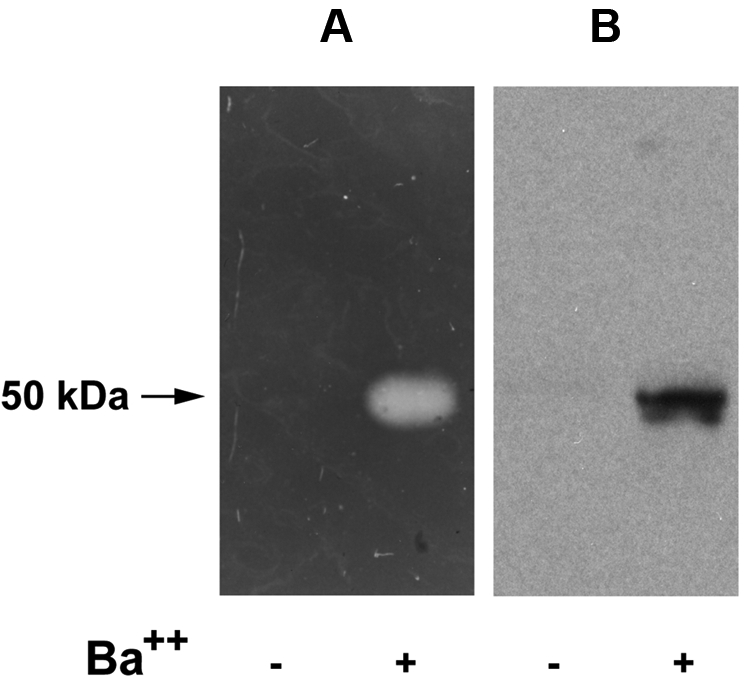
Reverse fibrin zymography of catecholamine storage vesicle proteins. PC12 cells were incubated at 37°C for 30 minutes in release buffer in the presence (+) or absence (−) of the secretagogue, BaCl2 (2 mM). The releasates were electrophoresed on 10% SDS gels and subjected either to reverse fibrin zymography as described in “Reverse fibrin zymography” (A), or Western analysis using an IgG fraction of rabbit anti–rat PAI-1 (B).
Immunoelectron microscopy of PAI-1 in secretory vesicles of PC12 cells and bovine adrenomedullary chromaffin granules
The localization of PAI-1 antigen within secretory vesicles was further examined by immunoelectron microscopy. PAI-1 antigen was detected using specific rabbit anti–rat PAI-1 IgG, followed by a goat anti–rabbit IgG conjugated to 10-nm colloidal gold particles. In PC12 cells, immunoelectron microscopy revealed the gold-labeled PAI-1 antigen within dense core catecholamine storage vesicles, as shown in Figure 5A, as well as in Figure 5B at higher magnification. Control experiments (Figure 5C) showed a paucity of immunolabeling when normal rabbit IgG was used as the primary antibody.
Figure 5.

Immunoelectron microscopy of PAI-1 in catecholamine storage vesicles. (A-C) PC12 cells were prepared for immunoelectron microscopy as described in “Immunoelectron microscopy of PAI-1 in PC12 cells and chromaffin granules.” PAI-1 antigen was detected in PC12 cells as gold particles in catecholamine storage vesicles using specific rabbit anti–rat PAI-1 IgG and goat anti–rabbit IgG conjugated to 10-nm colloidal gold particles (arrowheads in panels A and B). Controls (C) indicated a paucity of immunolabeling when normal rabbit IgG was used as the primary antibody. The bar in panel A corresponds to 200 nm, the bars in panels B & C to 100 nm. Magnification: A, 48 000; B-C, 110 250. (D-E) Isolated bovine chromaffin vesicles were prepared for PAI-1 immunoelectron microscopy as described in “Cells and tissues.” PAI-1 antigen was detected in isolated bovine chromaffin vesicles as gold particles using specific rabbit anti–rat PAI-1 IgG and goat anti–rabbit IgG conjugated to 10-nm colloidal gold particles (D). Controls (E) indicated a paucity of immunogold staining when rabbit IgG was used as the primary antibody. The bars in panels D & E correspond to 100 nm. Magnification: 164 000.
In experiments with isolated bovine chromaffin granules, PAI-1 antigen was again demonstrated to be localized to secretory vesicles, as shown in Figure 5D, with specific immunogold labeling of PAI-1 over the dense core secretory granules. Minimal immunolabeling was detected in control experiments with normal rabbit IgG as primary antibody (Figure 5E).
In quantitative analyses of immunogold results, in PC12 cells (from n = 14 random electron micrographs), the density of specific anti–PAI-1 labeled gold particles associated with secretory vesicles was markedly increased compared with the density of gold particles overlying other (nonvesicular) subcellular regions (542 ± 24 gold particles/μm2 [125 secretory vesicles evaluated]), vs 14 ± 2 gold particles/μm2, P < .0000001 by Wilcoxon signed ranks test). Parallel studies with isolated bovine chromaffin granules (from n = 12 random electron micrographs) also showed a marked increase in the density of anti–PAI-1 labeled gold particles associated with dense core secretory vesicles (621 ± 45 particles/μm2 [126 secretory vesicles evaluated], vs 34 ± 3 gold particles/μm2, P < .000 01 by Wilcoxon signed ranks test). In specificity control studies with nonspecific IgG as primary antibody, minimal gold labeling was noted and no significant increase in gold particle density was observed in association with secretory vesicles for either PC12 cells or bovine chromaffin granules. For PC12 cells, the density of nonspecific IgG gold particles associated with secretory vesicles was 39 ± 5 particles/μm2 (134 vesicles evaluated), vs 30 ± 4 gold particles/μm2 in nonvesicular regions, P = NS. For bovine chromaffin cells, the density of nonspecific IgG gold particles associated with secretory vesicles was 23 ± 5 particles/μm2 (120 vesicles evaluated), vs 18 ± 5 gold particles/μm2 in nonvesicular regions, P = NS.
Effects of modulation of cellular PAI-1 on catecholamine secretion
Previously, we found that local activation of plasminogen to plasmin, and plasmin-mediated processing of the neurosecretory cell protein, chromogranin A (CgA), to generate secretion-inhibitory CgA peptides, substantially modulates nicotine-mediated catecholamine release.16–18,29 Therefore, we examined the effect of modulation of local PAI-1 on nicotine-mediated catecholamine release from PC12 cells. In the presence of the anti–PAI-1 IgG (to inhibit endogenously released PAI-1), nicotine-mediated catecholamine secretion was markedly decreased, compared with secretion from cells treated with control normal rabbit IgG (P < .001; Figure 6). Compared with control IgG, anti–PAI-1 IgG had no significant effect on basal catecholamine secretion (in the absence of nicotine; P = .318; Figure 6).
Figure 6.

Blockade of released PAI-1 from PC12 cells: effect on catecholamine secretion. PC12 cells were labeled with [3H]–norepinephrine, washed with release buffer, and incubated at 37°C for 45 minutes in release buffer with either rabbit anti–PAI-1 IgG (open bars) or normal rabbit IgG (solid bars) in the presence of 2 μM plasminogen. After incubation, the cells were treated with 60 μM nicotine (+) or buffer (−). Results are mean ± SEM; n = 3 for each group. Nicotine-mediated catecholamine secretion was significantly decreased from cells treated with anti–PAI-1 IgG compared with secretion from cells treated with nicotine plus normal rabbit IgG (P < .001).
Discussion
PAI-1 is a SERPIN and the major physiologic inhibitor of plasminogen activation through inactivation of the plasminogen activators tPA and uPA.1 In this report, we investigated the expression of PAI-1 and its trafficking in catecholaminergic neurosecretory cells. Our study shows that PAI-1 is prominently expressed in these cells, where it is sorted into catecholamine storage vesicles, and is released with catecholamines in response to secretagogue stimulation.
Secretory proteins expressed in neuronal and neuroendocrine cells are sorted into constitutive or regulated pathways.11,12 Proteins entering the constitutive pathway are not stored but are transported directly to the cell surface and secreted in the absence of any extracellular signal. By contrast, proteins entering the regulated pathway are concentrated and stored in vesicles and subsequently released on stimulation by a secretagogue or other specific extracellular stimuli. Catecholamine storage vesicles within chromaffin cells are prototypic examples of regulated secretory vesicles.10,12,14,28 The primary sequence of PAI-1 includes a 23-amino acid signal sequence,30,31 suggesting its ability to enter the secretory pathway. However, the intracellular trafficking of PAI-1, and its mechanism of release (via regulated vs constitutive secretory pathways) from these cells, have not been examined. Thus, our results demonstrate that PAI-1 is targeted to the regulated secretory pathway in these cells. Furthermore, these studies identify a specific subcellular compartment (the catecholamine storage vesicle) into which PAI-1 is sorted and from which PAI-1 is released by specific stimuli.
Trafficking of PAI-1 to the regulated secretory pathway was demonstrated in chromaffin cells using biochemical, functional, and morphologic studies. First, sucrose gradient fractionation studies revealed colocalization of PC12 cell PAI-1 and catecholamines to the same subcellular fractions, at a buoyant density peak (1.4M sucrose) consistent with that of catecholamine storage vesicles.14,15,26 Second, functional secretagogue release studies showed regulated secretion of PAI-1 in PC12 cells and bovine chromaffin cells in response to several well-established chromaffin cell exocytotic secretagogues, which resulted in corelease of PAI-1 in parallel with catecholamines. Third, immunoelectron microscopy of PAI-1 in PC12 cells and in experiments with bovine chromaffin granules further confirmed that PAI-1 is targeted into catecholamine storage vesicles. Thus, our current results provide the first evidence that endogenous neurosecretory cell PAI-1 is sorted into the regulated secretory pathway.
Recent studies suggest that plasma PAI-1 concentrations are substantially increased during mental and physiologic stress in humans and in rodent models, and that PAI-1 is a crucial mediator of the observed increase in thrombotic events associated with stress.9 Thus, our results suggest that stress-mediated sympathoadrenal activation with resultant exocytotic release of PAI-1 from catecholamine storage vesicles may be an important mechanism for the observed increase in circulating PAI-1 concentrations, with important implications for stress-induced thrombotic episodes.
Interestingly, plasma PAI-1 concentrations are elevated in essential hypertension as well as in the insulin resistance/metabolic syndrome,32–34 disorders that occur commonly in the adult population. These disorders are associated with an increase in body adiposity, and release of PAI-1 from adipose tissue appears to be an important source of plasma PAI-1.9,35,36 However, these disorders are also characterized by substantially augmented sympathoadrenal activity.37,38 In the current study, we observed significant increases in plasma PAI-1 concentrations that were in parallel with increases in plasma catecholamines in response to acute sympathoadrenal activation in mice in vivo. These results are consistent with direct release from the same intracellular pool, or, alternatively, may reflect PAI-1 release from another compartment (eg, vascular endothelium or adipose tissue) that is mediated by the actions of catecholamines. However, the increases in plasma PAI-1 in response to sympathoadrenal activation were highly correlated with the increases in plasma catecholamines (r = 0.825, P = .001). Thus, taken together our results suggest that release of PAI-1 from catecholamine storage vesicles may also contribute to the increased plasma PAI-1 concentrations, as well as the increased risk of thrombotic events, including myocardial infarction and deep vein thrombosis, that are observed in the above disorders. Because PAI-1 is expressed by a variety of tissues, the relative contributions of such tissues to perturbations in plasma concentrations of PAI-1, and to overall fibrinolytic balance, in response to stress, is a complex issue, as has been pointed out by other investigators.9,39
In previous studies, we demonstrated that the plasminogen activator, t-PA is also sorted into and released from catecholamine storage vesicles.15,40 On a quantitative basis, by ELISA, the cellular content of PAI-1 was similar to values which we previously noted for t-PA antigen in these cells (tPA cell content of 9.4 ± 0.5 ng/106 cells and 4.7 ± 0.7 ng/106 cells for PC12 and bovine chromaffin cells, respectively).15 Thus, the net effect on plasminogen activation and fibrinolysis from this source (as well as local effects on neurosecretory cell function [vide infra]) will depend on a variety of factors that affect local PAI-1/t-PA balance, including the relative rates of synthesis of PAI-1 and t-PA, trafficking to the vesicles, as well as the potential formation of t-PA/PAI-1 complexes within the vesicle and on release. Previous studies have demonstrated that sympathoadrenal and sympathoneural tissues may represent substantial sources contributing to changes in plasma t-PA concentrations.41,42 Further studies will be required to determine what conditions and factors may alter local PAI-1/t-PA balance in these cells.
In addition to providing a potential source of circulating PAI-1, the presence of PAI-1 within catecholamine storage vesicles and the release of PAI-1 from these organelles have important implications for neuroendocrine function. Previously we have provided evidence for the presence of a local, chromaffin cell plasminogen activation system, which includes secretory vesicle t-PA, and high affinity cell surface receptors for plasminogen and t-PA, to promote plasminogen activation at the cell surface.15,16,18,29,40,43 In addition, there is evidence for lower levels of expression of other fibrinolytic components, including uPA,44 the uPA receptor (uPAR),45 and plasminogen,46,47 by these cells. Furthermore, this system crucially participates in cell-associated neuroendocrine prohormone processing to generate bioactive peptides that modulate the secretory characteristics of the cells and regulate catecholamine secretion.16–18 We have demonstrated that perturbation of the local chromaffin cell plasminogen activation system markedly influences catecholamine secretion.16–18 In the current study, modulation of local PAI-1 by specific anti–PAI-1 IgG caused a marked decrease in nicotine-mediated catecholamine release. This result is consistent with our previous results demonstrating that the local plasminogen activation system participates in local prohormone processing of chromogranin A to liberate a bioactive peptide that inhibits catecholamine release and provides an important negative feedback regulatory role on these cells.16,17 Thus, in this paradigm, neutralization of the inhibitory effect of locally secreted PAI-1 by specific anti–PAI-1 antibody would be expected to enhance this local negative feedback system, resulting in decreased catecholamine release, as observed. However, it is possible that other local cellular effects of PAI-1 that are independent of effects on local proteolytic activity may contribute to this effect. Additional in vivo studies will be required to more definitively assess the importance of fibrinolytic components in prohormone processing and their specific role in this regulatory system.
We examined chromaffin cells for PAI-1 expression at several levels, including PAI-1 mRNA, PAI-1 antigen, and PAI-1 activity. Exposure of cells to NGF, a neurotrophic factor, which has been shown to activate gene expression of other chromaffin cell proteins that are targeted to catecholamine storage vesicles,20,48 resulted in substantial induction of endogenous PAI-1 gene expression (2.5- to 3-fold).
Chromaffin cell PAI-1 antigen was present in several forms, including free and in complex. In our Western blot studies investigating expression and release of PAI-1 from chromaffin cells, we observed the presence of 3 primary bands: a band at Mrapp 110 000, consistent with PAI-1 in a complex with t-PA (also consistent with our previous studies showing this complex by immunoprecipitation with anti–t-PA antisera and by fibrin zymography15), and a doublet at Mrapp ∼ 50 000, the more slowly migrating band within the doublet representing free PAI-1, and the faster migrating band representing cleaved substrate PAI-1 (Figure 3B). In the heavily loaded Western blots of sucrose density fractions from PC12 cells (Figure 2), these same 3 PAI-1 immunoreactive bands were present, along with an additional lower molecular weight minor band, consistent with cleavage of active PAI-1 as described.49 Thus, these results are consistent with the presence of both free and complexed forms of PAI-1 in chromaffin cells.
We also investigated neurosecretory cell releasates using reverse fibrin zymography. Plasminogen activator inhibitor activity was present and was of the appropriate size for active PAI-1 (and/or latent PAI-1 reactivated during zymography). Only trace amounts of plasminogen activator inhibitory activity were detected in the conditioned media of cells incubated with buffer alone, whereas a prominent zone of plasminogen activator inhibitory activity was observed after treatment of cells with secretagogue. Western blotting showed an additional lower molecular weight band in catecholamine storage vesicle fractions, suggesting that this represents inhibitor that was previously complexed with t-PA and, hence, had been present in the active form. In addition, the presence of PAI-1 activity can also be inferred from the presence of the t-PA–PAI-1 complex.
In summary, these results demonstrate that PAI-1 is expressed in neurosecretory catecholaminergic cells where it is targeted into the regulated secretory pathway (into catecholamine storage vesicles), and is exocytotically coreleased with catecholamines by chromaffin cell stimulation. Synthesis and release of PAI-1 from sympathoadrenal sources may be an important source of circulating PAI-1. Because PAI-1 is a crucial mediator of stress-induced hypercoagulability and thrombosis, these results suggest fundamental physiologic and pathologic links among PAI-1, sympathoadrenal activation, altered fibrinolytic activity, and increased thrombotic risk associated with stress. In addition, taken together with recent results demonstrating expression of other components of the fibrinolytic system in chromaffin cells, and with results showing that local activity of fibrinolytic molecules may participate in prohormone processing to influence catecholamine release, these results further define the presence of a local, catecholaminergic cell plasminogen activation system, which may be regulated at several levels, including modulation by local synthesis and release of PAI-1. Thus, these results suggest major interactions between catecholaminergic and fibrinolytic systems which may profoundly influence catecholamine release and have important implications for disease states characterized by altered sympathoadrenal activity.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants HL-50 398 (to R.J.P.), HL-45 934 and HL-081046 (to L.A.M.), and by the Department of Veterans Affairs (to R.J.P.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Q.J., N.A.G., and M.A.O. performed experiments, analyzed results, and made the figures; L.A.M. and R.J.P. designed the research; and Q.J., L.A.M., and R.J.P. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert J. Parmer, MD, Nephrology/Hypertension (9111-H), University of California, San Diego, 3350 La Jolla Village Dr, San Diego, CA 92161; e-mail: rparmer@ucsd.edu.
References
- 1.Yepes M, Loskutoff DJ, Lawrence DA. Plasminogen Activator Inhibitor-1. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Baltimore, MD: Lippincott Williams & Wilkins; 2006. pp. 365–380. [Google Scholar]
- 2.Wiman B, Hamsten A. The fibrinolytic enzyme system and its role in the etiology of thromboembolic disease. Semin Thromb Hemost. 1990;16(3):207–216. doi: 10.1055/s-2007-1002671. [DOI] [PubMed] [Google Scholar]
- 3.Hamsten A, Wiman B, de Faire U, Blomback M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. 1985;313(25):1557–1563. doi: 10.1056/NEJM198512193132501. [DOI] [PubMed] [Google Scholar]
- 4.Wiman B, Andersson T, Hallqvist J, et al. Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor-1 complex and von Willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol. 2000;20(8):2019–2023. doi: 10.1161/01.atv.20.8.2019. [DOI] [PubMed] [Google Scholar]
- 5.Sobel BE, Taatjes DJ, Schneider DJ. Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(11):1979–1989. doi: 10.1161/01.ATV.0000091250.53231.4D. [DOI] [PubMed] [Google Scholar]
- 6.Pralong G, Calandra T, Glauser MP, et al. Plasminogen activator inhibitor 1: a new prognostic marker in septic shock. Thromb Haemost. 1989;61(3):459–462. [PubMed] [Google Scholar]
- 7.Jern C, Eriksson E, Tengborn L, et al. Changes of plasma coagulation and fibrinolysis in response to mental stress. Thromb Haemost. 1989;62(2):767–771. [PubMed] [Google Scholar]
- 8.Raikkonen K, Lassila R, Keltikangas-Jarvinen L, Hautanen A. Association of chronic stress with plasminogen activator inhibitor-1 in healthy middle-aged men. Arterioscler Thromb Vasc Biol. 1996;16(3):363–367. doi: 10.1161/01.atv.16.3.363. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto K, Takeshita K, Shimokawa T, et al. Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci U S A. 2002;99(2):890–895. doi: 10.1073/pnas.022608799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winkler H, Westhead E. The molecular organization of adrenal chromaffin granules. Neuroscience. 1980;5(11):1803–1823. doi: 10.1016/0306-4522(80)90031-7. [DOI] [PubMed] [Google Scholar]
- 11.Kelly RB. Pathways of protein secretion in eukaryotes. Science. 1985;230(4721):25–32. doi: 10.1126/science.2994224. [DOI] [PubMed] [Google Scholar]
- 12.Moore HP. Factors controlling packaging of peptide hormones into secretory granules. Ann N Y Acad Sci. 1987;493:50–61. doi: 10.1111/j.1749-6632.1987.tb27180.x. [DOI] [PubMed] [Google Scholar]
- 13.Greene LA, Tischler AS. Establishment of a noradrenergic clonal cell line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73(7):2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parmer RJ, Xi X-P, Wu H-J, Helman LJ, Petz LN. Secretory protein traffic: chromogranin A contains a dominant targeting signal for the regulated pathway. J Clin Invest. 1993;92(2):1042–1054. doi: 10.1172/JCI116609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parmer RJ, Mahata M, Mahata S, et al. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway: catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J Biol Chem. 1997;272(3):1976–1982. doi: 10.1074/jbc.272.3.1976. [DOI] [PubMed] [Google Scholar]
- 16.Parmer RJ, Mahata M, Gong Y, et al. Processing of chromogranin A by plasmin provides a novel mechanism for regulating catecholamine secretion. J Clin Invest. 2000;106(7):907–915. doi: 10.1172/JCI7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Q, Taupenot L, Mahata SK, et al. Proteolytic cleavage of chromogranin A (CgA) by plasmin. Selective liberation of a specific bioactive CgA fragment that regulates catecholamine release. J Biol Chem. 2001;276(27):25022–25029. doi: 10.1074/jbc.M101545200. [DOI] [PubMed] [Google Scholar]
- 18.Miles LA, Andronicos NM, Baik N, Parmer RJ. Cell-surface actin binds plasminogen and modulates neurotransmitter release from catecholaminergic cells. J Neurosci. 2006;26(50):13017–13024. doi: 10.1523/JNEUROSCI.2070-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miles LA, Hawley SB, Parmer RJ. Chromaffin cell plasminogen receptors. Ann N Y Acad Sci. 2002;971:454–459. doi: 10.1111/j.1749-6632.2002.tb04508.x. [DOI] [PubMed] [Google Scholar]
- 20.Mahata SK, Mahata M, Wu H, Parmer RJ, O'Connor DT. Neurotrophin activation of catecholamine storage vesicle protein gene expression: signaling to chromogranin a biosynthesis. Neuroscience. 1999;88(2):405–424. doi: 10.1016/s0306-4522(98)00225-5. [DOI] [PubMed] [Google Scholar]
- 21.Tjurmina OA, Armando I, Saavedra JM, Goldstein DS, Murphy DL. Exaggerated adrenomedullary response to immobilization in mice with targeted disruption of the serotonin transporter gene. Endocrinology. 2002;143(12):4520–4526. doi: 10.1210/en.2002-220416. [DOI] [PubMed] [Google Scholar]
- 22.Kubovcakova L, Tybitanclova K, Sabban EL, et al. Catecholamine synthesizing enzymes and their modulation by immobilization stress in knockout mice. Ann N Y Acad Sci. 2004;1018:458–465. doi: 10.1196/annals.1296.056. [DOI] [PubMed] [Google Scholar]
- 23.Loskutoff DJ, Schleef RR. Plasminogen activators and their inhibitors. Methods Enzymol. 1988;163:293–302. doi: 10.1016/0076-6879(88)63028-x. [DOI] [PubMed] [Google Scholar]
- 24.Miles LA, Dahlberg CM, Plow EF. The cell-binding domains of plasmimogen and their function in plasma. J Biol Chem. 1988;263(24):11928–11934. [PubMed] [Google Scholar]
- 25.Martelli AM, Baldini G, Tabellini G, et al. Rab3A and Rab3D control the total granule number and the fraction of granules docked at the plasma membrane in PC12 cells. Traffic. 2000;1(12):976–986. [PubMed] [Google Scholar]
- 26.Schubert D, Klier FG. Storage and release of acetylcholine by a clonal cell line. Proc Natl Acad Sci U S A. 1977;74:5184–5188. doi: 10.1073/pnas.74.11.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roda LG, Nolan JA, Kim SU, Hogue-Angeletti RA. Isolation and characterization of chromaffin granules from a pheochromocytoma (PC12) cell line. Exp Cell Res. 1980;128(1):103–109. doi: 10.1016/0014-4827(80)90392-4. [DOI] [PubMed] [Google Scholar]
- 28.Schweitzer ES, Kelly RB. Selective packaging of human growth hormone into synaptic vesicles in a rat neuronal (PC12) cell line. J Cell Biol. 1985;101(2):667–676. doi: 10.1083/jcb.101.2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, Yasothornsrikul S, Taupenot L, Miles LA, Parmer RJ. The local chromaffin cell plasminogen/plasmin system and the regulation of catecholamine secretion. Ann N Y Acad Sci. 2002;971:445–449. doi: 10.1111/j.1749-6632.2002.tb04506.x. [DOI] [PubMed] [Google Scholar]
- 30.Ny T, Sawdey M, Lawrence D, Millan JL, Loskutoff DJ. Cloning and sequence of a cDNA coding for the human beta-migrating endothelial-cell-type plasminogen activator inhibitor. Proc Natl Acad Sci U S A. 1986;83(18):6776–6780. doi: 10.1073/pnas.83.18.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ginsburg D, Zeheb R, Yang AY, et al. cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J Clin Invest. 1986;78(6):1673–1680. doi: 10.1172/JCI112761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang TJ, Gona P, Larson MG, et al. Multiple biomarkers and the risk of incident hypertension. Hypertension. 2007;49(3):432–438. doi: 10.1161/01.HYP.0000256956.61872.aa. [DOI] [PubMed] [Google Scholar]
- 33.Juhan-Vague I, Alessi MC. PAI-1, obesity, insulin resistance and risk of cardiovascular events. Thromb Haemost. 1997;78(1):656–660. [PubMed] [Google Scholar]
- 34.Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost. 2008;99(6):995–1000. doi: 10.1160/TH07-11-0682. [DOI] [PubMed] [Google Scholar]
- 35.Loskutoff DJ, Samad F. The adipocyte and hemostatic balance in obesity: studies of PAI-1. Arterioscler Thromb Vasc Biol. 1998;18(1):1–6. doi: 10.1161/01.atv.18.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Shimomura I, Funahashi T, Takahashi M, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996;2(7):800–803. doi: 10.1038/nm0796-800. [DOI] [PubMed] [Google Scholar]
- 37.Parmer RJ, Cervenka JH, Stone RA, O'Connor DT. Autonomic function in hypertension. Are there racial differences. Circulation. 1990;81(4):1305–1311. doi: 10.1161/01.cir.81.4.1305. [DOI] [PubMed] [Google Scholar]
- 38.Abate NI, Mansour YH, Tuncel M, et al. Overweight and sympathetic overactivity in black Americans. Hypertension. 2001;38(3):379–383. doi: 10.1161/01.hyp.38.3.379. [DOI] [PubMed] [Google Scholar]
- 39.O'Rourke J, Jiang X, Hao Z, Cone RE, Hand AR. Distribution of sympathetic tissue plasminogen activator (tPA) to a distant microvasculature. J Neurosci Res. 2005;79(6):727–733. doi: 10.1002/jnr.20366. [DOI] [PubMed] [Google Scholar]
- 40.Parmer RJ, Miles LA. Targeting of tissue plasminogen activator to the regulated pathway of secretion. Trends Cardiovasc Med. 1998;8(7):306–312. doi: 10.1016/s1050-1738(98)00025-5. [DOI] [PubMed] [Google Scholar]
- 41.Peng T, Jiang X, Wang YF, et al. Sympathectomy decreases and adrenergic stimulation increases the release of tissue plasminogen activator (t-PA) from blood vessels: Functional evidence for a neurologic regulation of plasmin production within vessel walls and other tissue matrices. Journal of Neuroscience Research. 1999;57(5):680–692. [PubMed] [Google Scholar]
- 42.Jiang X, Wang YF, Hand AR, et al. Storage and release of tissue plasminogen activator by sympathetic axons in resistance vessel walls. Microvascular Research. 2002;64(3):438–447. doi: 10.1006/mvre.2002.2441. [DOI] [PubMed] [Google Scholar]
- 43.Parmer RJ, Mahata SK, Jiang Q, et al. Tissue plasminogen activator and chromaffin cell function. Adv Exp Med Biol. 2000;482:179–192. doi: 10.1007/0-306-46837-9_14. [DOI] [PubMed] [Google Scholar]
- 44.Theodorou G, Bizelis I, Rogdakis E, Politis I. The ovine urokinase plasminogen activator and its receptor cDNAs: molecular cloning, characterization and expression in various tissues. Gene. 2009;443(1–2):158–169. doi: 10.1016/j.gene.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Farias-Eisner R, Vician L, Silver A, et al. The urokinase plasminogen activator receptor (UPAR) is preferentially induced by nerve growth factor in PC12 pheochromocytoma cells and is required for NGF-driven differentiation. J Neurosci. 2000;20(1):230–239. doi: 10.1523/JNEUROSCI.20-01-00230.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Seiffert D, Fowler BJ, et al. Plasminogen has a broad extrahepatic distribution. Thromb Haemost. 2002;87(3):493–501. [PubMed] [Google Scholar]
- 47.Gutierrez-Fernandez A, Parmer RJ, Miles LA. Plasminogen gene expression is regulated by nerve growth factor. J Thromb Haemost. 2007;5(8):1715–1725. doi: 10.1111/j.1538-7836.2007.02636.x. [DOI] [PubMed] [Google Scholar]
- 48.Laslop A, Tschernitz C. Effects of nerve growth factor on the biosynthesis of chromogranin A and B, secretogranin II and carboxypeptidase H in rat PC12 cells. Neuroscience. 1992;49:443–450. doi: 10.1016/0306-4522(92)90109-f. [DOI] [PubMed] [Google Scholar]
- 49.Ngo TH, Zhou Y, Stassen JM, Declerck PJ. Importance of N-terminal residues in plasminogen activator inhibitor 1 on its antibody induced latency transition. Thromb Haemost. 2002;88(2):288–293. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}