

Abstract
Cytochrome c (cyt c) release upon oxidation of cardiolipin (CL) in the mitochondrial inner membrane (IM) under oxidative stress occurs early in the intrinsic apoptotic pathway. We postulated that CL oxidation mobilizes not only cyt c but also CL itself in the form of hydroperoxide (CLOOH) species. Relatively hydrophilic CLOOHs could assist in apoptotic signaling by translocating to the outer membrane (OM), thus promoting recruitment of the pro-apoptotic proteins truncated Bid (tBid) and Bax for generation of cyt c-traversable pores. Initial testing of these possibilities showed that CLOOH-containing liposomes were permeabilized more readily by tBid plus Ca2+ than CL-containing counterparts. Moreover, CLOOH translocated more rapidly from IM-mimetic to OM-mimetic liposomes than CL and permitted more extensive OM permeabilization. We found that tBid bound more avidly to CLOOH-containing membranes than to CL counterparts, and binding increased with increasing CLOOH content. Permeabilization of CLOOH-containing liposomes in the presence of tBid could be triggered by monomeric Bax, consistent with tBid/Bax cooperation in pore formation. Using CL-null mitochondria from a yeast mutant, we found that tBid binding and cyt c release were dramatically enhanced by transfer acquisition of CLOOH. Additionally, we observed a pre-apoptotic IM-to-OM transfer of oxidized CL in cardiomyocytes treated with the Complex III blocker, antimycin A. These findings provide new mechanistic insights into the role of CL oxidation in the intrinsic pathway of oxidative apoptosis.
Keywords: Lipid Oxidation, Membrane Lipids, Mitochondrial Apoptosis, Oxidative Stress, Oxygen Toxicity, Phospholipid, Phospholipid Vesicle, Cardiolipin Oxidation, Lipid Peroxidation, Lipid Translocation
Introduction
Cytochrome c (cyt c)2 dissociation from the mitochondrial inner membrane (IM), movement into the intermembrane space, and then release into cytosol is recognized as a key early event in the intrinsic (mitochondrion-initiated) pathway of oxidative stress-induced apoptosis (1–3). How this occurs is still not completely understood, but accumulating evidence suggests that oxidative modification of cardiolipin (CL), e.g. conversion to hydroperoxide species (CLOOHs), plays an important role (4–7). CL (diphosphatidylglycerol) is located primarily in the mitochondrial IM of normal eukaryotic cells, where it interacts with and supports the functions of cyt c, Complex III (cytochromes b and c1), and Complex IV (cytochrome c oxidase) (8). Unlike most other phospholipids in which only the sn-2 fatty acyl group is unsaturated, both sn-1 and sn-2 groups of natural CL are typically unsaturated. In mammalian heart, for example, tetra-linoleoyl CL composes 75–80% of overall CL molecular species (9). Consequently, CL oxidizability and -OOH content per average oxidized molecule are typically much higher than for more conventional phospholipids such as those in the phosphatidylcholine and phosphatidylethanolamine families. Model studies with bovine heart CL in thin film or liposomal form have shown that its normally strong interaction with some fraction of IM cyt c is progressively weakened with increasing -OOH content (4), suggesting possible involvement in pro-apoptotic cyt c release under oxidative stress conditions (5–7). Additional support for this idea was derived from studies based on manipulation of lipid hydroperoxide (LOOH) detoxifying enzymes (7, 10–12). Apart from its accepted function of tethering cyt c to the IM (13, 14), CL is reported to be necessary for binding of truncated BH3-interacting domain death agonist (tBid) to the outer membrane (OM), which in turn promotes binding and oligomerization of monomeric B-cell lymphoma 2-associated X protein (Bax) (15–17). This results in formation of megapores in the OM through which cyt c and other proapoptotic proteins can pass for the activation of cytosolic caspases (17–21). However, this scenario raises a question about accessibility because CL in non-stressed cells resides in the IM, whereas tBid and Bax are recruited to the OM. How would CL materialize in the OM if the mechanism described for permeabilization is applicable? Moreover, if the CL required for OM pore formation were in its native state, this would be inconsistent with the reported loss of CL during apoptosis (22–24). In addressing these questions, various investigators have suggested that CL or some modified form of CL existing at IM-OM contact sites is sufficient for apoptotic tBid recruitment (25, 26). It has also been proposed that CL and/or its lyso-derivatives can redistribute from IM to OM in cells exposed to apoptotic stresses (27, 28). To date, however, there has been little unequivocal supporting evidence for these possibilities, and they remain unsettled.
Using model systems, we showed previously that LOOHs can translocate between membranes much more rapidly than parent lipids and that this can be further accelerated by the nonspecific lipid transfer protein (nsLTP) (29–32). On the basis of this evidence, we postulated that CL peroxidation in oxidatively stressed mitochondria results not only in cyt c mobilization but also mobilization of CL itself, this fostering its movement to the OM for targeting of tBid. In the study to be described we provide initial supporting evidence for this novel hypothesis.
EXPERIMENTAL PROCEDURES
Materials
DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine), DOPC, LacPE, POPC, POPE, TOCL (1,1,2′,2′-tetraoleoyl cardiolipin), bovine liver PI, and heart CL were from Avanti Polar Lipids (Alabaster, AL). PerkinElmer Life Sciences supplied the [4-14C]Ch, glycerol tri-[9,10-3H]oleate, and 1-palmitoyl-2-[1-14C]oleoyl-sn-3-phosphocholine. Peroxidized CL (CLOOH) was prepared by dye-sensitized photooxidation (33) and separated by reverse-phase HPLC with 232-nm UV detection, as described (34). POPC hydroperoxides (PCOOHs) were prepared similarly and separated from parent lipid by normal-phase HPLC (33, 35). [14C]CL generated by metabolic radiolabeling of H9c2 cardiomyocytes (36) was used for preparing [14C]CLOOH. For recovery of liposomal or mitochondrial lipids, samples were mixed with butylated hydroxytoluene, EDTA, and DFO (final concentrations, 20, 100, and 100 μm, respectively), then extracted with ice-cold deoxygenated chloroform/methanol (2:1, v/v) (37). Phospholipid hydroperoxides (PLOOHs) were standardized with respect to both peroxyl and phosphoryl content using an iodometric (38) and phosphate assay (39), respectively. CLOOH used in liposome preparation contained on average one peroxyl group per molecule (two phosphoryl groups). ANTS, butylated hydroxytoluene, Ch, CHAPS, DFO, DPX, FD-10, RCA120 agglutinin, and Triton X-100 were obtained from Sigma. The 13.2-kDa human recombinant nsLTP (also known as sterol carrier protein-2) was prepared and assayed for transfer activity as described (30). Rabbit anti-cytochrome c oxidase subunit IV antibody was kindly provided by Dr. Margaret Wong-Riley as a gift. Mouse anti-pigeon cyt c antibody was obtained from Pharmingen, and peroxidase-conjugated anti-rabbit IgG was from MP Biochemicals (Aurora, OH). Mouse anti-human Bax monoclonal antibody was from Trevigen (Gaithersburg, MD). R&D Systems (San Diego, CA) supplied the rat anti-mouse Bid monoclonal antibody, His-tagged caspase-8, and caspase-8-cleaved recombinant mouse Bid. The latter consists of a proapoptotic 15-kDa C-terminal fragment (truncated Bid or tBid) and an inactive 7-kDa N-terminal fragment (40).
Expression and Isolation of Bax and tBid
Expression systems for human Bax and mouse full-length Bid, both His-tagged at their N termini, were kindly provided by Dr. Bruno Antonsson (Serono Pharmaceutical Research Institute, Geneva, Switzerland) as gifts. Expressed Bax protein was isolated by affinity chromatography using a nickel-nitrilotriacetic acid-agarose column (GE Healthcare) with a 25–250 mm imidazole linear elution gradient (40). Final purification was achieved by anion-exchange chromatography on a Q-Sepharose column (GE Healthcare). For preparing monomeric Bax, 1% (w/v) CHAPS was included in the elution buffer; for preparing oligomeric Bax, 1% (w/v) octylglucoside was included. Protein purification was monitored by SDS-PAGE with silver staining, and identity was confirmed by immunoblotting. Other details were as described previously (41). Expressed Bid protein was selectively cleaved by treating with His-tagged caspase-8 (42), giving the His-tagged N-terminal fragment and non-tagged tBid. The latter was separated from the tagged components by nickel-nitrilotriacetic acid chromatography, concentrated by spin-filtering, and checked for purity by SDS-PAGE with silver staining and identity by immunoblotting. The recombinant proteins in glycerol-containing buffer solutions were stored at −80 °C.
Liposome Preparation
Unilamellar liposomes were fabricated by an extrusion process (29, 33) using polycarbonate filters of specified pore size and either a syringe-type extruder from Avanti Polar Lipids or a larger scale device from Lipex Biomembranes (Vancouver, BC, Canada). The membranes were prepared so as to contain a specified mol % of CL or CLOOH.
For assessing intermembrane transfer of CL or CLOOH, we used small unilamellar vesicles (50 nm SUVs) as donors and large unilamellar vesicles (100 nm LUVs) as acceptors. Stock SUV preparations consisted of 0.7 mm POPC, 0.1 mm LacPE, and 0.2 mm CL or 0.1 mm each of CL and CLOOH in PBS (Chelex-treated PBC (25 mm sodium phosphate, 125 mm NaCl (pH 7.4))), 0.1 mm DFO (pH 7.4). The PBS was Chelex-treated to remove redox metal ions; DFO, an iron chelator/redox inhibitor (43), was used for added protection against any possible CL oxidation or CLOOH turnover. A small amount of [14C]Ch (2 mol %, ∼0.1 μCi/ml) was periodically included in SUV preparations as an internal standard, its transfer properties being well established (29). Stock LUVs consisted of 1.0 mm POPC in PBS, 0.1 mm DFO (pH 7.4). The LUVs typically contained a trace of non-transferrable [4H]triolein to assess the extent of recovery after separation from SUVs by agglutination (see below). Donor and acceptor liposomes were stored under argon at 4 °C and used for experiments within 3 days.
For studying the effects of transfer-acquired CL or CLOOH on membrane permeabilization by tBid, we again used 50 nm SUV donors and 100 nm LUV acceptors. Stock SUVs of two different compositions were prepared: (i) 0.95 mm POPC and 0.05 mm CL or CLOOH; (ii) 0.8 mm POPC and 0.2 mm CL or CLOOH. Both types were in 25 mm HEPES, 125 mm NaCl, 50 μm DFO (pH 7.4). Stock LUVs consisting of 1.0 mm POPC alone in 25 mm HEPES, 50 mm NaCl, 50 μm DFO (pH 7.4) were fabricated in the presence of 12.5 mm ANTS and 45 mm DPX. Encapsulated ANTS served as a fluorescent permeability indicator, and DPX served as a quencher (44, 45). Removal of non-encapsulated molecules was accomplished by passing the LUVs through a Sephadex G-75 column (1 × 75 cm) using 25 mm HEPES, 125 mm NaCl, 50 μm DFO (pH 7.4) for elution. Liposome-containing fractions were pooled and checked for phospholipid content using a modified Bartlett assay (39). Preparations were stored under argon at 4 °C and used for experiments within 48 h.
To assess how CL and CLOOH affect tBid and Bax-induced membrane permeabilization, we used 200-nm LUVs containing encapsulated 10-kDa FITC-dextran (FD-10) as an indicator. The membranes were constituted of DOPC/CL (95:5, mol/mol), DOPC/CLOOH (95:5, mol/mol), or DOPC alone. Dried lipid films were dispersed in 20 mm HEPES, 150 mm NaCl, 50 μm DFO (pH 7.4) containing FD-10 (100 mg/ml). After 20 cycles of freeze-thawing, the reconstituted lipids (5 mm in all) were extruded 30 times through two 0.2-μm polycarbonate membranes. Non-encapsulated FD-10 was removed by size exclusion chromatography using a Sephadex G-200 column (1 × 20 cm).
Multilamellar vesicles (MLVs) intended for tBid binding experiments were prepared in 10 mm HEPES, 20 mm KCl, 5 mm MgCl2, 0.1 mm DFO (pH 7.4), using a bath sonicator with ten 10-min pulses. Sonications were carried out on ice under an argon stream.
CLOOH-containing SUVs of overall lipid composition mimicking that of mitochondrial inner membrane of mammalian cells, i.e. POPC/POPE/PI/CLOOH/Ch (41:27:9:16:7, by mol) were prepared by extrusion into MS buffer (200 mm mannitol, 50 mm sucrose, 1 mm KH2PO4, 2 mm MgCl2, 1 mm EGTA, 0.05 mm DFO, 5 mm MOPS (pH 7.2)). These SUVs were used as CLOOH donors to YZD5 mitochondria (see below) for assessing tBid/Bax-dependent cyt c release. The extent of peroxide transfer was assessed by using [14C]CLOOH in the SUVs and measuring radioactivity in the mitochondrial compartment by scintillation counting.
Isolation of Mitochondria
A CRD1-null mutant (YZD5) of Saccharomyces cerevisiae lacking CL along with a CL-sufficient wild type (DL1) were used as mitochondrial sources. The two strains were kindly provided by Dr. William Dowhan (University of Texas-Houston). Yeast were grown at 30 °C in YPEG medium (1% Bacto yeast extract, 2% Bacto peptone, 3% glycerol, 1% ethanol) (46). Mitochondria were isolated as described (46) and finally suspended in 10 mm MOPS, 250 mm sucrose, 1 mm EDTA (pH 7.2). Total protein content was determined by a Bradford assay (47). For experimental work including measurement of respiratory capacity, the mitochondria were transferred to a working (MS) buffer: 200 mm mannitol, 50 mm sucrose, 1 mm KH2PO4, 2 mm MgCl2, 1 mm EGTA, 0.05 mm DFO, 5 mm MOPS (pH 7.2).
Measurement of CL and CLOOH Translocation
Reaction mixtures for assessing intermembrane CL transfer contained POPC/CL/LacPE (7:2:1 by mol) SUV donors and POPC-only LUV acceptors in PBS, 0.1 mm DFO. Mixtures for assessing CLOOH transfer contained POPC/CL/CLOOH/LacPE (7:1:1:1 by mol) SUVs and POPC LUVs in PBS, 0.1 mm DFO. Total SUV and LUV lipid concentration in bulk phase was 0.12 and 0.6 mm, respectively. Mixtures were incubated at 37 °C both in the absence and presence of nsLTP (typically 50 μg/ml). At various time points, transfer was quenched by mixing 25 μl of reaction mixture with 100 μl of ice-cold PBS containing 50 μg of RCA120 agglutinin. After a 10-min incubation on ice to allow SUV agglutination (LacPE binding by RCA120 (30)), the mixture was centrifuged at 16,000 × g for 10 min. A 50-μl aliquot of each LUV-containing supernatant was diluted to 0.25 ml with cold PBS, 1 mm EDTA and extracted with 0.4 ml of chloroform/methanol (2:1, v/v) (36). The lipid fraction was recovered, dried under nitrogen, dissolved in 25 μl of isopropyl alcohol, and analyzed by normal-phase HPLC with UV detection (34), CL being monitored by absorbance at 205 nm and CLOOH by absorbance at 232 nm, the latter reflecting the presence of conjugated dienes. For some experiments, CLOOH was assessed by HPLC with mercury cathode electrochemical detection using conditions similar to those described previously (33). Total CL or CLOOH in each system was determined by cooling and extracting samples directly, i.e. without agglutination; this was typically done immediately before and after a given period of transfer incubation. When included in SUV preparations as a transfer standard, [14C]Ch was monitored by scintillation counting.
Determination of ANTS/DPX-bearing Liposome Permeabilization by tBid
Two different systems were studied: (i) ANTS/DPX-containing LUVs with preexisting CL or CLOOH; (ii) a SUV/LUV mixture in which the LUVs had been transfer-enriched in CL or CLOOH by incubating with SUV donors in the absence versus presence of nsLTP. A 50-μl sample from each system was diluted to 1.5 ml with 25 mm HEPES buffer (pH 7.4) in a quartz fluorescence cuvette with slow magnetic stirring. The mixture was brought to 10 mm in CaCl2 to destabilize the membranes and promote tBid binding (45). A QM-7SE spectrofluorimeter from Photon Technology International (London, ON, Canada) was used for monitoring permeabilization. Background fluorescence due to any free ANTS was recorded as a function of incubation time at 37 °C (λex 360 nm; λem 530 nm; 8-nm bandwidth). After a specified interval, tBid (40–50 nm) was introduced, and increasing fluorescence due to membrane permeabilization and release/dequenching of ANTS was tracked. Maximum fluorescence was determined by lysing the vesicles with 0.1% (w/v) Triton X-100.
Determination of F-10-bearing Liposome Permeabilization by tBid/Bax
Reaction mixtures contained LUVs freed of non-encapsulated FD-10 (20 μm total lipid) in 1.2 ml of 20 mm HEPES, 150 mm KCl, 1 mm EDTA, 0.05 mm DFO (pH 7.4). Reactions were carried out at 37 °C with slow magnetic stirring. Increase of FD-10 fluorescence (λex 490 nm; λem 515 nm; 5 nm slits) was monitored, this due to loss of self-quenching of the fluorophore as it was released. The effects of sequentially added tBid and monomeric Bax on the rate of FD-10 release were monitored. Percent release was calculated as
where It is the measured fluorescence of protein-treated LUVs at time t, Io is the initial fluorescence before proteins are added, and Imax is maximal fluorescence in the presence of Triton X-100 (0.1%).
Determination of tBid Binding to Liposomes
tBid binding to CLOOH-containing MLVs was assessed by an adaptation of a procedure previously described for CL-containing liposomes (15). [14C]POPC MLVs containing various mol % levels of CLOOH were studied along with CL- and PCOOH-containing counterparts for comparison. The MLVs were incubated with tBid (0.5–1.0 μm) for a specified period at 30 °C then centrifuged at 350,000 × g for 10 min using a Beckman TL-100 ultracentrifuge with TLS-55 rotor. Lipid recovery in sedimented vesicles was checked by scintillation counting. tBid bound to pelleted vesicles was determined by Western blot analysis using an anti-Bid antibody.
Determination of CLOOH/tBid/Bax-dependent cyt c Release from Mitochondria
For examining the effects of CLOOH transfer uptake by mitochondria on tBid binding and tBid/Bax-induced cyt c release, mixtures of yeast YZD5 or DL1 mitochondria (∼5 mg protein/ml), IM-mimetic SUVs (∼1 mm total lipid), and nsLTP (50 μg/ml) in MS buffer were incubated at 30 °C for a specified period. Two types of control were run alongside; that is, one containing everything except nsLTP and the other containing mitochondria, nsLTP, and SUVs lacking CLOOH. After centrifugation at 4 °C, the mitochondria were washed, resuspended to ∼0.6 mg of protein/ml in MS buffer, and incubated at 30 °C for a specified period in the presence of tBid alone (40 nm), monomeric Bax alone (100 nm), or both tBid and Bax. After rapid pelleting at 4 °C and 1 wash, the mitochondria were analyzed for (i) recovered protein and (ii) acquired radioactivity to assess nsLTP effectiveness in terms of [14C]CLOOH transfer relative to a control lacking nsLTP. Immunoblotting was used for determining released cyt c in supernatant fractions and retained cyt c and bound tBid in mitochondrial pellets (18, 19). Pellet blots were also probed for cytochrome c oxidase, which served as a loading standard.
RESULTS
Comparative Effects of CL and CLOOH in Sensitizing LUVs to Permeabilization by tBid
In initial experiments, we tested whether CLOOH-containing membranes can recruit and be permeabilized by tBid using a liposomal model. Our preparations of pure tBid behaved identically to a commercial preparation of caspase-8-cleaved Bid that contained the N-terminal fragment. LUVs encapsulating a small fluorophore reporter (ANTS) and a quencher (DPX) were employed. Previous studies showed that tBid permeabilizes ANTS/DPX-containing liposomes in a Ca2+- and CL-dependent manner, resulting in the release and dequenching of ANTS (44, 45). The rate at which the fluorescence signal intensifies reflects the leakage rate of vesicle contents. Using this approach, we found that LUVs containing 5 mol % of non-oxidized CL (Fig. 1A) or HPLC-purified moderately peroxidized CL, averaging ∼1 -OOH per 2 Pi (Fig. 1B), were permeabilized by tBid in the presence of 10 mm CaCl2. Neither tBid nor CaCl2 individually had any significant effect on either LUV type (results not shown). The Ca2+-dependent leakage rate for CLOOH-LUVs was ∼70% greater than that for CL-LUVs, suggesting better tBid recognition by the former. Importantly, LUVs containing 5 mol % of peroxidized POPC (∼1 -OOH per Pi) instead of CLOOH were not affected by tBid plus Ca2+ (Fig. 1C), indicating that recognition is highly specific for CL or CLOOH. Interestingly, decreasing the CL content of POPC/CL LUVs to 2 mol % slowed the rate of tBid-dependent leakage proportionately to CL, i.e. by ∼2.5-fold, whereas decreasing the CLOOH content of POPC/CLOOH LUVs to 2 mol % had no effect on leakage (supplemental Fig. S1). This again indicates that CLOOH was more effective than CL in sensitizing membrane permeabilization.
FIGURE 1.
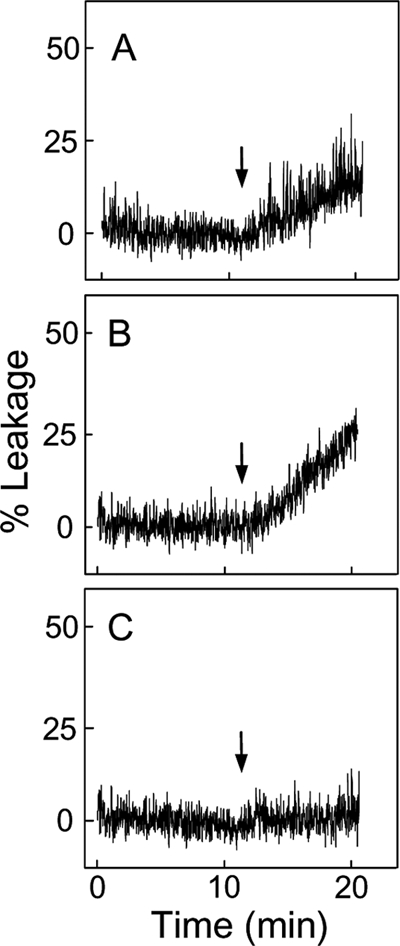
Effect of preexisting CL or CLOOH on LUV susceptibility to tBid permeabilization. shown is ANTS/DPX leakage from POPC/CL (95:5 mol/mol) (A), POPC/CLOOH (95:5 (mol/mol) (B), and POPC/POPC-OOH (95:5 mol/mol) (C) LUVs. Each reaction mixture, 2.0 ml in a fluorescence cuvette at 37 °C, contained 50 μm total lipid and 10 mm CaCl2 in 25 mm HEPES buffer (pH 7.4). Mixtures were subjected to gentle magnetic stirring throughout. tBid (40 nm) in a negligible volume was introduced at the indicated time points (arrows). Fluorescence emission was monitored at 530 nm using 360-nm excitation. Maximal fluorescence was determined by lysing the LUVs with 0.1% Triton X-100. Traces shown are representative of three separate experiments with identical results.
Effect of Transfer-acquired CL or CLOOH on LUV Susceptibility to tBid Permeabilization
We postulated that in mitochondria under oxidative pressure, movement of CLOOH to the outer membrane would sensitize the latter to permeabilization by tBid and Bax. To begin testing this we used a model system consisting of POPC/CLOOH (8:2, mol/mol) SUV donors, and POPC LUV acceptors, the latter containing trapped ANTS/DPX. POPC/CL (8:2, mol/mol) donors were also used to compare the responses to CLOOH and parental CL. Complete reaction systems included recombinant nsLTP, which is known to facilitate intermembrane transfer of LOOHs as well as unoxidized lipids (29, 30). For CLOOH-bearing SUV donors, the sensitivity of LUV acceptors to permeabilization by tBid in the presence of Ca2+ was found to be strongly enhanced after an 18-h co-incubation with nsLTP (Fig. 2, compare panels C and D). Although CL-bearing donors also produced an effect, it was much smaller than that of the CLOOH counterparts (Fig. 2, compare panels B and D). Rates of tBid/Ca2+-induced ANTS/DPX leakage for these systems after 10 and 18 h of incubation in the presence versus absence of nsLTP are shown in Table 1. As can be seen, nsLTP increased the leakage rate by ∼24-fold for the 18-h CLOOH system, and this rate was ∼17 times greater than that for the corresponding CL system. One might ask whether trivial CL/CLOOH acquisition via SUV/LUV fusion could account for any of the observed responses to tBid. Although we have not checked directly for possible fusion effects, they can tentatively be ruled out on the basis of the nsLTP effects (Fig. 2, Table 1) because it is unlikely that this protein promotes membrane fusion.
FIGURE 2.
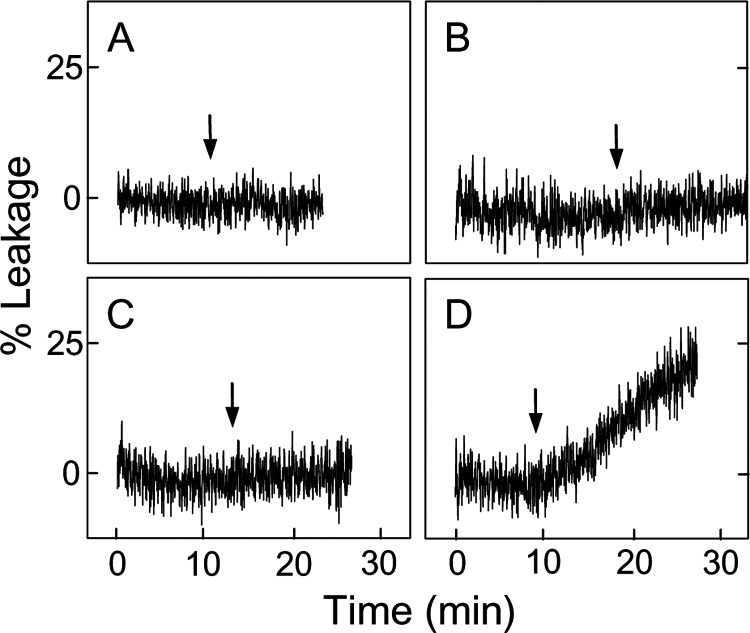
Effect of CL or CLOOH transfer uptake on LUV susceptibility to tBid permeabilization. POPC LUV acceptors were mixed with SUV donors, each at a final concentration of 50 μm total lipid in 25 mm HEPES buffer containing 10 mm CaCl2 (pH 7.4). The SUVs consisted of POPC/CL (8:2, mol/mol) (A and B) and POPC/CLOOH (8:2, mol/mol) (C and D). Reaction mixtures were incubated in the absence (A and C) or presence (B and D) of nsLTP (50 μg/ml) for 18 h at 37 °C. tBid (40 nm) was introduced at the indicated times during incubation (arrows). Other details were as described in Fig. 1. Data from this and a duplicate experiment are also represented in Table 1.
TABLE 1.
Kinetics of liposome permeabilization by tBid
| Donor SUV | Leakage ratea |
|||
|---|---|---|---|---|
| −nsLTP |
+nsLTP |
|||
| 10 h | 18 h | 10 h | 18 h | |
| % per min × 102 | ||||
| POPC/CL | NDb | ND | 2.04 ± 0.18 | 8.34 ± 0.42 |
| POPC/CLOOH | 0.26 ± 0.02 | 6.12 ± 0.36 | 34.7 ± 0.6 | 145 ± 1 |
a Rate of tBid-induced ANTS and DPX leakage from 50 μm POPC LUVs in 25 mm HEPES/10 mm CaCl2 (pH 7.4) during incubation with 50 μm POPC/CL or POPC/CLOOH (95:5 mol/mol) donor SUVs for 10 or 18 h at 37 °C in the absence or presence of nsLTP (50 μg/ml). The means ± deviation of values from two separate experiments are shown; data from the experiment represented in Fig. 2 are included.
b ND, not detectable.
We reasoned that SUV-to-LUV transfer of CL or CLOOH was necessary for the tBid-permeabilizing effects shown in Fig. 2 and Table 1. To confirm this, we incubated POPC/CL//LacPE or POPC/CL/CLOOH/LacPE SUVs with POPC LUVs in 5-fold total lipid molar excess, removed the SUVs by RCA120 agglutination at various time points, and analyzed the remaining LUVs for acquired CL or CLOOH by HPLC using A205 and A232 for detecting CL and CLOOH, respectively. Although A232 would detect conjugated dienes in cardiolipin hydroxides (CLOHs) as well as CLOOHs, independent determination of the latter by electrochemical detection (not shown) revealed that no significant reductive loss occurred under the incubation conditions used. As shown in Fig. 3A, spontaneous intermembrane transfer of CL was extremely slow, the apparent first-order rate constant being ∼2.2 × 10−3 h−1 (t½ ∼13 days). The SUVs we used contained 20 mol % CL at the outset. When half of this was replaced by CLOOH (∼1 -OOH/2 Pi), the transfer rate constant for the latter was found to be ∼7.3 × 10−3 m−1 (Fig. 3B), i.e. 3.3 times greater than that for non-oxidized CL (Fig. 3A). It is important to note that SUVs with 20 mol % CLOOH could not be used because of bilayer instability, whereas those containing 10 mol % each of CLOOH and CL (i.e. the same total CL backbone content) were sufficiently stable for studying CLOOH transfer. In addition, the 10% CL, 10% CLOOH SUVs more closely mimic an IM stress situation in which half of the CL has been oxidized. When lipid transfer protein nsLTP (50 μg/ml) was included in the reaction mixture, the rate constant for CL transfer increased to ∼1.3 × 10−2 h−1, i.e. 6-fold over background (Fig. 3A), whereas that for CLOOH transfer increased to ∼9.8 × 10−2 h−1, i.e. 13-fold over background (Fig. 3B). Thus, nsLTP accelerated transfer of both CL and CLOOH, but the rate constant enhancement for CLOOH was more than twice that for CL, suggesting a more favorable interaction of CLOOH with nsLTP. As anticipated (30), nsLTP-enhanced rates of CL and CLOOH transfer increased proportionately with protein concentration; e.g. at 50 μg/ml, the CLOOH rate was 2.5 times greater than that at 20 μg/ml (supplemental Fig. S2). HPLC analysis of non-RCA120-treated samples from reaction mixtures at various incubation times indicated that there was no significant change in overall CL or CLOOH content under the conditions used (data not shown). Because there was no net loss of either analyte, it is apparent that the decay plots shown in Fig. 3 can only reflect intermembrane translocation. Clearly, therefore, nsLTP-facilitated CL/CLOOH transfer was necessary for the tBid targeting described in Fig. 2 and at least part of the reason why CLOOH was more effective than CL in this regard is that it translocated more rapidly.
FIGURE 3.
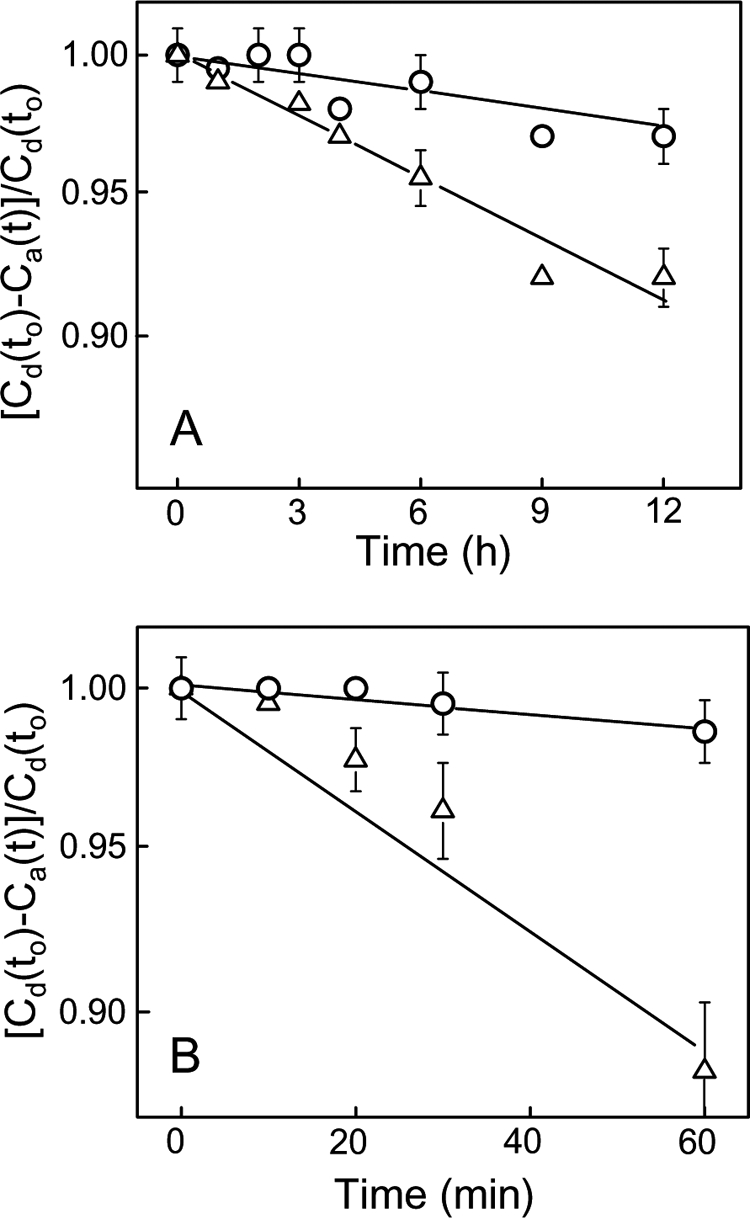
Comparative kinetics of CL and CLOOH transfer between liposomes; spontaneous versus nsLTP-facilitated transfer. A, spontaneous transfer of CL (○) and CLOOH (△) from SUVs to LUVs is shown. Reaction mixtures at 37 °C contained POPC/CL/LacPE (7:2:1 by mol) or POPC/CL/CLOOH/LacPE (7:1:1:1 by mol) SUV donors and POPC LUV acceptors (1:5 lipid mol ratio). At the indicated incubation times, the membranes were separated by agglutination, and CL/CLOOH in the LUV fraction was analyzed by HPLC using A205 and A232 for CL and CLOOH detection, respectively. B, nsLTP-enhanced CL (○) and CLOOH (△) transfer. Reaction mixtures were the same as in A except for the inclusion of nsLTP (50 μg/ml). Timed samples were quenched on ice and analyzed for LUV-acquired CL or CLOOH as described in A. Incubations in the absence and presence of nsLTP were carried out simultaneously. Plotted values in A and B are the means ± S.E. of values from three separate experiments. Cd(to) and Ca(t) denote analyte concentration in donor SUVs at time 0 and in acceptor LUVs at time t, respectively. Calculated apparent first-order rate constants for transfer are as follows: A, 2.2 ± 0.3 × 10−3 h−1 (CL), 7.3 ± 1.5 × 10−3 h−1 (CLOOH); B, 1.3 ± 0.2 × 10−2 h−1 (CL), 9.8 ± 0.2 × 10−2 h−1 (CLOOH).
Cooperative Effects of tBid and Bax in Permeabilizing CLOOH-sensitized LUVs to a Macromolecular Probe
To more closely model an actual mitochondrial setting, we used LUVs with trapped FD-10, a macromolecular fluorophore almost as large as cyt c (10 versus 12 kDa). Our aim was to examine the concerted permeabilizing action of tBid and monomeric Bax in CLOOH-containing membranes. To do this, we first established a level of tBid and Bax individually that would cause minimal permeabilization of DOPC/CLOOH (95:5 mol/mol) membranes. Then using these levels of tBid and Bax combined, we again monitored FD-10 release, comparing it with that from DOPC/CL (95/5 mol/mol) LUVs and from all-DOPC LUVs. Incubation of CLOOH-containing LUVs with either 40 nm tBid alone or 100 nm monomeric Bax alone resulted in no more than ∼2 or ∼5% release, respectively, after 30 min (results from three replicate experiments). However, 40 nm tBid followed by 100 nm Bax produced a substantial increase in membrane permeabilization, freeing ∼40% of the FD-10 after 30 min (Fig. 4). The release rate was approximately the same when CL-containing LUVs were used (Fig. 4), whereas those lacking CL or CLOOH failed to respond to tBid and Bax, in agreement with previous findings (18, 44). Unlike CLOOH, PCOOH (not shown) did not permit any significant permeabilization by tBid and Bax (cf. Fig. 1). Thus, the action of these proteins was strongly dependent on the presence of CL species, and CLOOH with ∼1-OOH/2 Pi appeared to be at least as effective as parental CL. CL is known to induce negative curvature when present in membrane bilayers (45), and this structural deformation appears to play a key role in tBid binding required for pore formation (16, 17). The observed CLOOH sensitization of LUV membranes to tBid/Bax-dependent poration (Fig. 4) can be explained on similar grounds.
FIGURE 4.

CLOOH sensitization of LUVs to cooperative permeabilization by tBid and Bax. Reaction mixtures contained FD-10-bearing DOPC/CL (95:5, mol/mol), DOPC/CLOOH (95:5, mol/mol), or DOPC-only LUVs in 20 mm HEPES, 150 mm KCl, 1 mm EDTA, 0.05 mm DFO (pH 7.4). Each mixture (20 μm in total lipid) was gently stirred at 37 °C. At the indicated time points (arrows), tBid (40 nm) and monomeric Bax (100 nm) were added sequentially. FD-10 fluorescence emission was monitored at 520 nm using 490-nm excitation. Maximal fluorescence was determined by including 0.1% Triton X-100.
In a previous study (48), it was found that CLOOH-containing LUVs, upon exposure to tBid and Bax, released a trapped FITC-dextran much more rapidly than CL-containing counterparts. This contrasts with our results in Fig. 4. However, the LUVs used in the earlier work differed from ours in size, lipid composition, and CLOOH content (10 versus 5 mol%). Also, the CLOOH employed contained on average more than one -OOH group per molecule, whereas ours had only one. Any or all of these factors might explain why CLOOH in the previous study (48) sensitized greater poration than CL.
tBid Binding by CLOOH- Versus CL-containing LUVs
Prior studies have shown that permeabilization of CL-bearing membranes by tBid requires avid binding of the latter (16–20). It was of interest to know how increasing CL peroxidation would affect this binding. We studied this for [14C]POPC/CL and [14C]POPC/CLOOH MLVs prepared with 0–30 mol % CL or CLOOH using [14C]POPC-only and [14C]POPC/PCOOH counterparts as controls. Relatively dense MLVs were used to easily separate bound from unbound tBid via centrifugation. Immunoblot-assessed tBid binding was found to increase progressively with CL or CLOOH content from 10–30 mol %, and this was substantially greater for CLOOH than CL (Fig. 5). There was no significant binding increment when PCOOH was substituted for CLOOH (Fig. 5), thus confirming a high degree of specificity for the CL backbone. These results are consistent with the leakage results for LUVs with pre-existing (Fig. 1) or transfer-acquired (Fig. 2) CLOOH or CL, assuming that leakage was directly proportional to the amount of tBid bound and inserted into the bilayer. The situation for Fig. 4 was more complex, i.e. membrane poration depended on both tBid and Bax recruitment, and involved a much larger efflux marker (FD-10), possibly explaining why it was more difficult to distinguish between CLOOH and CL sensitization in this case.
FIGURE 5.
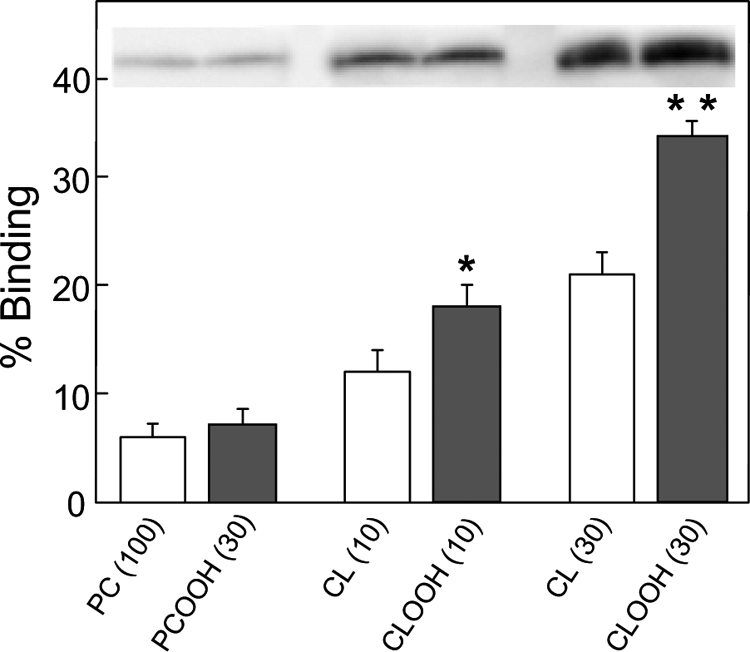
tBid binding by CLOOH-containing versus CL-containing liposomes. [14C]phosphocholine (PC)-only and [14C]PC/PCOOH, [14C]PC/CL, and [14C]PC/CLOOH MLVs of the indicated mol % CL, PCOOH, or CLOOH (∼1 -OOH/Pi for PCOOH; ∼1 -OOH/2 Pi for CLOOH; 1 mm total lipid; 0.1 μCi/ml) were prepared by sonication in 10 mm HEPES, 20 mm KCl, 5 mm MgCl2, 0.1 mm DFO (pH 7.4). The MLVs were incubated with 1 μm tBid for 10 min at 30 °C and then sedimented by centrifugation; lipid recovery was monitored by scintillation counting. Bound tBid was determined by immunoblotting using the same amount of total MLV lipid for each sample lane. tBid bands and densitometrically measured band intensities for each condition are shown. Values are the means ± S.E. (n = 3). *, p < 0.01 versus 10% CL; **, p < 0.001 versus 30% CL.
Effect of Transfer-acquired CLOOH on tBid Binding by YZD5 Versus DC1 Mitochondria
In followup experiments we advanced to the mitochondrial level, asking how CLOOH uptake via translocation would affect tBid binding. To minimize basal signals, we used mitochondria from a CL-null mutant yeast strain (YZD5), comparing their responsiveness with that of mitochondria from CL-sufficient wild type (DL1). The mitochondria were incubated with CLOOH-containing SUVs in the presence of nsLTP and after pelleting/washing were incubated with tBid, then pelleted/washed again, and analyzed for bound tBid by immunoblotting. CLOOH transfer from SUV donors was found to sensitize YZD5 mitochondria to progressively greater tBid binding, which increased ∼2.5-fold after 1 h with nsLTP present (supplemental Fig. S3). Similar treatment of DL1 mitochondria elicited a smaller net increase in tBid binding (∼1.5-fold) above a higher background (supplemental Fig. S3). The higher background was attributed to available preexisting CL in the DL1 membranes. These results clearly demonstrated that tBid targeting to mitochondria could be substantially enhanced by the presence of translocated CLOOH.
Effect of Transfer-acquired CLOOH on tBid/Bax-dependent cyt c Release from YZD5 Mitochondria
Additional support for our hypothesis was sought by examining the effects of CLOOH transfer uptake on tBid recruitment by isolated mitochondria. To optimize responsiveness, we used mitochondria from a CL-null mutant yeast strain (YZD5). IM-mimetic SUVs were used as CLOOH donors. As shown in Fig. 6, incubation of YZD5 mitochondria with CLOOH-containing SUVs and nsLTP for 1 h followed by tBid plus monomeric Bax for 1 h resulted in cyt c release that was substantially greater than that in a control preincubated with CLOOH-free SUVs plus nsLTP. We determined that nsLTP was effective in the Fig. 6 experiment by showing that 64 ± 4% of SUV [14C]CLOOH was taken up by mitochondria in 1 h (means ± S.E.; n = 3). At the level used (40 nm), tBid alone had no significant effect on CLOOH-enriched mitochondria (not shown), whereas Bax alone (100 nm) caused some net cyt c release but much less than when combined with tBid (Fig. 6). Western analyses also showed that YZD5 mitochondria that had been transfer-enriched in CLOOH retained much more tBid after washing than non-CLOOH controls (Fig. 6), in agreement with previous evidence that some form of CL is required for synergistic pro-apoptotic binding of tBid and Bax to the mitochondrial OM (15–20).
FIGURE 6.

Transfer-acquired CLOOH sensitization of YZD5 mitochondria to tBid/Bax-dependent poration. Suspensions containing POPC/POPE/PI/[14C]CLOOH/Ch (41:27:9:16:7 by mol) SUVs (1 mm total lipid; ∼10 nCi/ml), YZD5 mitochondria (3.3 mg of protein/ml), and nsLTP (50 μg/ml) in MS buffer were incubated for 1 h at 30 °C, then centrifuged and washed once. A control system without CLOOH, i.e. with POPC/POPE/PI/Ch (57:27:9:7 by mol), SUVs was run alongside. Mitochondria were recovered, resuspended in protease inhibitor-containing MS buffer to 0.6 mg of protein/ml, then incubated in the presence of 40 nm tBid alone, 100 nm monomeric Bax alone, or tBid plus Bax for 1 h at 30 °C. After centrifugation, mitochondrial fractions were recovered and analyzed for protein content and the extent of [14C]CLOOH translocation uptake, as determined by scintillation counting. Western blots for released cyt c in the supernatant fractions and remaining cyt c and bound tBid in the pellet fractions are shown. Numbers below the released cyt c bands for each system represent integrated band intensities relative to the control and normalized to cytochrome c oxidase (Cox) as a loading standard. Data from one experiment representative of three are shown.
IM-to-OM Translocation of Oxidized CL in Antimycin A-treated Cardiomyocytes
We next sought to obtain direct evidence for IM-to-OM transfer of oxidized CL in mitochondria of cells exposed to an IM-damaging oxidative stress. To attempt this, we used (i) cardiomyocytes that had been grown in the presence of [14C]acetate to radiolabel CL and other membrane lipids and (ii) antimycin A (AA), which generates reactive oxidants by blocking mitochondrial electron transport at Complex III (49). Radiolabeled cells were incubated in the absence (control) versus presence of 0.1 mm antimycin A for 1.5 h, after which homogenates were prepared, mitochondria were recovered, and OM- and IM-containing fractions were isolated for lipid analysis by two-dimensional high performance TLC (see the supplemental Experimental Procedures). Western blot analysis beforehand, using an antibody against IM cytochrome c oxidase, revealed little if any contamination of the OM fraction with IM (supplemental Fig. S4A). Likewise, OM contamination of IM was negligible (results not shown). Two-dimensional high performance TLC chromatograms for four different sample types, IM (control and AA) and OM (control and AA), showed that CL species were clearly detectable and sufficiently resolved from other phospholipids (supplemental Fig. S4A). Quantitative phosphorimaging indicated that for the control most of the mass of CL species was in the IM, as expected. After AA treatment, there was a shift in location such that substantially more mass of CL species now resided in the OM, with a corresponding loss from the IM (supplemental Fig. S4A). No significant redistribution of other major phospholipids, e.g. PC and PE, was apparent under the reaction conditions used (data not plotted). It is important to point out that oxidized forms of CL, including CLOOHs, were not resolved from parent CL by the chromatographic approach used. Although oxidation status was not established, it is likely that the “CL species” described for AA-treated cells consisted of CLOOHs and possibly other CL oxides, based on our results described up to this point. We showed additionally that similar treatment of cardiomyocytes with AA resulted in a significant activation of caspase-3/7 above the control level, beginning after the IM-to-OM redistribution of oxidized CL was measured (supplemental Fig. S4B). Collectively, these results support the idea that the CLOOH translocation observed in model systems (Figs. 2, 3, 6, and supplemental Fig. 2) also applies at the cellular level and that it serves as a key early event in oxidative apoptosis, i.e. upstream of tBid/Bax recruitment for membrane pore formation and release of cyt c and other apoptogenic proteins.
DISCUSSION
Numerous studies over the past decade have revealed that CL and Bcl-2 homology (BH) domain proteins such as BH3-only Bid and BH1–3 Bax play a key role in the intrinsic (mitochondrion-initiated) pathway of oxidative stress-induced apoptosis (16–20). Recent fluorescence resonance energy transfer-based studies have shown that membrane permeabilization by tBid and Bax requires an ordered uptake and interaction of these proteins and that antiapoptotic BH1–4 Bcl-xL antagonizes this by binding tBid (50). During respiration and oxidative phosphorylation, cyt c on the outer face of the mitochondrial IM shuttles electrons from Complex III to Complex IV, but under stress conditions it becomes mobilized and moves into the cytosol, where it participates in formation of the caspase-9-activating apoptosome complex (1–3, 19–21). cyt c mobilization has been linked to oxidative modification of underlying IM lipids, particularly CL, which tethers cyt c via hydrophobic and electrostatic interactions (51, 52). Bearing four fatty acyl chains, all typically unsaturated (9), natural CL is much more susceptible to oxidation than phospholipids with two fatty acyl chains. Model studies with CL in thin film or liposomal form, for example, have shown that its normally tight association with cyt c is progressively weakened with increasing CL peroxidation (4). Later work based on ELISA-type analyses (7, 53) confirmed and extended this by showing that cyt c binds well to unoxidized CL or enzymatically reduced CLOOH (i.e. CLOH), but not to CLOOH, presumably due to the latter's altered configuration and/or increased hydrophilicity. According to recent studies (54–56), cyt c in oxidatively stressed mitochondria can switch from being an electron transporter to a peroxidase. This would result in selective oxidation of associated CL to CLOOH species, thereby promoting the protein release.
In addition to tethering cyt to the IM under natural conditions and releasing it under oxidative pressure, CL is known to be required for permeabilization of the OM via recruitment of cytosolic tBid and monomeric Bax (15, 19). Pre-oligomerized Bax can permeabilize the OM in the absence of CL and tBid (57, 58), but this appears to be physiologically irrelevant vis à vis apoptosis. Substantial evidence from various laboratories suggests that CL or some metabolite thereof is presented at the OM surface in early response to a stress signal (15–20). This facilitates binding of cytosolic tBid, which serves as a nexus for Bax binding and oligomerization, giving transmembrane megapores through which cyt c, Smac/DIABLO, AIF, and other apoptogenic proteins can pass (18–21). Although some monomeric Bax may bind in the absence of CL and tBid, both of the latter appear to be necessary for full Bax activation with pore formation (57, 59). Recent evidence has revealed that OM CL also provides an activating platform for caspase-8, which processes Bid to tBid (60). Although generally accepted, this model is confronted with the following crucial question: How does CL, which originates in the mitochondrial IM, make its way to the OM for recruitment of procaspase-8 and tBid/Bax? Of added importance is whether this CL is modified in some way. If so, this would agree with numerous reports indicating that there is a net loss of cellular CL during oxidative apoptosis (22–24). Some of this could be accounted for by formation of CLOOHs and other oxidation products. Various hydrolyzed species, including monolyso-CL and dilyso-CL have also been identified (20, 28), consistent with phospholipids being more susceptible to phospholipase action after being oxidized (61). Another highly relevant property of CL is its tendency, like cyt c, to become mobilized under oxidative pressure and to translocate to other membrane sites. For example, TNF-α treatment of HL-60 tumor cells was reported to cause a redistribution of nonyl acridine orange-detectable CL from the inner to outer leaflet of the IM, which occurs well in advance of cell death via apoptosis (62). Of related interest is the fact that oxidized phosphatidylserine on the plasma membrane inner face of stressed cells readily translocates or “flips” to the outer face, where recognition by phagocytic cells occurs (63). In the case of CL, translocation on a broader scale has also been described. For example, Fas-challenged monocytes were found to contain significant amounts of tBid-recognizable CL and lyso-CL in the plasma membrane and other non-mitochondrial compartments (20, 26). In addition, CL antibodies have been detected in individuals with autoimmune diseases and other pathologic conditions (64), implying that CL species are mobilized and somehow move to cell surfaces, where they elicit immune responses.
Proapoptotic binding of tBid has been reported to occur mainly at IM-OM contact sites (25, 26). In these zones, CL (or some active CL metabolite) would somehow move from the IM inner leaflet to OM outer leaflet, thus presenting itself to tBid. It was recently reported that an electrostatic interaction occurs between lysyl residues in the αH6 helix domain of tBid and contact site CL (65). Although the contact site model has been widely invoked, it has certain weaknesses. First, these sites are linked mainly by proteins rather than IM and OM lipids, and the major resident proteins, VDAC (voltage-dependent anion channel protein) and ANT, are not known to have phospholipid transfer activity (66). On the other hand, it has been reported (67) that interliposomal phospholipid transfer can be enhanced by mitochondrial creatine kinase or nucleotide diphosphokinase, each known to reside at contact sites; however, the mechanism and biological importance of this remains to be established. Second, substantial tBid binding has been observed at non-contact loci (25), demonstrating that recognizable OM CL is not restricted to contact sites. Third, tomographic examination of mitochondria in a relatively unperturbed state (68, 69) has revealed far fewer contact sites than previously estimated by electron microscopy.
In the present study, we provide the first evidence in support of a more general translocation mechanism, i.e. one not restricted to contact sites, and based on CLOOHs (and possibly other CL oxides) as mobile pro-apoptotic signaling species. According to this mechanism, CLOOHs might emerge from several different sites along the IM, the most obvious possibilities being outer leaflet sites where tethered cyt c can function as a CL peroxidase (54, 55). The feasibility of IM-to-OM CLOOH transfer in stressed mitochondria is favored by several different factors, including (i) the high starting concentration of CL in the IM, (ii) the CL high degree of unsaturation, making it readily peroxidizeable, (iii) the relatively short average distance between IM and OM (50–70 A°), and (iv) an initial steep negative CLOOH concentration gradient, which would favor diffusion from IM to OM. Using liposome/liposome and liposome/mitochondria pairings to model IM-to-OM lipid movement, we showed that CLOOH translocates much more rapidly than parental CL. The CLOOH used in these experiments contained only ∼1 peroxyl group per molecule. A greater peroxyl density (up to four per molecule) is feasible for natural CL under high oxidative pressure, and this would be expected to increase transfer rate proportionately due to greater hydrophilicity. In addition to relatively simple modeling of intermembrane translocation, we showed that imposing a mitochondrial stress on cardiomyocytes resulted in a large IM-to-OM redistribution of CL-derived species, which was followed by caspase-3/7 activation. A similar effect has been described by others for HL-60 cells exposed to proapoptotic staurosporine (54). Based on our other findings (Figs. 3 and 6), it is likely that the translocated material described in supplemental Fig. S4 consisted of CL oxidation products such as CLOOHs rather than CL itself, although actual product identification will require more extensive analysis.
As indicated above, peroxidized CL in stressed cells is susceptible to phospholipase-mediated hydrolysis to lyso derivatives (61). Lacking one or more fatty acyl chains, these species would likely be even more hydrophilic than CLOOH precursors. Thus, lysolipid formation could further enhance CL movement between membranes. Employing a liposomal system such as described in Fig. 3, we are in the process of examining CLOOH transfer kinetics as a function of peroxyl group density on the one hand and deleted fatty acyl chains on the other. We showed in the present study that relatively rapid CLOOH transfer between liposomes or liposomes and mitochondria is substantially accelerated by nsLTP, a nonspecific lipid transfer protein also known as SCP-2 (29). This protein exists in mitochondria of certain cells (70), but whether it might function there as depicted in our IM-to-OM model is not known. In any event there is a profound interest in how CL (in native or modified form) appears on the OM, and several different mechanisms based on protein-facilitated transfer have been proposed. One of these stems from evidence that full-length cytosolic Bid has lipid transfer activity such that during mitochondrial phospholipid recycling, it can deliver CL to the OM extramitochondrially for tBid/Bax targeting (71). Also, as indicated above, oligomeric creatine kinase and nucleoside diphosphate kinase, known to exist at contact sites, are reported to facilitate IM-to-OM phospholipid transfer with a preference for CL in a liposomal model (67). A more recent study (72) tested the hypothesis that mitochondrial phospholipid scramblase-3 (PLS3) plays a crucial role in TNF-α-induced apoptosis of HeLa cells by mediating CL movement to the mitochondrial surface. This was affirmed by showing that PLS3 overexpression increased the percentage of CL in the OM, whereas PLS3 knockdown decreased it, tBid binding being altered in parallel. However, scramblases typically move lipids between leaflets of a given membrane, so how PLS3 might catalyze IM-to-OM translocation of CL as described (72) is unclear. None of the transfer-based studies described (67, 71, 72) has specifically addressed the prospect of intermembrane CLOOH transfer as done in the present study.
In summary, we describe translocation via the aqueous intermembrane space as a novel means by which CLOOHs and other CL oxidation products can move from the IM to OM of oxidatively stressed mitochondria, thus sensitizing the OM for proapoptotic recruitment of tBid and Bax.
Supplementary Material
Acknowledgments
We thank Dr. Kyoung Joon Oh (Rosalind Franklin Institute) for graciously providing us with samples of recombinant monomeric Bax as a gift, allowing us to check for consistency in results obtained with our own expressed monomeric Bax. The technical assistance of Vlad Levchenko and Jared Schmitt is greatly appreciated.
This work was supported, in whole or in part, by National Institutes of Health Grants HL85677 and CA72630 (USPHS). This work was also supported by Polish Ministry of Science Grant 3P05A-5523.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- cyt c
- cytochrome c
- AA
- antimycin A
- ANTS
- 8-aminonaphthalene-1,3,6-trisulfonic acid
- Bax
- B-cell lymphoma 2-associated X protein
- Bid
- BH3-interacting domain death agonist
- tBid
- truncated Bid
- Ch
- cholesterol
- CL
- cardiolipin
- CLOOH
- cardiolipin hydroperoxide
- DFO
- desferrioxamine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPX
- p-xylene-bis(N-pyridinium bromide)
- FD-10
- fluorescein isothiocyanate-conjugated dextran of average Mr 10
- IM
- inner membrane
- LacPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-lactosyl
- LOOH
- lipid hydroperoxide
- nsLTP
- non-specific lipid transfer protein
- OM
- outer membrane
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- PI
- phosphatidylinositol
- LUV
- large unilamellar vesicle
- MLV
- multilamellar vesicle
- SUV
- small unilamellar vesicle
- PLS3
- phospholipid scramblase-3
- BH
- Bcl-2 homology.
REFERENCES
- 1. Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996) Cell 86, 147–157 [DOI] [PubMed] [Google Scholar]
- 2. Newmeyer D. D., Ferguson-Miller S. (2003) Cell 112, 481–490 [DOI] [PubMed] [Google Scholar]
- 3. Garrido C., Galluzzi L., Brunet M., Puig P. E., Didelot C., Kroemer G. (2006) Cell Death Differ. 13, 1423–1433 [DOI] [PubMed] [Google Scholar]
- 4. Shidoji Y., Hayashi K., Komura S., Ohishi N., Yagi K. (1999) Biochem. Biophys. Res. Commun. 264, 343–347 [DOI] [PubMed] [Google Scholar]
- 5. Petrosillo G., Ruggiero F. M., Pistolese M., Paradies G. (2001) FEBS Lett. 509, 435–438 [DOI] [PubMed] [Google Scholar]
- 6. Petrosillo G., Ruggiero F. M., Paradies G. (2003) FASEB J. 17, 2202–2208 [DOI] [PubMed] [Google Scholar]
- 7. Nomura K., Imai H., Koumura T., Kobayashi T., Nakagawa Y. (2000) Biochem. J. 351, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M., Mileykovskaya E., Dowhan W. (2002) J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 9. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. (2002) Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 10. Kriska T., Korytowski W., Girotti A. W. (2002) Free Radic. Biol. Med. 33, 1389–1402 [DOI] [PubMed] [Google Scholar]
- 11. Enoksson M., Fernandes A. P., Prast S., Lillig C. H., Holmgren A., Orrenius S. (2005) Biochem. Biophys. Res. Commun. 327, 774–779 [DOI] [PubMed] [Google Scholar]
- 12. Chang T. S., Cho C. S., Park S., Yu S., Kang S. W., Rhee S. G. (2004) J. Biol. Chem. 279, 41975–41984 [DOI] [PubMed] [Google Scholar]
- 13. Vik S. B., Georgevich G., Capaldi R. A. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 1456–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Speck S. H., Neu C. A., Swanson M. S., Margoliash E. (1983) FEBS Lett. 164, 379–382 [DOI] [PubMed] [Google Scholar]
- 15. Lutter M., Fang M., Luo X., Nishijima M., Xie X., Wang X. (2000) Nat. Cell Biol. 2, 754–761 [DOI] [PubMed] [Google Scholar]
- 16. Gonzalvez F., Pariselli F., Dupaigne P., Budihardgo I., Lutter M., Antonsson B., Diolez P., Manon S., Martinou J-C., Goubern M., Wang X., Bernard S., Petit P. X. (2010) PLoS One 5, e9342 [DOI] [PubMed] [Google Scholar]
- 17. Gonzalvez F., Gottlieb E. (2007) Apoptosis 12, 877–885 [DOI] [PubMed] [Google Scholar]
- 18. Uren R. T., Dewson G., Bonzon C., Lithgow T., Newmeyer D. D., Kluck R. M. (2005) J. Biol. Chem. 280, 2266–2274 [DOI] [PubMed] [Google Scholar]
- 19. Kuwana T., Mackey M. R., Perkins G., Ellisman M. H., Latterich M., Schneiter R., Green D. R., Newmeyer D. D. (2002) Cell 111, 331–342 [DOI] [PubMed] [Google Scholar]
- 20. Esposti M. D. (2002) Cell Death Differ. 9, 234–236 [DOI] [PubMed] [Google Scholar]
- 21. Terrones O., Antonsson B., Yamaguchi H., Wang H. G., Liu J., Lee R. M., Herrmann A., Basañez G. (2004) J. Biol. Chem. 279, 30081–30091 [DOI] [PubMed] [Google Scholar]
- 22. Morin C., Zini R., Tillement J. P. (2003) Biochem. Biophys. Res. Commun. 307, 477–482 [DOI] [PubMed] [Google Scholar]
- 23. Lesnefsky E. J., Slabe T. J., Stoll M. S., Minkler P. E., Hoppel C. L. (2001) Am. J. Physiol. Heart Circ. Physiol. 280, H2770–H2778 [DOI] [PubMed] [Google Scholar]
- 24. Kirkland R. A., Adibhatla R. M., Hatcher J. F., Franklin J. L. (2002) Neuroscience 115, 587–602 [DOI] [PubMed] [Google Scholar]
- 25. Kim T. H., Zhao Y., Ding W. X., Shin J. N., He X., Seo Y. W., Chen J., Rabinowich H., Amoscato A. A., Yin X. M. (2004) Mol. Biol. Cell 15, 3061–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cristea I. M., Degli Esposti M. (2004) Chem. Phys. Lipids 129, 133–160 [DOI] [PubMed] [Google Scholar]
- 27. Epand R. F., Martinou J. C., Montessuit S., Epand R. M. (2003) Biochemistry 42, 14576–14582 [DOI] [PubMed] [Google Scholar]
- 28. Sorice M., Circella A., Cristea I. M., Garofalo T., Di Renzo L., Alessandri C., Valesini G., Esposti M. D. (2004) Cell Death Differ. 11, 1133–1145 [DOI] [PubMed] [Google Scholar]
- 29. Vila A., Korytowski W., Girotti A. W. (2001) Biochemistry 40, 14715–14726 [DOI] [PubMed] [Google Scholar]
- 30. Vila A., Levchenko V. V., Korytowski W., Girotti A. W. (2004) Biochemistry 43, 12592–12605 [DOI] [PubMed] [Google Scholar]
- 31. Kriska T., Levchenko V. V., Korytowski W., Atshaves B. P., Schroeder F., Girotti A. W. (2006) J. Biol. Chem. 281, 23643–23651 [DOI] [PubMed] [Google Scholar]
- 32. Girotti A. W. (2008) Free Radic. Biol. Med. 44, 956–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korytowski W., Niziolek M., Girotti A. W. (2005) Anal. Biochem. 343, 136–142 [DOI] [PubMed] [Google Scholar]
- 34. Lesnefsky E. J., Stoll M. S., Minkler P. E., Hoppel C. L. (2000) Anal. Biochem. 285, 246–254 [DOI] [PubMed] [Google Scholar]
- 35. Korytowski W., Geiger P. G., Girotti A. W. (1999) Methods Enzymol. 300, 23–33 [DOI] [PubMed] [Google Scholar]
- 36. Jiang Y. J., Lu B., Xu F. Y., Gartshore J., Taylor W. A., Halayko A. J., Gonzalez F. J., Takasaki J., Choy P. C., Hatch G. M. (2004) J. Lipid Res. 45, 244–252 [DOI] [PubMed] [Google Scholar]
- 37. Kriska T., Girotti A. W. (2004) Anal. Biochem. 327, 97–106 [DOI] [PubMed] [Google Scholar]
- 38. Girotti A. W., Korytowski W. (2000) Methods Enzymol. 319, 85–100 [DOI] [PubMed] [Google Scholar]
- 39. Itoh Y. H., Itoh T., Kaneko H. (1986) Anal. Biochem. 154, 200–204 [DOI] [PubMed] [Google Scholar]
- 40. Gross A., Yin X. M., Wang K., Wei M. C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S. J. (1999) J. Biol. Chem. 274, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 41. Montessuit S., Mazzei G., Magnenat E., Antonsson B. (1999) Protein Expr. Purif. 15, 202–206 [DOI] [PubMed] [Google Scholar]
- 42. Terrones O., Etxebarria A., Landajuela A., Landeta O., Antonsson B., Basañez G. (2008) J. Biol. Chem. 283, 7790–7803 [DOI] [PubMed] [Google Scholar]
- 43. Keberle H. (1964) Ann. N.Y. Acad. Sci. 119, 758–768 [DOI] [PubMed] [Google Scholar]
- 44. Ellens H., Bentz J., Szoka F. C. (1985) Biochemistry 24, 3099–3106 [DOI] [PubMed] [Google Scholar]
- 45. Epand R. F., Martinou J. C., Fornallaz-Mulhauser M., Hughes D. W., Epand R. M. (2002) J. Biol. Chem. 277, 32632–32639 [DOI] [PubMed] [Google Scholar]
- 46. Zhang M., Mileykovskaya E., Dowhan W. (2005) J. Biol. Chem. 280, 29403–29408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 48. Marí M., Colell A., Morales A., Caballero F., Moles A., Fernández A., Terrones O., Basañez G., Antonsson B., García-Ruiz C., Fernández-Checa J. C. (2008) Gastroenterology 134, 1507–1520 [DOI] [PubMed] [Google Scholar]
- 49. Turrens J. F., Alexandre A., Lehninger A. L. (1985) Arch. Biochem. Biophys. 237, 408–414 [DOI] [PubMed] [Google Scholar]
- 50. Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., Andrews D. W. (2008) Cell 135, 1074–1084 [DOI] [PubMed] [Google Scholar]
- 51. Rytömaa M., Mustonen P., Kinnunen P. K. (1992) J. Biol. Chem. 267, 22243–22248 [PubMed] [Google Scholar]
- 52. Rytömaa M., Kinnunen P. K. (1995) J. Biol. Chem. 270, 3197–3202 [DOI] [PubMed] [Google Scholar]
- 53. Kriska T., Korytowski W., Girotti A. W. (2005) Arch. Biochem. Biophys. 433, 435–446 [DOI] [PubMed] [Google Scholar]
- 54. Kagan V. E., Tyurin V. A., Jiang J., Tyurina Y. Y., Ritov V. B., Amoscato A. A., Osipov A. N., Belikova N. A., Kapralov A. A., Kini V., Vlasova I. I., Zhao Q., Zou M., Di P., Svistunenko D. A., Kurnikov I. V., Borisenko G. G. (2005) Nat. Chem. Biol. 1, 223–232 [DOI] [PubMed] [Google Scholar]
- 55. Belikova N. A., Vladimirov Y. A., Osipov A. N., Kapralov A. A., Tyurin V. A., Potapovich M. V., Basova L. V., Peterson J., Kurnikov I. V., Kagan V. E. (2006) Biochemistry 45, 4998–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tyurin V. A., Tyurina Y. Y., Osipov A. N., Belikova N. A., Basova L. V., Kapralov A. A., Bayir H., Kagan V. E. (2007) Cell Death Differ. 14, 872–875 [DOI] [PubMed] [Google Scholar]
- 57. Iverson S. L., Enoksson M., Gogvadze V., Ott M., Orrenius S. (2004) J. Biol. Chem. 279, 1100–1107 [DOI] [PubMed] [Google Scholar]
- 58. Polcic P., Su X., Fowlkes J., Blachly-Dyson E., Dowhan W., Forte M. (2005) Cell Death Differ. 12, 310–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lucken-Ardjomande S., Montessuit S., Martinou J. C. (2008) Cell Death Differ. 15, 929–937 [DOI] [PubMed] [Google Scholar]
- 60. Gonzalvez F., Schug Z. T., Houtkooper R. H., MacKenzie E. D., Brooks D. G., Wanders R. J., Petit P. X., Vaz F. M., Gottlieb E. (2008) J. Cell Biol. 183, 681–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Van Kuijk F. J. G. M., Sevanian A., Handelman G. J., Dratz E. A. (1987) Trends Biochem. Sci. 12, 31–34 [Google Scholar]
- 62. Garcia Fernandez M., Troiano L., Moretti L., Nasi M., Pinti M., Salvioli S., Dobrucki J., Cossarizza A. (2002) Cell Growth Differ. 13, 449–455 [PubMed] [Google Scholar]
- 63. Kagan V. E., Gleiss B., Tyurina Y. Y., Tyurin V. A., Elenström-Magnusson C., Liu S. X., Serinkan F. B., Arroyo A., Chandra J., Orrenius S., Fadeel B. (2002) J. Immunol. 169, 487–499 [DOI] [PubMed] [Google Scholar]
- 64. McIntyre J. A., Wagenknecht D. R., Faulk W. P. (2003) Prog. Lipid Res. 42, 176–237 [DOI] [PubMed] [Google Scholar]
- 65. Gonzalvez F., Pariselli F., Jalmar O., Dupaigne P., Sureau F., Dellinger M., Hendrickson E. A., Bernard S., Petit P. X. (2010) PLoS One 5, e9342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thomson M. (2003) Bioessays 25, 252–258 [DOI] [PubMed] [Google Scholar]
- 67. Epand R. F., Schlattner U., Wallimann T., Lacombe M. L., Epand R. M. (2007) Biophys. J. 92, 126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mannella C. A., Pfeiffer D. R., Bradshaw P. C., Moraru I. I., Slepchenko B., Loew L. M., Hsieh C. E., Buttle K., Marko M. (2001) IUBMB Life 52, 93–100 [DOI] [PubMed] [Google Scholar]
- 69. Mannella C. A. (2006) Biochim. Biophys. Acta 1762, 140–147 [DOI] [PubMed] [Google Scholar]
- 70. Gallegos A. M., Atshaves B. P., Storey S. M., Starodub O., Petrescu A. D., Huang H., McIntosh A. L., Martin G. G., Chao H., Kier A. B., Schroeder F. (2001) Prog. Lipid Res. 40, 498–563 [DOI] [PubMed] [Google Scholar]
- 71. Esposti M. D., Erler J. T., Hickman J. A., Dive C. (2001) Mol. Cell. Biol. 21, 7268–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu J., Epand R. F., Durrant D., Grossman D., Chi N. W., Epand R. M., Lee R. M. (2008) Biochemistry 47, 4518–4529 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.