

Abstract
An intriguing mystery about tryptophan 2,3-dioxygenase is its hydrogen peroxide-triggered enzyme reactivation from the resting ferric oxidation state to the catalytically active ferrous form. In this study, we found that such an odd Fe(III) reduction by an oxidant depends on the presence of l-Trp, which ultimately serves as the reductant for the enzyme. In the peroxide reaction with tryptophan 2,3-dioxygenase, a previously unknown catalase-like activity was detected. A ferryl species (δ = 0.055 mm/s and ΔEQ = 1.755 mm/s) and a protein-based free radical (g = 2.0028 and 1.72 millitesla linewidth) were characterized by Mössbauer and EPR spectroscopy, respectively. This is the first compound ES-type of ferryl intermediate from a heme-based dioxygenase characterized by EPR and Mössbauer spectroscopy. Density functional theory calculations revealed the contribution of secondary ligand sphere to the spectroscopic properties of the ferryl species. In the presence of l-Trp, the reactivation was demonstrated by enzyme assays and by various spectroscopic techniques. A Trp-Trp dimer and a monooxygenated l-Trp were both observed as the enzyme reactivation by-products by mass spectrometry. Together, these results lead to the unraveling of an over 60-year old mystery of peroxide reactivation mechanism. These results may shed light on how a metalloenzyme maintains its catalytic activity in an oxidizing environment.
Keywords: Electron Paramagnetic Resonance (EPR), Enzyme Mechanisms, Heme, Metalloenzymes, Mossbauer Spectroscopy, Radicals, Spectroscopy, Activation
Introduction
Hemoproteins perform a wide range of biological functions, including oxygen transport, storage, electron transfer, monooxygenation, and reduction of dioxygen. However, they rarely express dioxygenase activity as their native biological function. Tryptophan 2,3-dioxygenase (TDO)3 is the first described exception (1–3). This enzyme employs a b-type ferrous heme prosthetic group to catalyze the oxidative cleavage of the indole ring of l-Trp, converting it to N-formylkynurenine (NFK) (Scheme 1). This is the first and rate-limiting step of the kynurenine pathway of l-Trp metabolism, which oxidizes over 99% of l-Trp in mammalian intracellular and extracellular pools (2, 4–9). The kynurenine pathway constitutes the major steps in biosynthesis of NAD, an essential redox cofactor in all living systems (5).
SCHEME 1.
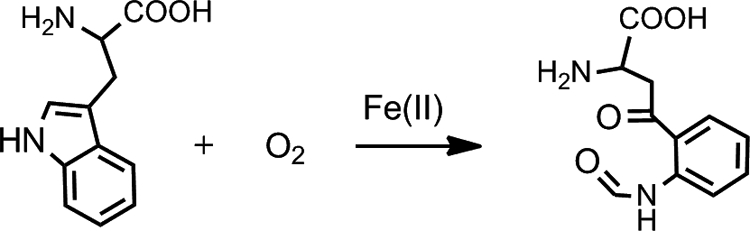
Chemical reaction catalyzed by TDO.
TDO is a hepatic enzyme first discovered in rat liver extracts in 1936 (1). An analogous enzyme, indoleamine 2,3-dioxygenase (IDO), was isolated 31 years later from tissues other than the liver (10). Although both enzymes catalyze the same reaction, TDO is highly substrate-specific with l-Trp, whereas IDO presents a more relaxed specificity. TDO is a homotetramer with a total mass of ∼134 kDa, whereas IDO is a monomeric protein. The two enzymes share only 14% sequence identity but conserve similar active site architectures (11–13). In addition to humans, TDO has also been found in other mammals, such as rats and mice, as well as in mosquitoes and bacteria (2, 5, 14–18). Recently, a potential heme-dependent dioxygenase enzyme superfamily has been proposed (19). Moreover, several other heme-based proteins, such as myoglobin and hemoglobin, express dioxygenase activities from their mutant proteins under certain circumstances (20, 21). In general, TDO has been considered as a prototypical member and model system for studying the chemistry of heme-based dioxygenases.
The ferrous heme in TDO is the catalytic center that binds and activates molecular oxygen. Like many other Fe(II)-dependent enzymes, TDO becomes auto-oxidized when its primary substrate is absent. In previous studies, hydrogen peroxide (H2O2) was implicated as an activator by the finding that the resting ferric TDO becomes active toward l-Trp after treatment with H2O2 (22). This was later confirmed by independent optical spectroscopic studies from various laboratories (14, 17, 23) and by the observations that the overall enzyme reactivation was inhibited by catalase and that the addition of peroxide relieved the reactivation inhibition (14). However, the mechanism by which ferric TDO is reduced to its ferrous form by reacting with H2O2 remains elusive after over 60 years.
Here, we provide unequivocal evidence in support of the formation of the ferrous form of the enzyme by reaction of ferric TDO with peroxide and l-Trp. A previously unknown two-phase enzyme reactivation mechanism is proposed based on the chemical identification of nearly all of the intermediates and products. In the first phase, the enzyme is oxidized by peroxide to generate a compound ES-type ferryl intermediate. In the second phase, the ferryl and the protein-based free radical intermediates are each reduced by l-Trp. We further hypothesize that the physiological significance of the peroxide reactivity is to allow for the reactivation of the enzyme in an oxidizing environment.
EXPERIMENTAL PROCEDURES
Reagents
l-Trp (99.5%) was purchased from Sigma. H216O2 (30%, v/v) was obtained from Fisher. The concentration of H2O2 was calculated based on the extinction coefficient of ϵ240 nm = 43.6 m−1 cm−1. H218O2 (2% v/v solution) and H218O were purchased from Icon Isotopes at 90.0 and 97.6% isotope enrichment, respectively. All experiments were performed in 50 mm Tris-HCl buffer, pH 7.4, unless otherwise specified. 57Fe (95% enrichment) was obtained from Science Engineering and Education Co. (Edina, MN).
Expression and Purification of Ferric TDO
The construction of the plasmid encoding full-length Cupriavidus metallidurans TDO (CmTDO) has been described elsewhere (13). The protein was purified by using a 100-ml HiLoad nickel-affinity column and a Superdex 200 size-exclusion column on an ÄKTA FPLC system as described in an earlier spectroscopic study of the enzyme (24). The optical absorption spectrum of the as-isolated TDO used in this work displays a 405:280 nm ratio of ∼1.4–1.5:1 (supplemental Fig. S1), corresponding to 60–65% heme occupancy based on the determination of protein concentration and iron content using inductively coupled plasma optical emission spectroscopy and EPR spin quantitation technique. The purified enzyme demonstrated a specific activity of 25 μmol/min/mg. The ferrous enzyme used as control samples in the UV-visible and Mössbauer experiments was obtained by dithionite reduction of ferric enzyme under anaerobic conditions.
Catalase-like Activity Assay
Oxygen production was measured in a sealed reaction chamber (3 ml) with an integrated oxygen electrode unit (Oxygraph System, Hansatech Instruments) at 25 °C. The oxygraph experiments were initiated in aerated buffer in the absence and presence of l-Trp. The production of oxygen was monitored as a function of time. The kinetic data were fitted to Equation 1,
where v is the steady state velocity; [E] is the concentration of TDO; [S] is the concentration of H2O2; kcat is the apparent catalytic turnover constant; Km is the Michaelis-Menten constant; and n is the Hill coefficient or cooperativity index. TDO is known to exhibit a homotropic cooperativity during l-Trp binding (25).
UV-visible Spectroscopy
All TDO samples were prepared in 50 mm Tris-HCl, pH 7.4. The absorption spectra were obtained by using an Agilent 8453 UV-visible spectrophotometer at room temperature. The enzyme reactivation by peroxide was performed under anaerobic conditions using a homemade long arm sealed cuvette (1 ml). All the reagents had been degassed and purged with argon prior to the experiments. l-Trp stock solution was prepared in the reaction buffer in a warm bath (80 °C). Both l-Trp and H2O2 were freshly prepared in 50 mm Tris-HCl buffer, pH 7.4, that had been previously degassed and purged with argon. Unless otherwise stated, the final concentration of TDO in the reaction system was 5 μm. The enzyme was made anaerobic in a vial containing concentrated ferric TDO by repeated evacuation and refilling with argon. A gas-tight microsyringe was used for addition of various amounts of argon-saturated oxygen-free H2O2 to the enzyme-substrate complex. NFK concentration was determined by the known extinction coefficient at 321 nm (ϵ321 nm = 3,150 m−1 cm−1) (26). The apparent rates of the dioxygenation reaction were determined from the initial velocity of the NFK formation. The aerobic reaction of H2O2 with TDO in presence of l-Trp was performed similarly with exclusion of the steps associated with the oxygen removal.
To test whether the ferric form of TDO can react with H2O2 and directly produce NFK through an unknown shunt pathway, we performed the enzyme reactivation in the presence of carbon monoxide (CO). In these experiments, CO was introduced to the reaction system either prior to the reaction or in the middle of the reaction (for comparison) by direct bubbling of CO gas into the reaction solution containing TDO, H2O2, and l-Trp.
Mass Spectrometry (MS)
All reagents were prepared using anaerobic 50 mm Tris-HCl buffer, pH 7.4, which was bubbled and purged with argon prior to the TDO reaction with peroxide experiments. The reactions were performed on ice with stirring using septum-sealed reaction vials. Ferric TDO (100 μm) was allowed to react with H2O2 in the presence of l-Trp (5 mm), in which either H216O2 or H218O2 was added to a total concentration of 4 mm through a stepwise addition. After reacting for 20 min, TDO was removed from the reaction system using Centriprep-10 at 3,000 × g for 10 min, and the filtrate was collected for electrospray ionization-mass spectrometry (ESI-MS) analysis.
ESI spectrometric analyses were conducted on a Waters ESI-Q-TOF micro mass spectrometer equipped with a Waters alliance 2695 HPLC system (Milford, MA) in a positive mode. Samples were analyzed through either a direct infusion or through HPLC separation on a Waters 2695 alliance HPLC system before MS analysis. The TDO reaction samples were mixed with 50% acetonitrile in water containing 0.1% formic acid before analysis. In LC-MS analysis, collision energy was set to 30 eV. HPLC separation was achieved on a Restek Allure C18 column (100 × 2-mm inner diameter, 3 μm). Mobile phase A was composed of water and 0.1% formic acid. Mobile phase B was composed of acetonitrile and 0.1% formic acid. The elution gradients were performed as follows: starting at 100% A for 5 min; falling to 0% A over 10 min; staying at 0% A for 15 min; and then rising to 100% A over 20 min at a constant flow rate of 200 μl/min. MassLynx 4.1 software was used for instrument control and data acquisition.
Solvent Exchange of the Dioxygenation Product
16O-NFK was prepared according to the enzyme-based procedures described above. A concentrated sample of 16O-NFK was dissolved in H218O (75% 18O) to exchange with solvent on ice for 20 min prior to the ESI-MS analysis. In a parallel experiment, a concentrated 16O-NFK sample was first dissolved in H218O (75% 18O) for 20 min and then concentrated again by rapidly evaporating the solvent under a vacuum. Finally, it was re-diluted with H216O. The final ratio of H216O/H218O was determined to be 60:1. This experiment was intended to examine if the solvent exchange would be fully reversible.
Electron Paramagnetic Resonance (EPR) Spectroscopy
TDO EPR samples were made in reaction vials containing 50 mm Tris-HCl buffer, pH 7.4, with 10% glycerol, transferred to EPR tubes, and quickly frozen in cold isopentane (−140 °C) or liquid nitrogen after the desired reaction time. The 12- and 30-s samples were made in EPR tubes by using a System 1000 rapid-freeze quenching apparatus (Update Instruments Inc.) with a cold isopentane bath (−140 °C). Typically, 10 EPR samples with 0.15–0.50 mm TDO were made in each set of experiments with 1–6 eq of peroxide in different experiments. Multiple sets of the EPR experiments were conducted to optimize the enzyme/peroxide ratio for production of reactive intermediates while minimizing loss of the heme cofactor. X-band EPR first derivative spectra were recorded in perpendicular mode on a Bruker EMX spectrometer at 100-kHz modulation frequency using a 4119HS resonator. The EPR measurement temperature was maintained with an ESR910 liquid helium cryostat and an ITC503 temperature controller. A calibrated frequency meter was used to aid the g value determination. Spin concentration was determined by double integration of the EPR signals obtained under low microwave power conditions and comparing with that of a copper standard (0.5 mm CuSO4, 5 mm EDTA) obtained under identical conditions.
The relaxation properties of the free radical at different temperatures were analyzed from EPR spectra obtained by varying microwave powers in the range 0.0002–200 milliwatts. The values of half-saturation parameters (P½) were obtained by fitting the data according to Equation 2.
where I is the EPR signal amplitude, b is the inhomogeneous broadening factor, and P is microwave power.
Mössbauer Spectroscopy
The 57Fe-enriched protein was obtained by expressing TDO in Escherichia coli using 57Fe-enriched culture medium as described previously (24). The Mössbauer samples were prepared from the as-isolated 57Fe(III)-TDO and frozen in liquid nitrogen. In the Fe(II) formation Mössbauer experiments, a final concentration of 1.0 mm (heme concentration) 57Fe-TDO was used. To generate the high valent Fe(IV) intermediate, 1.6 mm (heme concentration) 57Fe-TDO was used to react with 6 eq of H2O2 (9.6 mm) and frozen in liquid nitrogen at 20 or 50 s after reaction. Mössbauer spectra were recorded on a constant acceleration instrument with an available temperature range of 1.5 to 200 K. Isomer shifts are reported relative to Fe(IV) metal at 298 K. Least square fitting of the spectra was performed with the WMOSS software package (WEB Research, Edina, MN). The low temperature Mössbauer spectra of resting TDO were fit with the standard spin Hamiltonian (Equation 3),
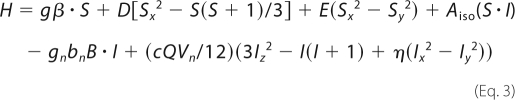 |
Computational Modeling
The hybrid functional B3LYP (27) with a Wachter's basis (62111111/3311111/3111) for Fe(IV) (28), 6–311G* for all the other heavy atoms, and 6–31G* for hydrogens was used to predict the values of Mössbauer quadrupole splitting and isomer shift, the same approach used in the previous work for various iron-containing proteins and models (see supplemental material for more details) (29, 30). In all the investigated models, the heme group is represented by a porphyrin with original β substituent replaced by methyl groups, and the axial histidine group is truncated to be 5-methylimidazole. Geometries of all the structural models investigated in this work were optimized (see supplemental Table S1–S14 for the optimized coordinates) with the terminal atoms fixed at the x-ray crystal structure (Protein Data Bank entry 2NW7; see Ref. 12) positions to mimic the protein environment effect, using the density functional theory method BPW91 with the above basis set (31, 32), which is the same approach used previously to investigate other oxyferryl species (29, 30).
RESULTS
Reaction of Oxidized TDO and H2O2 in the Absence of l-Trp
The reaction of ferric TDO with H2O2 was examined with an oxygen electrode in a stirred cell at 25 °C. Fig. 1A shows that the addition of 50 eq of H2O2 to the oxidized protein resulted in an immediate increase in the oxygen concentration of the reaction chamber. Another addition of H2O2 in the same amount led to a similar increase in the oxygen concentration, indicating that O2 generation is reproducible (Fig. 1A). When ferric TDO was added to the buffer containing H2O2, similar O2 production was observed (Fig. 1B), indicating no dependence on the order of additions. In contrast, addition of either the protein or peroxide alone, as shown in Fig. 1, did not produce O2. These results demonstrate that O2 is produced from H2O2 and lead to the conclusion that Fe(III)-TDO possesses a catalase-like activity with H2O2 in the absence of l-Trp. The kcat, Km, and kcat/Km values of the TDO catalase-like activity determined from steady state analysis according to Equation 1 are 13 ± 2 s−1, 16 ± 3 mm, and 850 ± 65 m−1 s−1, respectively (supplemental Fig. S2). The kinetic data show no cooperative behavior (n = 1) during O2 production from H2O2.
FIGURE 1.

A, H2O2 decomposition and O2 production mediated by Fe(III)-TDO in a stirred O2 electrode cell in response to discrete additions of H2O2. B, reaction initiated by TDO. Arrows indicate the time points in which ferric TDO (5 μm) and H2O2 (250 μm or 1 mm in A and 1 mm in B) were added to the reaction cell.
At 4.2 K, the as-isolated 57Fe-TDO shows a six-line magnetic pattern in Mössbauer spectrum (Fig. 2). The simulation overlaid on the experimental data (Fig. 2, solid line) is calculated for an S = 5/2 iron site with δ = 0.42 mm/s, ΔEQ = 1.53 mm/s, D = 13 cm−1, E/D = 0.01, and Aiso = 195 kG (Fig. 2B). These values are indicative of a high spin ferric heme (24). Another sample of this protein solution was treated with 6 eq of H2O2 (20 s reaction plus 10 s frozen time) prior to the spectroscopic characterization (Fig. 2A). The Mössbauer spectrum of this sample is composed of four species. One species is recognized as the high spin ferric TDO that accounts for 25% of the iron in the sample. Fig. 2C shows the difference spectrum of A − 0.25B. This difference spectrum is composed of three overlapping doublets as indicated on the figure. The fit to the three doublets (Fig. 2C, solid lines) gives Fe(IV) parameters and relative amounts of the following: 1) δ = 0.055 mm/s, ΔEQ = 1.755 mm/s, 33%; 2) δ = 0.350 mm/s, ΔEQ = 0.703 mm/s, 17%; 3) δ = 0.585 mm/s, ΔEQ = 1.5 mm/s, 25%. Species 1 is assigned to an S = 1 Fe(IV) heme, tentatively in the Fe(IV)=O form. The parameter ranges of known S = 1 Fe(IV)-oxo heme species are δ = 0–0.15 and ΔEQ = 1.0–1.6 mm/s. The other two species appear to be degradation products of the reaction with peroxide. Species 2 has parameters in the range of high spin ferric hemes, but the diamagnetic doublet indicates that the hemes are forming μ-oxo bridges (33). Species 3 is typical of nondescript Fe(III) formation (34), presumably due to loss of the iron ion from heme. The same difference spectrum was unchanged in character when recorded at 100 K. Thus, the addition of 6 eq of H2O2, all at once, resulted in a 42% loss of the heme and/or 57Fe from TDO, 25% remained unchanged, and 33% of the iron formed an iron(IV)-oxo heme species. The loss of heme is consistent with the observed tendency of the protein to lose the b-type heme cofactor. Similar results were observed for two repeats of the 57Fe-enriched TDO with H2O2 and observation with Mössbauer spectroscopy. At longer reaction times (50-s reaction plus 10-s frozen time), species 1 was decreased from 33 to 20%.
FIGURE 2.

Mössbauer spectra of 57Fe-TDO. A, reaction of H2O2 (9.6 mm) with TDO (1.6 mm); B, TDO protein in resting state prior to the reaction with H2O2, and C, difference spectrum of A − 0.25B. All spectra were recorded at 4.2 K in an applied field of 45 mT parallel to the direction of the beam of γ rays. The solid lines are least squares fits with parameters given in the text.
The reaction of TDO with H2O2 was also studied by EPR spectroscopy. The as-isolated ferric TDO displays a nearly axial EPR signal at g = 6 (supplemental Fig. S3) and a weak resonance at g = 2 (Fig. 3), typical of a high spin ferric ion in a heme environment. Fig. 3 shows that this ferric EPR signal decreases in intensity upon addition of H2O2, concomitantly with the formation of a g = 2.0028 free radical signal. The amplitude of the EPR signal for the radical is shown on a reduced scale for comparison with the ferric heme signal. At 30 s, the radical species has a spin concentration of 18% of the initial ferric heme concentration. A plot of the concentrations of the high spin heme and radical species as a function of time is present in supplemental Fig. S4. At 12 s, the free radical species has a spin concentration of 40% of the initial iron concentration. The sharp EPR signal of the 12-s sample at g = 2.0028, shown in supplemental Fig. S5, is omitted from Fig. 3 for clarity. The EPR-active radical intermediate is present in addition to the postulated Fe(IV)-oxo species characterized by Mössbauer spectroscopy.
FIGURE 3.

Change in the EPR signals of TDO during the room temperature reaction of TDO (150 μm) with H2O2 (900 μm). The times at which the various EPR samples were frozen after addition of H2O2 are listed on the figure. The g = 2 signal is 20% of the original experimental data. EPR parameters for obtaining spectra are as follows: temperature, 10 K; microwave frequency, 9.44 GHz; microwave power, 1 milliwatt; and modulation amplitude, 0.8 mT.
At later reaction times, the radical species decays, and the high spin ferric EPR signal gradually increases in intensity. At 10 min, the concentration of the high spin heme species is about 70% of its initial concentration due to 30% loss of the heme under these conditions. The detected loss of heme in the EPR samples is lower than the ratio obtained by Mössbauer spectroscopy, presumably due to the low concentration of H2O2 used in the EPR experiments. During the reaction time of Fig. 3, the shape of the high spin heme EPR signal subtly changed to a more axial species (supplemental Fig. S3), which suggests that the electronic environment of the heme changes during the reaction with peroxide. In the 12-s sample, the ferric heme signal at g = 6 is about 25% of the initial intensity prior to the reaction with H2O2 (supplemental Fig. S4). This observation suggests that although there are “inequivalent” hemes in the enzyme, they all react with H2O2.
The peak-to-peak line width of the radical signal is 1.72 mT (supplemental Fig. S5), which is too large for a peroxide-based free radical (<1 mT)) but typical for a protein-derived aromatic radical (35, 36). The microwave power saturation behavior of the free radical signal at g = 2.0028 was measured at 10 and 100 K, respectively (supplemental Fig. S5). The fit to the curves using Equation 2 led to P½ values of 0.11 and 1.16 milliwatts for 10 and 100 K, respectively. These values suggest a weak interaction of the protein radical with the Fe(IV) ion. At approximately the same reaction time as the Mössbauer sample, the spin concentration of the protein radical is found to be comparable with the concentration of the Fe(IV)=O heme species. Together, these results suggest the formation of an intermediate composed of an Fe(IV)=O heme in close proximity to the free radical, similar to the so-called compound ES description based on the initial characterization from cytochrome c peroxidase (37, 38). Compound ES of cytochrome c peroxidase is a semi-stable enzyme intermediate that contains an Fe(IV)=O heme and a Trp radical (39).
Reaction of Oxidized TDO and H2O2 with l-Trp
The as-isolated TDO exhibits visible absorbance characteristics of a histidine-ligated ferric heme protein with a Soret band at 405 nm (supplemental Fig. S1). In the presence of l-Trp, the Soret band shifts to 406 nm (Fig. 4). The intensity of this 406-nm band decreases, and new features at 432 and 321 nm develop during the addition of H2O2 to a reaction mixture containing ferric TDO and excess (1,000 eq, 5 mm) l-Trp (Fig. 4). The 321-nm spectral feature resembles the optical data for the dioxygenation reaction of ferrous TDO using O2 as the oxidant, and the absorbance in the range of 310–330 nm has previously been used to measure the formation of NFK (2, 40). Hence, the absorption at 321 nm is tentatively assigned to NFK production. When H2O2 was incubated with l-Trp in the absence of TDO, the development of the 321-nm chromophore did not occur, indicating that NFK formation is an enzymatic process. As shown below, the NFK formation is due to generation of the ferrous form of enzyme. When the peroxide reaction was carried out in the presence of hydroxyurea, a known scavenger of protein-based free radicals, the NFK production in the anaerobic reaction with peroxide is significantly inhibited (Fig. 5), suggesting the protein radical detected by EPR spectroscopy is indeed an intermediate in enzyme reactivation. In contrast, NFK production in the normal catalytic cycle of Fe(II)-TDO and O2 is not affected by the presence of hydroxyurea (supplemental Fig. S6). The inset (top panel) in Fig. 4C shows a slightly less than 1:2 ratio of [NFK]/[H2O2]stoichiometry. When the peroxide reaction was carried out under aerobic conditions, the amount of NFK formed was not associated with the concentration of peroxide because the reaction mediated by the ferrous enzyme had multiple sources of O2 (supplemental Fig. S7).
FIGURE 4.

Optical spectra of TDO. A, Soret bands of ferric and ferrous CmTDO (5 μm) in the presence of l-Trp (5 mm) are observed at 406 and 432 nm, respectively. The Soret band of the ferric sample splits into two parts 30 s after addition of H2O2 (30 μm). B, ferric TDO (black), after sequential addition of l-Trp (red), CO (green), and H2O2 (blue). The red and green traces are nearly identical to each other. C, optical spectra of TDO and l-Trp taken from the sequential additions of equal amount of H2O2 (see text for experiment details). The inset is the calculated NFK concentration as a function of the added H2O2 under anaerobic conditions. The concentration of H2O2 was determined using ϵ240 nm = 43.6 m−1 cm−1. NFK concentration was determined using ϵ321 nm = 3150 m−1 cm−1 after subtraction of the initial spectrum.
FIGURE 5.
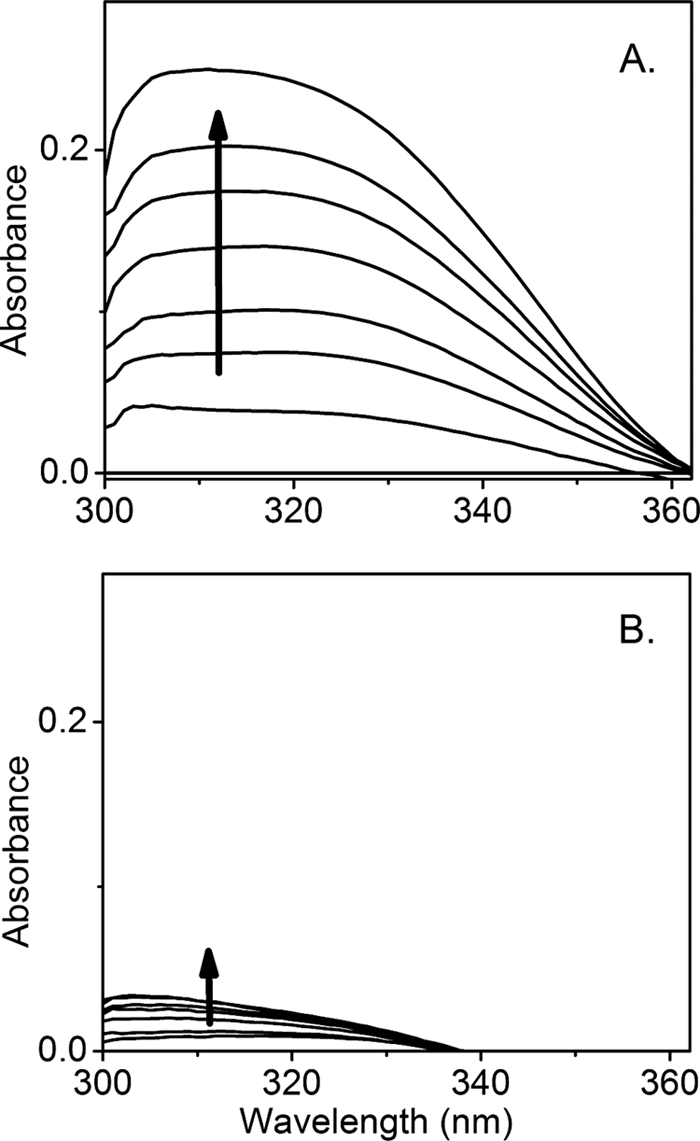
Formation of NFK as a result of the peroxide reaction with ferric TDO (5 μm) and l-Trp (5 mm). The spectra were taken from the anaerobic titration of 35 eq of H2O2 (total 175 μm) in the absence (A) and presence (B) of hydroxyurea (10 mm). Each trace was obtained after the reaction was complete and subtracted against a spectrum of ferric TDO at the same concentration (5 μm).
The fully reduced TDO, generated by chemical reduction by dithionite, presents a Soret band at 432 nm when l-Trp is bound (Fig. 4A). During the course of the reaction of ferric TDO with H2O2 and l-Trp, the Soret band decreases, whereas an additional spectral shoulder feature emerges at 432 nm (Fig. 4). The 432-nm chromophore matches the Soret band of ferrous TDO complex with l-Trp, suggesting the formation of the ferrous TDO from a fraction of the ferric form of the enzyme. To confirm the formation of Fe(II) heme, CO was introduced to stabilize the presumed ferrous species in a separate test. In our previous study of ferrous TDO, the CO adduct exhibits a Soret band at 421 nm in the presence of l-Trp, and it is stable in the presence of O2 (24). A similar 421-nm peak has also been reported for the ferrous-CO adduct of human TDO (41). When CO was bubbled into a solution containing ferric TDO and l-Trp, the addition of CO does not cause an observable shift of the Soret band. However, upon further addition of H2O2, a 421-nm band corresponding to the ferrous-CO adduct of TDO is generated (Fig. 4B).
The formation of the active Fe(II) form of TDO from the ferric state by peroxide was further verified by Mössbauer spectroscopy. Fig. 6A shows the Mössbauer spectrum of substrate-bound ferric TDO before addition of CO and peroxide. CO gas was bubbled through an anaerobic sample of ferric TDO with l-Trp for 5 min, after which 2 eq of H2O2 were added, and the sample was immediately frozen in liquid nitrogen. The spectrum of the H2O2-treated sample is shown in Fig. 6B. The difference spectrum (Fig. 6C), generated by subtracting 50% of the unreacted enzyme-substrate complex, shows a doublet indicating a diamagnetic species. The Mössbauer parameters of this species are the same as those of ferrous-CO adduct characterized in our recent work (24).
FIGURE 6.

Mössbauer spectra of 57Fe-TDO. A, ferric TDO (1 mm) with 50 eq of l-Trp; B, after treatment with CO and H2O2 (2 mm); and C, difference spectrum of B − 0.5A. All spectra were recorded at 4.2 K in an applied field of 45 mT parallel to the direction of the beam of γ rays. The solid line is the least squares fits with parameters given in the text.
It is known that CO reacts with the ferrous heme of TDO and that the ferrous-CO complex is catalytically inactive (41). Like O2, CO does not bind to ferric hemoproteins (42). With this knowledge in hand, CO was again employed as a probe to test if the ferrous heme generated by addition of H2O2 is catalytically active. Fig. 7 shows that the NFK production is inhibited by CO, indicating the dioxygenation product is generated by the Fe(II) heme. When the reaction was initiated by addition of ferric TDO, the initial rate of the CO-treated reaction system dropped by ∼6.5-fold compared with that of the untreated sample under the same conditions (Fig. 7A). In a separate set of experiments, CO was introduced into the system 15 s after the reaction was initiated. An inhibition of the NFK formation was observed (Fig. 7B), demonstrating depletion of catalytically active Fe(II) enzyme by forming a stable but inactive Fe(II)-CO complex. Before CO was bubbled into the system, the formation of Fe(II) heme was visualized at 432 nm. After a small lag phase upon addition of CO, a sharp decrease of [Fe(II)] was observed (Fig. 7C). The absorbance at 421 nm was concomitantly increased due to the formation of the ferrous-CO adduct (Fig. 7B). These experiments further show that the formation of NFK is catalyzed by ferrous TDO, and an enzyme reactivation mechanism must exist during the reaction with peroxide under appropriate conditions discussed later.
FIGURE 7.

Effect of CO on the enzyme reactivation and formation of NFK. A, preincubation of CO into a solution of l-Trp (5 mm) and H2O2 (10 μm) (solid trace) compared with that in the absence of CO (dotted trace). The reaction was initiated by addition of ferric TDO (1 μm). B, CO bubbling 15 s after the reaction, which was initiated by addition of TDO (solid trace). The dotted trace shows a control experiment in the absence of CO. C, change of the Soret absorbance as a function of reaction time monitored at 421 nm (corresponding to Fe(II)-CO adduct) and 432 nm (ferrous heme in TDO), respectively. CO gas was bubbled into the solution after 50 s of the reaction. The experiments were carried out under aerobic conditions.
Source of Oxygen in NFK
Mass spectrometric analyses of the reaction of ferric TDO and l-Trp with H216O2 and H218O2 were conducted. The enzyme was removed by filtration after 5 min of reaction with peroxide and prior to the mass spectrometry analysis as described under “Experimental Procedures.” Fig. 8A shows that the substrate l-Trp presents an ion of mass-to-charge ratio (m/z) 205, corresponding to the anticipated [M + H]+ form. The ion at m/z 243 in this control sample is tentatively assigned to the dimeric form of Tris (Mr 242). A new major ion at m/z 237 is present in the TDO reaction mixture with H216O2 as the oxidant (Fig. 8B). This new ion is absent in the control sample where TDO was omitted (Fig. 8A). The 32-dalton mass shift of the m/z 237 ion compared with the substrate is consistent with production of NFK in which two oxygen atoms have been incorporated into l-Trp. In addition, a peak at m/z 409 was observed in the reaction and is tentatively assigned to an l-Trp dimer (supplemental Fig. S8). It was undetectable when either TDO or H2O2 was absent or in the regular dioxygen reaction of ferrous TDO and O2. The m/z 409 ion was invariant when 18O-enriched peroxide was employed in the reaction.
FIGURE 8.

ESI-MS characterization of l-Trp (A), the reaction product of l-Trp and H216O2 catalyzed by ferric TDO (B), and the TDO reaction using isotope-labeled H218O2 as the oxygen donor (C). A 16O-NFK sample, generated from the TDO reactivation process, was dissolved in an 18O-enriched water (16O/18O ratio of 3:5) after concentrating the reaction solution and removing the enzyme by filtration (D). A copy of sample D was further diluted by 16O water to reach a final 16O/18O ratio of 60:1 (1.6% 18O) (E). TDO reaction was carried out in H218O-based Tris-HCl buffer (F).
When H218O2 was used instead of H216O2, an ion of m/z 241 was detected (Fig. 8C). This ion is consistent with the incorporation of two atoms of 18O into the product from H218O2. An ion at m/z 237 was also observed due to the presence of the unlabeled 16O fraction of the peroxide reagent (90% 18O enrichment). Although the experiment was carried out under O2-free conditions, oxygen leak could have occurred during the process of filtering the enzyme out of the reaction system, thus generating a small fraction of NFK containing unlabeled oxygen. The amplitude of m/z 237 ion is estimated to correspond to less than 20% of the sum of the total product ions. In Fig. 8C a significant ion of m/z 239 is also present, which corresponds to the incorporation of one atom of 16O and one atom of 18O into l-Trp. To investigate the cause of the 16O/18O scrambling, a sample of 16O-NFK was prepared that presents the ion of m/z 237 of Fig. 8B. The 16O-NFK sample was then re-dissolved in a solvent containing 18O-enriched water (78.1 atom %) with H216O/H218O ratio of 1:4. The final 16O/18O ratio was ∼3:5. Fig. 8D shows that a new ion at m/z 239 is generated from the nonlabeled NFK in a post-enzymatic reaction process in the presence of 18O-enriched water, which indicates that one 18O from solvent is exchanged into 16O-NFK.
The 18O water-exchanged sample containing both the m/z 237 and 239 ions was then added to 16O water to reach a H216O/H218O ratio of 60:1 (1.6% 18O atom) and was analyzed again by ESI-MS after a few minutes of solvent exchange. Fig. 8E shows that the m/z 239 peak is substantially reduced, whereas the original m/z 237 ion becomes the predominating species, which indicates a reversible solvent exchange for NFK. No m/z 241 peak was observed in either of the exchange experiments, indicating that one and only one oxygen site in the product NFK is solvent-exchangeable.
A parallel set of experiments was performed using unlabeled H2O2 as the oxidant in 18O-based solvent. The m/z 237 and 239 ions are present in the H216O2/H218O sample (Fig. 8F), consistent with solvent exchange after the catalytic reaction to form the m/z 239 ion. Collectively, these results suggest that the two new oxygen atoms incorporated into NFK are both derived from H2O2, and one of the oxygen atoms is readily exchangeable with solvent.
Identification of a Minor but Reproducible Mono-oxygenated Tryptophan Product
In addition to the dioxygenation product NFK, two ions at m/z 220 and 221 are observed in the reaction of ferric TDO + l-Trp with unlabeled H2O2 (Fig. 8B). As described below, the m/z 220 ion is shown in the MS/MS experiments to be a fragment of NFK. The m/z 221 ion is, however, 16 daltons greater than l-Trp, which is consistent with the insertion of one oxygen atom into l-Trp. In the isotope-labeling experiments using H218O2 as the oxidant, a corresponding m/z 223 ion, 18 daltons greater than l-Trp, is observed at the expense of the m/z 221 ion (Fig. 8C). The m/z 221 ion was not observed in the control mass spectrometry measurements of normal turnover using ferrous TDO, l-Trp, and O2.
Fig. 9 shows the results of an LC-MS experiment using H216O2 with H218O solvent to further characterize the two ions at m/z 220 and 221. The substrate (m/z 205) shows the greatest retention time of 16.6 min. The m/z 237 and 239 ions with a retention time of 6.1 min are due to the dioxygenation product NFK (Fig. 9D). The former has two 16O atoms inserted, and the latter has a 16O and an 18O atom inserted into the substrate. The m/z 243 ion with a 2.9-min retention time is attributed to the Tris-HCl buffer used in the reaction, and the m/z 214 ion is a fragment of the Tris-HCl buffer (Fig. 9B). The ions at m/z 221 and 223 have a retention time of 5.3 min (Fig. 9C). It should be noted that the much longer solvent exchange took place in the LC-MS experiments. The observation of the m/z 223 ion in H218O indicates that the presumed monooxygenated by-product is solvent-exchangeable but at a much slower rate. When 18O-enriched peroxide was used in the experiments without LC separations, the m/z 223 ion was observed, although m/z 221 ion was nearly absent (Fig. 9C).
FIGURE 9.

LC-MS characterization of the TDO reaction with H2O2 and l-Trp. A, HPLC of the reaction product of H216O2-dependent oxygenation mediated by ferric TDO performed in H218O. MS spectra at retention time of 2.88 (B), 5.25 (C), and 6.10 min (D).
ESI-MS/MS experiments were subsequently conducted to characterize the major products of m/z 237 and 239 ions. The supplemental Fig. S9 confirms that m/z 220 and 222 are the fragments of NFK (m/z 237 and 239, respectively), as a consequence of each losing a –16OH or –18OH group during ionization. The absence of the m/z 221 (223 with 18O) ion in the MS/MS spectrum of the m/z 237 (239 with 18O) ion is consistent with the LC-MS and isotope labeling results and thus confirms that the m/z 221 ion is not a fragment of the NFK. The presence of the m/z 223 ion in the 18O sample suggests an isotope equivalent of the presumed monooxygenated tryptophan (Trp-O). From these results, it can be concluded that the m/z 221 ion (223 with 18O) has arisen from Trp-O. In a post-reaction treatment experiment after removing the enzyme by filtration, the sample containing the Trp-O was incubated with H2O2 overnight at 4 °C in the absence of TDO. The m/z 221 (or 223 with 18O) ion was unchanged in the spectrum after the peroxide treatment.
Modeling Study
The TDO ferryl species observed in our Mössbauer study exhibits an unusually large quadrupole splitting parameter of 1.755 mm/s at the physiologically relevant pH 7.4. This is greater than that of any other reported Fe(IV)-oxo species of hemoproteins but is smaller than that of protonated Fe(IV)-oxo (see under “Discussion”). Density functional theory calculations on the TDO ferryl intermediate were performed to evaluate the possible structural influences on the Mössbauer parameters, including protonation of the oxo group, hydrogen bonding to the oxo group from a distal histidine and a conserved Ser-Gly pair, and conformational change of the proximal histidine ligand (Table 1). The data presented suggest that the relatively large positive value of quadrupole splitting of parameter is a result of the H-bonding to the oxo group (see under “Discussion” and supplemental material).
TABLE 1.
Results of various models for TDO ferryl species
HB represents the Ser124–Gly125 residues hydrogen-bonded to the oxo group. In 7A, the terminal atoms in the distal histidine are fixed at x-ray-determined positions, and in 8A, those atoms are allowed to be optimized. In 9A, all atoms of the HB group (CmTDO Ser124–Gly125) are also allowed to be optimized compared with 8A.
| Model | RFeO | ΔEQ | δFe | |
|---|---|---|---|---|
| Å | mm/s | mm/s | ||
| TDO | Experimental | 1.755 | 0.055 | |
| 1A | FeIV(porphyrin)2−(His)0(O)2− | 1.654 | 1.54 | 0.14 |
| 1B | Twisted His | 1.648 | 1.97 | 0.13 |
| 2A | FeIV(porphyrin)2−(His)0(OH)1− | 1.799 | 3.02 | 0.08 |
| 2B | Twisted His | 1.795 | 3.19 | 0.11 |
| 3A | FeIV(porphyrin)2−(His)0(O···HB)2− | 1.663 | 1.78 | 0.12 |
| 3B | Twisted His | 1.657 | 2.20 | 0.11 |
| 4A | FeIV(porphyrin)2−(His···H2O)0(O)2− | 1.656 | 1.44 | 0.14 |
| 4B | Twisted His | 1.646 | 2.14 | 0.12 |
| 5A | FeIV(porphyrin)2−(His···H2O)0(O···HB)2− | 1.665 | 1.66 | 0.11 |
| 5B | Twisted His | 1.660 | 2.11 | 0.11 |
| 6A | FeIV(porphyrin)2−(His)0(OH···HB)1− | 1.792 | 3.07 | 0.10 |
| 7A | FeIV(porphyrin)2−(His)0(O···HB)2−(distal His) − 1 | 1.664 | 1.82 | 0.12 |
| 8A | FeIV(porphyrin)2−(His)0(O···HB)2−(distal His) − 2 | 1.672 | 1.97 | 0.09 |
| 9A | FeIV(porphyrin)2−(His)0(O···HB)2−(distal His) − 3 | 1.670 | 2.01 | 0.10 |
DISCUSSION
Reactivation of Ferric TDO
This work describes an extensive effort to uncover the long standing reactivation mystery of the reactivation of ferric TDO by an oxidant. Although the activation of ferric TDO by H2O2 in the presence of l-Trp has been known since 1950 (22), the mechanism of the reactivation was not resolved. The optical and Mössbauer spectroscopic data shown in this work confirm that the addition of H2O2 to the ferric form of TDO in the presence of l-Trp results in reduction of the enzyme to produce ferrous heme. The enzyme assay demonstrates that the ferrous enzyme generated by peroxide is catalytically active and is inhibited by CO. A plausible mechanistic model is presented in Scheme 2 that brings together the EPR, Mössbauer, optical, and mass spectrometry data. The spectroscopic observations of each of the proposed intermediates and products that have been presented here, including the ferryl intermediate, protein-based free radical, O2 production, Trp-Trp dimer, monooxygenated Trp, and the normal product NKF, provide unequivocal support for this model.
SCHEME 2.

Mechanism of enzyme reactivation by hydrogen peroxide in tryptophan 2,3-dioxygenase. The reactivation pathway branches at the compound ES-type ferryl intermediate. In the absence of l-Trp, a catalase-like activity is present. The enzyme reactivation occurs when the protein radical and the ferryl species are each reduced by l-Trp. Intermediates shown in parentheses are predicted but not detected experimentally.
In the proposed reactivation model, the first step of the reaction involves a peroxide-dependent process to generate compound ES (Scheme 2). The second step involves the following two branching pathways that deplete compound ES: (a) a catalase-like reaction leading to O2 production, and (b) reduction of the protein radical and Fe(IV)=O species by l-Trp resulting in Trp-O and ferrous TDO. The second branching reactions consume the two oxidizing equivalents stored in compound ES intermediate and, consequently, lead to enzyme reactivation and l-Trp dimerization. The reduction of the protein radical by the presence of l-Trp is a necessary step for the Fe(IV)=O to oxidize the substrate and become reduced to ferrous in branch B, because the high valent Fe(IV) ion alone can no longer perform the catalase-like function. We performed a set of experiments in which the protein radical is quenched by a scavenger, such as hydroxyurea, and we found that the catalase-like reaction is stalled. Consequently, O2 production is inhibited and so is the NFK production in the anaerobic experiments (Fig. 5).
The reactivated enzyme turns over l-Trp with O2 to produce NFK, which is observed both optically and with mass spectrometry. Under anaerobic conditions, the source of oxygen for the Fe(II)-dependent dioxygenation reaction arises solely from the catalase-like catalytic cycle. Under aerobic conditions the oxygen is not limited to that produced by the catalase-like activity and l-Trp is quickly converted to NFK. High concentration of peroxide can inhibit the enzyme reactivation in the aerobic experiments. This is due to the depletion of l-Trp and oxidation of the newly generated ferrous TDO (supplemental Fig. S7).
Although the presence of both H2O2 and l-Trp will cause TDO reactivation, there are two prerequisites for spectral detection of Fe(II) heme from the ferric enzyme as follows: 1) l-Trp must be present in large excess relative to enzyme, and 2) l-Trp/H2O2 ratio must be greater than 2. The branched pathways shown in Scheme 2 are the competing reactions. Branch A is [H2O2]-dependent, although it is independent of [l-Trp]. Conversely, branch B is [l-Trp]-dependent. H2O2 is also required for branch B to take place, so it is not independent of [H2O2]. A large excess of l-Trp must be present for branch B to effectively compete with branch A. The branched pathways are also internally connected. When the concentration of H2O2 is increased, the rate of O2 formation also increases; at the same time, the NFK formation should decrease as branch A competes with branch B. However, the O2 generated from branch A would become a substrate of the dioxygenase reaction in branch B, hence masking this effect. O2 production was also detected in the reaction of TDO and H2O2 in the presence of l-Trp, but at a much slower and variable rate depending on the reaction conditions, e.g. the concentration and ratio of H2O2 and l-Trp. Because the Fe(II)-TDO consumes O2, the change of O2 concentration is a net effect of the catalase-like activity and the NFK formation. Thus, the classic kinetic measurements would not be very informative unless the exact concentration of the ferrous enzyme is characterized by spectroscopic methods at all times and under each of those conditions. Nevertheless, the observation of O2 production in the presence of l-Trp suggests that the catalase-like reaction is not affected by the presence of the enzyme-bound l-Trp. Hence, the catalase-like activity is probably an intrinsic property of TDO.
An intriguing aspect of the reactivation of TDO is the reduction of Fe(III) to Fe(II) heme in the absence of a reducing agent. H2O2 is a common oxidant with a standard reduction potential 1.32 V for the couple H2O2/2H2O at pH 7.0 (43), which is significantly higher than that of IDO (−30 mV) (44), and it is also expected to be much higher than that of CmTDO based on the reported data of Xanthomonas campestris TDO (<+150 mV) (12). Scheme 2 reveals that the reducing power in the TDO reactivation is ultimately derived from l-Trp.
A full conversion of Fe(III) to Fe(II) in TDO reactivation was never observed in this work. We believe this is due to the presence of competing reactions and because the ferrous heme can be re-oxidized. The amount of ferrous heme generated by this method depends on the concentrations of peroxide and l-Trp. In the presence of CO, the ferrous heme is stabilized against oxidation. The presence of CO also inhibits the production of NFK, indicating that the formation of NFK is through the normal enzyme cycle catalyzed by ferrous TDO and O2 rather than a short circuit or peroxide shunt described for cytochrome P450 enzymes (45–48). The experimental results presented here do not include evidence for a ferryl intermediate in the presence of l-Trp, but the observation of the ferryl intermediate in the absence of l-Trp suggests that the protein active site is capable of forming the ferryl intermediate. Previously, a high valent ferryl intermediate has never been trapped in TDO. Hence, it is likely that the decay rate of the high valent Fe(IV) intermediate is greater than the formation rate when the primary substrate l-Trp is available. The Fe(IV)-oxo intermediate would thus never accumulate in the presence of l-Trp.
Potential Physiological Relevance
H2O2 is naturally produced by enzymes such as oxidases in organisms as a by-product of aerobic respiration. Basal levels of H2O2 are present in most cells. In healthy individuals, H2O2 is produced in sufficient quantity to counteract unwanted bacterial invaders (49). During oxidative stress of the organism, reactive oxygen species, including H2O2, may be overproduced (50). In this study, we show the first clear spectroscopic observation that H2O2 is able to react with ferric TDO and l-Trp to produce the catalytically active form of the enzyme. The H2O2-based mechanism of enzyme reactivation may be physiologically important because TDO is a hepatic enzyme, and hepatocytes are known to be an oxidizing environment that may cause inactivation of TDO by oxidizing its iron ion. In contrast, the catalytic activity of IDO is known to be inhibited by H2O2 (51). It is worth noting that IDO exists in tissues other than the liver and is unlikely to become oxidized under normal cellular conditions, suggesting that the H2O2-triggered reactivation mechanism found in TDO would not be necessary for IDO. Under normal physiological conditions, H2O2 is present at low levels in cells. However, we find that a small amount of peroxide is sufficient to cause enzyme reactivation under aerobic conditions and when the primary substrate l-Trp is present. This is significant as amino acids are neither stored nor excreted in the human body. They have to be degraded. TDO is the key enzyme responsible for tryptophan degradation. In general, the discovery of such an enzyme reactivation mechanism by peroxide is important for understanding strategies how a ferrous enzyme maintains its catalytic activity in an oxidizing environment.
By-products of the Enzyme Reactivation
Two minor by-products were detected after enzyme reactivation as follows: Trp-Trp and a Trp-O species. An m/z of 409 ions corresponding to the l-Trp dimer is observed in our mass spectrometric study, which is absent in the control samples described under “Results.” The dimerization of l-Trp is tentatively attributed to the result of reduction of the protein radical by l-Trp. Our isotope labeling analyses show that Trp-Trp dimer is insensitive to 18O-enriched peroxide. Thus, its formation is not linked with the oxo group of the ferryl intermediate. The Fe(IV)-oxo intermediate oxidizes an enzyme-bound l-Trp to generate a monooxygenated product via a two-electron oxidation. This monooxygenated product is experimentally detected by mass spectrometry (m/z 221). An 18O-enriched form (m/z 223) is also observed. LC-MS experiments provided further evidence for the presence of a monooxygenated product.
The minor Trp-O product is a by-product of reactivation and is expected to be only equivalent to the ferrous heme concentration. Because of the limitation of l-Trp solubility, one cannot increase the enzyme concentration to the millimolar range for performing the reactivation reaction. The yield of Trp-O is unfortunately insufficient for further structural characterizations by other means such as NMR spectroscopy. Thus, its precise chemical structure is presently unknown. The most likely candidate is an epoxide, derived from O-insertion of the indole. An alternative candidate is 6-hydroxytryptophan, which is observed in the reaction of l-Trp, H2O2, and a triple mutant of myoglobin (53). The Trp-O by-product survives during the enzyme reactivation. A similar Trp-O product was not observed in the dioxygenase cycle of the ferrous TDO reaction with O2 as the oxidant nor is it shown in the reaction of peroxide with ferrous TDO. The structure of Trp-O is probably insignificant in this work because this minor product is not generated from the ferrous heme-dependent catalytic cycle of the dioxygenase reaction. Nonetheless, the finding of the Trp-O by-product has helped us understand how the reactivation proceeds through the involvement of l-Trp.
Catalase Activity of TDO
We have identified a previously unknown catalase-like activity for TDO by two sets of experiments. The first entails direct observation of O2 formation from H2O2, and the second is the spectroscopic study of an Fe(IV)=O species necessary for a catalase-like catalytic mechanism. Similar to other heme enzymes for which this catalase-like function is not native, the data presented here indicate that this activity is detrimental to the function of the enzyme. The addition of concentrated peroxide without l-Trp results in radical formation and irreversible partial loss of enzymatic activity as shown by the loss of heme and iron from the enzyme in our Mössbauer experiments. It has been shown that the heme-based catalases and the enzymes with a promiscuous catalase-like catalytic activity have a wide range of catalytic efficiencies (Table 2). The catalase-like activity observed from TDO, at 13 s−1, is significantly below those found in native catalases or the bifunctional catalase-peroxidase KatG. However, it is appreciable in comparison with the catalase activities of other hemoproteins whose primary biological activities are not catalase (Table 2). Whether the catalase-like activity has a physiological role in vivo is speculative. However, the reactivity of TDO with H2O2 is important for enzyme reactivation when the primary substrate is present.
TABLE 2.
The kinetic properties of catalase activity in hemoproteins
| Protein | kcat | Km | kcat/Km |
|---|---|---|---|
| s−1 | mm | m−1s−1 | |
| Horse liver catalase (52) | 3.8 × 107 | 1100 | 3.5 × 107 |
| E. coli catalase-peroxidase (76) | 1.6 × 104 | 3.9 | 4.1 × 106 |
| Mycobacterium tuberculosis KatG (77) | 1.0 × 104 | 5.2 | 1.9 × 106 |
| Recombinant KatG (52) | 2.3 × 103 | 30 | 7.7 × 104 |
| Periplasmic catalase-peroxidase (KatP) (78) | 1.8 × 104 | 27 | 6.4 × 105 |
| Nα-Acetylated microperoxidase-8 (79) | 4.1 | 40.9 | 100.2 |
| Catechol oxidase (80) | 0.063 | 1.2 | 63 |
| Hemoglobin (bovine) (81) | 1.92 | 24 | 80 |
| TDO (this work) | 13 | 16 | 850 |
In the mechanism of catalase, the ferric heme reacts with the first peroxide molecule to produce a reactive oxoferryl and a π-cationic porphyrin radical, which subsequently reacts with a second peroxide to produce an O2 molecule and water (54, 55). Our observation of an approximate ratio of 1:2 of [NFK]/[H2O2] stoichiometry under anaerobic conditions (Fig. 4, inset) is consistent with the catalase mechanism. The observed NFK/H2O2 ratio is slightly under 1:2, which is puzzling. This deviation may be explained by the nonproductive consumption of peroxide in the following processes: 1) the small amount of peroxide used to generate ferrous heme; 2) peroxide-induced heme degradation observed in our Mössbauer study; and 3) oxidation of the newly generated ferrous heme. The last process is inconsequential when O2 is the limiting reagent.
Ferryl Intermediate of TDO
We present clear spectroscopic evidence for the first experimental observation of a high valent Fe(IV) species in TDO. The detection of a monooxygenated product is consistent with the recent successful detection of an oxoferryl intermediate in the orthologous enzyme IDO by resonance Raman spectroscopy (56). Such a high valent Fe(IV) intermediate was expected to exist in the enzyme mechanism in a recent ONIOM study (57).
We show that the addition of H2O2 to ferric TDO, in the absence of l-Trp, generates an Fe(IV)-oxo species and a protein-based radical with a concomitant decrease in ferric TDO concentration. At approximately the same reaction time, the concentrations of the Fe(IV)-oxo and radical species are comparable. Thus, the generation of the Fe(IV)-oxo and radical species in nearly equal amounts is consistent with formation of a compound ES-type intermediate, rather than an Fe(IV)-oxo/porphyrin cation intermediate (compound I) observed in catalases. The presence of a compound ES species, which may be derived from a compound I-type intermediate in this case, may be critical for the subsequent enzyme reactivation to occur, because the Fe(IV)=O species and radical intermediate will have to be reduced by l-Trp through separate reactions.
The observed radical species has properties in common with those of protein-based aromatic radicals. The P½ value of the observed radical in TDO (1.2 milliwatts at 100 K) is significantly higher than that of isolated free radicals (36), for example 0.07 milliwatts at 90 K (58), indicating the presence of a relaxation mechanism. The P½ value for the protein-based Trp radical of compound ES species of cytochrome c peroxidase is 1.5 milliwatts at 100 K (59), and the Tyr radical of P450-ES is 1 milliwatt at 70 K (60). These higher P½ values have all been attributed to relaxation of the radical by the adjacent heme iron. The P½ value of the TDO radical is indicative of its close proximity to the metal center. The site of the radical in TDO is to be determined by future study. There are at least four tyrosine and tryptophan residues in the immediate vicinity of the enzyme active site. The identification of the radical site is challenging because a mutation of a tyrosine/tryptophan (fated to be a free radical) can result in the radical moving to a nearby tyrosine/tryptophan, as demonstrated in other heme-based enzymes such as prostaglandin H synthase (61).
Recent experimental studies and density functional theory calculations suggest that the ΔEQ value might correlate with the protonation state of some heme-based ferryl species (62, 63). The parameter range for protonated Fe(IV)-OH species is 2.00–2.5 mm/s (64), whereas the range for unprotonated Fe(IV)=O is 1.0–1.6 mm/s (62–66). The ΔEQ of the TDO Fe(IV)-oxo intermediate (1.755 mm/s determined at the physiologically relevant pH 7.4) lies between these ranges (Table 3). The quadrupole splitting parameter of the TDO intermediate is noticeably greater than those of any other heme-based ferryl species but is much smaller than those of protonated basic ferryl species. The nearest value is found in MauG, another enzyme that oxidizes l-Trp inside a protein (67). A bis-Fe(IV) intermediate has been trapped from MauG, and one of the hemes is described as an oxyferryl species with a ΔEQ value of 1.70 mm/s (67).
TABLE 3.
Comparison of the Mössbauer parameters of TDO Fe(IV) intermediate with known heme-based ferryl moieties
Abbreviations used in this table are as follows: CcP, cytochrome c peroxidase; HRP, horseradish peroxidase; Mb, myoglobin; JRP, Japanese radish peroxidase. P450-I was trapped and characterized from CYP119, the thermophilic P450 from Sulfolobus acidocaldarius (68).
| Intermediate | Iron species | Trans ligand | Spin | δ | ΔEQ | Refs. |
|---|---|---|---|---|---|---|
| mm/s | mm/s | |||||
| CcP-ES | [Fe4+=O2−]⨥ | Histidine | S = 1 | 0.05 | 1.55 | 38 |
| HRP-I | [Fe4+=O2−]⨥ | Histidine | S = 1 | 0.08 | 1.25 | 82, 83 |
| HRP-II | Fe4+=O2− | Histidine | S = 1 | 0.03 | 1.61 | 83 |
| Mb-II | Fe4+=O2− | Histidine | S = 1 | 0.09 | 1.43 | 84 |
| JRP-I | [Fe4+=O2−]⨥ | Histidine | S = 1 | 0.10 | 1.33 | 85 |
| JRP-II | Fe4+=O2− | Histidine | S = 1 | 0.03 | 1.59 | 84 |
| Mb (annealed) | Fe4+=O2− | Histidine | S = 1 | 0.10 | 1.49 | 86 |
| CPO-I | [Fe4+=O2−]⨥ | Cysteine | S = 1 | 0.13 | 0.96 | 87, 76 |
| CPO-II | Fe4+=O2− | Cysteine | S = 1 | 0.11(3) | 1.59 | 63 |
| P450-I | [Fe4+=O2−]⨥ | Cysteine | S = 1 | 0.11 | 0.90 | 68 |
| TDO | Fe4+=O2− | Histidine | S = 1 | 0.055 | 1.755 | This work |
| MauG | Fe4+=O2− (heme site 1) | Histidine | S = 1 | 0.06 | 1.70 | 67 |
| Basic CPO-II | [Fe4+=O2−]H+ | Cysteine | S = 1 | 0.10(3) | 2.06(3) | 63 |
| Basic P450BM3 | [Fe4+=O2−]H+ | Cysteine | S = 1 | 0.13 | 2.16 | 62 |
| Basic P450cam | [Fe4+=O2−]H+ | Cysteine | S = 1 | 0.14 | 2.06 | 62 |
Protein environments can conceivably provide a range of proton interactions with the oxyferryl heme. A possible interpretation of the atypical quadrupole splitting value was examined in this work by density functional theory calculations performed on 14 structural models (supplemental Tables S1–S14). Because the iron equatorial heme ligands in TDO are the same as those found with other heme proteins that display typical Mössbauer ΔEQ values for Fe(IV)=O species, these models were used to evaluate the structural contributions that can directly affect the iron axial ligands as follows: 1) protonation of the oxo group; 2) hydrogen bonding to the oxo group; 3) hydrogen bonding to the proximal His; and 4) conformation of the proximal His. All the models were generated on the basis of the x-ray crystal structure of the substrate-free TDO (Protein Data Bank code 2NW7). Geometries of the models (see supplemental Tables S1–S14 for the optimized coordinates) were optimized using the method developed previously for defining other oxoferryl species (29) with the terminal atoms fixed at the x-ray crystal structure positions to mimic the protein environment effect (supplemental Fig. S10). Both the Mössbauer quadrupole splitting and isomer shift parameters for these models were calculated using the density functional theory method, which enabled accurate predictions of these two properties in various iron proteins and models covering all iron spin states and coordination states (29, 30).
As shown in Table 1, the predicted Mössbauer isomer shifts (δ) of these models are all close to the experimental value with no significant difference, indicating its insensitivity to the secondary structural changes along the axial positions. In contrast, the predicted Mössbauer quadrupole splittings (ΔEQ) display a large range from 1.44 to 3.19 mm/s, suggesting its role as a sensitive structural probe. The best agreement with the experimental value was found by incorporation of the nearby hydrogen bonding residues Ser124–Gly125 (these two residues are fixed at their x-ray positions except for the peptide bond atoms CONH, which were allowed to be optimized). The predicted ΔEQ value of 1.78 mm/s for model 3A (i.e. FeIV(porphyrin)2−(His)0(O···HB)2−, see supplemental Table S5) is in excellent agreement with the experimental measurement of 1.755 mm/s described in this work. These calculations suggest that the ΔEQ value of the TDO ferryl species originates from the hydrogen bonding interaction provided by the unique protein environment, similar to the computational results obtained for the MauG Fe(IV) species (30).
An examination of the high resolution crystal structures of TDO from both Cupriavidus metallidurans and Xanthomonas campestris (Protein Data Bank entries 2NOX, 2NW7, and 2NW8) suggests that the conserved active site residues at the distal pocket, His72 and Gly125 (CmTDO numbering system), are ideal candidates for hydrogen bonding with the Fe(IV)-bound oxo group. These residues are also conserved in the human enzyme. The role of these two residues has already been investigated in recent experimental and computational studies (25, 56, 69, 70) by several laboratories. The consensus is that the heme site in the dioxygenase can indeed generate a high valent Fe(IV)-oxo species under appropriate conditions and that the protein microenvironment is critical for dictating the chemical and physical property of the intermediate.
Oxygen Exchange with Solvent in NFK
An unexpected minor finding of this study is that one of the oxygen atoms in the reaction product NFK is exchangeable with water in the time frame of minutes. Based on the well known ketonic oxygen exchange with water, and the fact that both the carboxylate oxygen and the amide group of NH-COOH can exchange with a buffered solvent slower than a ketone (71–74), we propose that the ketone carbonyl group exchanges its oxygen with solvent via a diol intermediate mechanism. Scheme 3 depicts a plausible mechanism for the solvent exchange. The nucleophilic attack at the ketone carbon by water generates a diol intermediate. This is facilitated by a transient state with a six-member ring structure. The finding of NFK solvent exchange may become important in the mechanistic studies of the enzyme with 18O. A previous 18O study was carried out by Hayaishi et al. (75) in the absence of the knowledge of solvent exchange described in this work. The less than theoretical 18O content was found in kynurenine, the hydrolysis product of NFK, and the exact contents vary in different sets of experiments (75). Furthermore, the results of 18O2 and 18O water are not mutually consistent. This was thought to be caused by either an exchange reaction during the isolation procedure or by preferential utilization of 16O over 18O by TDO (75). The previous observations can be fully explained by our proposed 18O-exchange mechanism. It is the ketonic oxygen exchange that causes less than one atom of 18O in kynurenine, rather than the preference of 16O over 18O hypothesized in the previous study. The exact 18O content in NFK and kynurenine is dependent on the sample preparation procedures, i.e. the longer solvent exchange time lowers 18O content.
SCHEME 3.

Proposed solvent exchange mechanism on the carbonyl group of NFK.
Supplementary Material
Acknowledgments
We thank Dr. Tadhg P. Begley for providing the TDO expression plasmid and the encouragement and discussions throughout the work on this project. We are grateful to Drs. Binghe Wang and Dabney Dixon for the invaluable discussion of the product 18O solvent-exchange mechanisms and C. Ian Davis for help on the preparation of the manuscript.
This work was supported by National Science Foundation Grant MCB-0843537 (to A. L.) and Georgia Cancer Coalition Distinguished Scholar Program (to A. L.). This work was also supported in part by National Institutes of Health Grants GM077387 (to M. P. H.) and GM085774 (to Y. Z.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S10, Tables S1–S14, and additional references.
- TDO
- tryptophan 2,3-dioxygenase
- CmTDO
- C. metallidurans TDO
- IDO
- indoleamine 2,3-dioxygenase
- NFK
- N-formylkynurenine
- ESI
- electrospray ionization
- mT
- millitesla.
REFERENCES
- 1. Kotake Y., Masayama I. (1936) Z. Physiol. Chem. 243, 237–244 [Google Scholar]
- 2. Yamamoto S., Hayaishi O. (1970) Methods Enzymol. 17, 434–438 [DOI] [PubMed] [Google Scholar]
- 3. Feigelson P., Greengard O. (1961) Biochim. Biophys. Acta 50, 200–202 [DOI] [PubMed] [Google Scholar]
- 4. Stone T. W., Darlington L. G. (2002) Nat. Rev. Drug Discov. 1, 609–620 [DOI] [PubMed] [Google Scholar]
- 5. Kurnasov O., Goral V., Colabroy K., Gerdes S., Anantha S., Osterman A., Begley T. P. (2003) Chem. Biol. 10, 1195–1204 [DOI] [PubMed] [Google Scholar]
- 6. Schwarcz R. (2004) Curr. Opin. Pharmacol. 4, 12–17 [DOI] [PubMed] [Google Scholar]
- 7. Robotka H., Toldi J., Vcsei L. (2008) Future Neurol. 3, 169–188 [Google Scholar]
- 8. Guillemin G. J., Meininger V., Brew B. J. (2005) Neurodegener. Dis. 2, 166–176 [DOI] [PubMed] [Google Scholar]
- 9. Guillemin G. J., Brew B. J. (2002) Redox Rep. 7, 199–206 [DOI] [PubMed] [Google Scholar]
- 10. Hayaishi O. (1993) Protein Sci. 2, 472–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugimoto H., Oda S., Otsuki T., Hino T., Yoshida T., Shiro Y. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2611–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forouhar F., Anderson J. L., Mowat C. G., Vorobiev S. M., Hussain A., Abashidze M., Bruckmann C., Thackray S. J., Seetharaman J., Tucker T., Xiao R., Ma L. C., Zhao L., Acton T. B., Montelione G. T., Chapman S. K., Tong L. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y., Kang S. A., Mukherjee T., Bale S., Crane B. R., Begley T. P., Ealick S. E. (2007) Biochemistry 46, 145–155 [DOI] [PubMed] [Google Scholar]
- 14. Tanaka T., Knox W. E. (1959) J. Biol. Chem. 234, 1162–1170 [PubMed] [Google Scholar]
- 15. Takikawa O. (2005) Biochem. Biophys. Res. Commun. 338, 12–19 [DOI] [PubMed] [Google Scholar]
- 16. Paglino A., Lombardo F., Arcà B., Rizzi M., Rossi F. (2008) Insect Biochem. Mol. Biol. 38, 871–876 [DOI] [PubMed] [Google Scholar]
- 17. Li J. S., Han Q., Fang J., Rizzi M., James A. A., Li J. (2007) Arch. Insect Biochem. Physiol. 64, 74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colabroy K. L., Begley T. P. (2005) J. Bacteriol. 187, 7866–7869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Laurentis W., Khim L., Anderson J. L., Adam A., Johnson K. A., Phillips R. S., Chapman S. K., van Pee K. H., Naismith J. H. (2007) Biochemistry 46, 12393–12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kühn H., Götze R., Schewe T., Rapoport S. M. (1981) Eur. J. Biochem. 120, 161–168 [DOI] [PubMed] [Google Scholar]
- 21. Rao S. I., Wilks A., Hamberg M., Ortiz de Montellano P. R. (1994) J. Biol. Chem. 269, 7210–7216 [PubMed] [Google Scholar]
- 22. Knox W. E., Mehler A. H. (1950) J. Biol. Chem. 187, 419–430 [PubMed] [Google Scholar]
- 23. Brady F. O., Forman H. J., Feigelson P. (1971) J. Biol. Chem. 246, 7119–7124 [PubMed] [Google Scholar]
- 24. Gupta R., Fu R., Liu A., Hendrich M. P. (2010) J. Am. Chem. Soc. 132, 1098–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukumura E., Sugimoto H., Misumi Y., Ogura T., Shiro Y. (2009) J. Biochem. 145, 505–515 [DOI] [PubMed] [Google Scholar]
- 26. Ishimura Y., Nozaki M., Hayaishi O., Nakamura T., Tamura M., Yamazaki I. (1970) J. Biol. Chem. 245, 3593–3602 [PubMed] [Google Scholar]
- 27. Becke A. D. (1993) J. Chem. Phys. 98, 5648–5652 [Google Scholar]
- 28. Wachters A. J. H. (1970) J. Chem. Phys. 52, 1033–1036 [Google Scholar]
- 29. Zhang Y., Oldfield E. (2004) J. Am. Chem. Soc. 126, 4470–4471 [DOI] [PubMed] [Google Scholar]
- 30. Ling Y., Davidson V. L., Zhang Y. (2010) J. Phys. Chem. Lett. 1, 2936–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Becke A. D. (1988) Phys. Rev. A 38, 3098–3100 [DOI] [PubMed] [Google Scholar]
- 32. Perdew J. P., Burke K., Wang Y. (1996) Phys. Rev. B 54, 16533–16539 [DOI] [PubMed] [Google Scholar]
- 33. Debrunner P. G. (1989) in Iron Porphyrins (Lever B., Gray H. B. eds) Vol. 3, pp. 137–234, VCH Publishers, Inc., NY [Google Scholar]
- 34. Münck E. (2000) in Physical Methods in Inorganic and Bioinorganic Chemistry (Que L., Jr. ed) pp. 287–319, University Science Books, Sausalito, CA [Google Scholar]
- 35. Stubbe J., van Der Donk W. A. (1998) Chem. Rev. 98, 705–762 [DOI] [PubMed] [Google Scholar]
- 36. Liu A. (2009) Wiley Encycl. Chem. Biol. 1, 591–601 [Google Scholar]
- 37. Yonetani T. (1965) J. Biol. Chem. 240, 4509–4514 [PubMed] [Google Scholar]
- 38. Lang G., Spartalian K., Yonetani T. (1976) Biochim. Biophys. Acta 451, 250–258 [DOI] [PubMed] [Google Scholar]
- 39. Sivaraja M., Goodin D. B., Smith M., Hoffman B. M. (1989) Science 245, 738–740 [DOI] [PubMed] [Google Scholar]
- 40. Mehler A. H., Knox W. E. (1950) J. Biol. Chem. 187, 431–438 [PubMed] [Google Scholar]
- 41. Basran J., Rafice S. A., Chauhan N., Efimov I., Cheesman M. R., Ghamsari L., Raven E. L. (2008) Biochemistry 47, 4752–4760 [DOI] [PubMed] [Google Scholar]
- 42. Dalosto S. D., Vanderkooi J. M., Sharp K. A. (2004) J. Phys. Chem. B 108, 6450–6457 [DOI] [PubMed] [Google Scholar]
- 43. Koppenol W. H. (1987) Bioelectrochem. Bioenerg. 18, 3–11 [Google Scholar]
- 44. Papadopoulou N. D., Mewies M., McLean K. J., Seward H. E., Svistunenko D. A., Munro A. W., Raven E. L. (2005) Biochemistry 44, 14318–14328 [DOI] [PubMed] [Google Scholar]
- 45. Porter T. D., Coon M. J. (1991) J. Biol. Chem. 266, 13469–13472 [PubMed] [Google Scholar]
- 46. White R. E., Coon M. J. (1980) Annu. Rev. Biochem. 49, 315–356 [DOI] [PubMed] [Google Scholar]
- 47. Ortiz de Montellano P. R. (ed) (1995) Cytochrome P450: Structure, Mechanism, and Biochemistry, 2nd Ed., pp. 245–303, Plenum Publishing Corp., New York [Google Scholar]
- 48. Sono M., Roach M. P., Coulter E. D., Dawson J. H. (1996) Chem. Rev. 96, 2841–2888 [DOI] [PubMed] [Google Scholar]
- 49. Van de Bittner G. C., Dubikovskaya E. A., Bertozzi C. R., Chang C. J. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 21316–21321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rana S. V. (1997) in Liver and Environmental Xenobiotics (Rana S. V., Taketa K. eds) pp. 114–134, Springer-Verlag, Heidelberg [Google Scholar]
- 51. Poljak A., Grant R., Austin C. J., Jamie J. F., Willows R. D., Takikawa O., Littlejohn T. K., Truscott R. J., Walker M. J., Sachdev P., Smythe G. A. (2006) Arch. Biochem. Biophys. 450, 9–19 [DOI] [PubMed] [Google Scholar]
- 52. Nagy J. M., Cass A. E., Brown K. A. (1997) J. Biol. Chem. 272, 31265–31271 [DOI] [PubMed] [Google Scholar]
- 53. Pfister T. D., Ohki T., Ueno T., Hara I., Adachi S., Makino Y., Ueyama N., Lu Y., Watanabe Y. (2005) J. Biol. Chem. 280, 12858–12866 [DOI] [PubMed] [Google Scholar]
- 54. Deisseroth A., Dounce A. L. (1970) Physiol. Rev. 50, 319–375 [DOI] [PubMed] [Google Scholar]
- 55. Gouet P., Jouve H. M., Williams P. A., Andersson I., Andreoletti P., Nussaume L., Hajdu J. (1996) Nat. Struct. Biol. 3, 951–956 [DOI] [PubMed] [Google Scholar]
- 56. Lewis-Ballester A., Batabyal D., Egawa T., Lu C., Lin Y., Marti M. A., Capece L., Estrin D. A., Yeh S. R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 17371–17376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chung L. W., Li X., Sugimoto H., Shiro Y., Morokuma K. (2010) J. Am. Chem. Soc. 132, 11993–12005 [DOI] [PubMed] [Google Scholar]
- 58. Schünemann V., Jung C., Trautwein A. X., Mandon D., Weiss R. (2000) FEBS Lett. 479, 149–154 [DOI] [PubMed] [Google Scholar]
- 59. Hoffman B. M., Roberts J. E., Kang C. H., Margoliash E. (1981) J. Biol. Chem. 256, 6556–6564 [PubMed] [Google Scholar]
- 60. Schünemann V., Lendzian F., Jung C., Contzen J., Barra A. L., Sligar S. G., Trautwein A. X. (2004) J. Biol. Chem. 279, 10919–10930 [DOI] [PubMed] [Google Scholar]
- 61. Tsai A. L., Kulmacz R. J. (2010) Arch. Biochem. Biophys. 493, 103–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Behan R. K., Hoffart L. M., Stone K. L., Krebs C., Green M. T. (2006) J. Am. Chem. Soc. 128, 11471–11474 [DOI] [PubMed] [Google Scholar]
- 63. Stone K. L., Hoffart L. M., Behan R. K., Krebs C., Green M. T. (2006) J. Am. Chem. Soc. 128, 6147–6153 [DOI] [PubMed] [Google Scholar]
- 64. Green M. T., Dawson J. H., Gray H. B. (2004) Science 304, 1653–1656 [DOI] [PubMed] [Google Scholar]
- 65. Horner O., Mouesca J. M., Solari P. L., Orio M., Oddou J. L., Bonville P., Jouve H. M. (2007) J. Biol. Inorg. Chem. 12, 509–525 [DOI] [PubMed] [Google Scholar]
- 66. Behan R. K., Green M. T. (2006) J. Inorg. Biochem. 100, 448–459 [DOI] [PubMed] [Google Scholar]
- 67. Li X., Fu R., Lee S., Krebs C., Davidson V. L., Liu A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8597–8600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rittle J., Green M. T. (2010) Science 330, 933–937 [DOI] [PubMed] [Google Scholar]
- 69. Batabyal D., Yeh S. R. (2009) J. Am. Chem. Soc. 131, 3260–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thackray S. J., Bruckmann C., Anderson J. L., Campbell L. P., Xiao R., Zhao L., Mowat C. G., Forouhar F., Tong L., Chapman S. K. (2008) Biochemistry 47, 10677–10684 [DOI] [PubMed] [Google Scholar]
- 71. Bender M. L., Kemp K. C. (1957) J. Am. Chem. Soc. 79, 116–120 [Google Scholar]
- 72. Ronsein G. E., Oliveira M. C., Miyamoto S., Medeiros M. H., Di Mascio P. (2008) Chem. Res. Toxicol. 21, 1271–1283 [DOI] [PubMed] [Google Scholar]
- 73. Byrn M., Calvin M. (1966) J. Am. Chem. Soc. 88, 1916–1922 [Google Scholar]
- 74. Byrn M., Calvin M. (1965) Lawrence Berkeley National Laboratory UCRL-16571, 1–26 [Google Scholar]
- 75. Hayaishi O., Rothberg S., Mehler A. H., Saito Y. (1957) J. Biol. Chem. 229, 889–896 [PubMed] [Google Scholar]
- 76. Claiborne A., Fridovich I. (1979) J. Biol. Chem. 254, 4245–4252 [PubMed] [Google Scholar]
- 77. Johnsson K., Froland W. A., Schultz P. G. (1997) J. Biol. Chem. 272, 2834–2840 [DOI] [PubMed] [Google Scholar]
- 78. Varnado C. L., Hertwig K. M., Thomas R., Roberts J. K., Goodwin D. C. (2004) Arch. Biochem. Biophys. 421, 166–174 [DOI] [PubMed] [Google Scholar]
- 79. Jeng W. Y., Tsai Y. H., Chuang W. J. (2004) J. Pept. Res. 64, 104–109 [DOI] [PubMed] [Google Scholar]
- 80. Gerdemann C., Eicken C., Magrini A., Meyer H. E., Rompel A., Spener F., Krebs B. (2001) Biochim. Biophys. Acta 1548, 94–105 [DOI] [PubMed] [Google Scholar]
- 81. Paco L., Galarneau A., Drone J., Fajula F., Bailly C., Pulvin S., Thomas D. (2009) Biotechnol. J. 4, 1460–1470 [DOI] [PubMed] [Google Scholar]
- 82. Schulz C., Chiang R., Debrunner P. G. (1979) J. Phys. Colloq. 40, 534–536 [Google Scholar]
- 83. Schulz C. E., Rutter R., Sage J. T., Debrunner P. G., Hager L. P. (1984) Biochemistry 23, 4743–4754 [DOI] [PubMed] [Google Scholar]
- 84. Harami T., Maeda Y., Morita Y., Trautwein A., Gonser U. (1977) J. Chem. Phys. 67, 1164–1169 [Google Scholar]
- 85. Maeda Y., Higashimura T., Morita Y. (1967) Biochem. Biophys. Res. Commun. 29, 362–367 [DOI] [PubMed] [Google Scholar]
- 86. Garcia-Serres R., Davydov R. M., Matsui T., Ikeda-Saito M., Hoffman B. M., Huynh B. H. (2007) J. Am. Chem. Soc. 129, 1402–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rutter R., Hager L. P., Dhonau H., Hendrich M., Valentine M., Debrunner P. (1984) Biochemistry 23, 6809–6816 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.