

Abstract
Among human N-formyl peptide chemoattractant receptors, FPR2/ALX and FPR3 share the highest degree of amino acid identity (83%), and trigger similar cell responses upon ligand binding. Although FPR2/ALX is a promiscuous receptor, FPR3 has only one specific high affinity ligand, F2L, and a more restricted tissue/cell distribution. In this study, we showed that FPR2/ALX behaved as the prototypical receptor FPR1. The agonist-dependent phosphorylation used a hierarchical mechanism with a prominent role of Ser329, Thr332, and Thr335. Phosphorylation of FPR2/ALX was essential for its desensitization but the lack of phosphorylation did not result in enhanced or sustained responses. In contrast, resting FPR3 displayed a marked level of phosphorylation, which was only slightly increased upon agonist stimulation. Another noticeable difference between the two receptors was their subcellular distribution in unstimulated cells. Although FPR2/ALX was evenly distributed at the plasma membrane FPR3 was localized in small intracellular vesicles. By swapping domains between FPR2/ALX and FPR3, we uncovered the determinants involved in the basal phosphorylation of FPR3. Experiments aimed at monitoring receptor-bound antibody uptake showed that the intracellular distribution of FPR3 resulted from a constitutive internalization that was independent of C terminus phosphorylation. Unexpectedly, exchanging residues 1 to 53, which encompass the N-terminal extracellular region and the first transmembrane domain, between FPR2/ALX and FPR3 switched localization of the receptors from the plasma membrane to intracellular vesicles and vice versa. A clathrin-independent, possibly caveolae-dependent, mechanism was involved in FPR3 constitutive internalization. The peculiar behavior of FPR3 most probably serves distinct physiological functions that remain largely unknown.
Keywords: Cell Surface Receptor, Endocytosis, G Protein-coupled Receptors (GPCR), Innate Immunity, Myeloid Cell, Protein Phosphorylation, Receptor Desensitization, Receptor Endocytosis, Formyl Peptide Receptors
Introduction
The human formyl peptide receptors (FPRs)2 are primarily myeloid cell receptors but are also expressed in nonmyeloid cells in multiple organs and tissues. In human, three genes encode three subtypes known as FPR1, FPR2/ALX, and FPR3 according to the recently recommended nomenclature (1). Formerly and most frequently in the literature, FPR1 was known as FPR, FPR2/ALX was referred to as FPRL1 (FPR-like receptor 1) or ALXR, and FPR3 was noted as FPRL2. Human FPRs belong to the G protein-coupled chemoattractant receptors, a subfamily of seven transmembrane domain G protein-coupled receptors (GPCRs). Evidence for an important role of FPRs both in infective and inflammatory processes is well documented in the literature. Multiple ligands, the majority of which are peptides, have been identified for the FPRs making them one of the most promiscuous receptor families (1). N-Formyl peptides that are breakdown products of bacterial and mitochondrial proteins (2) are ligands common to the three receptors. A large number of microbe- or host-derived nonformylated peptides, as well as library-derived synthetic peptides have been described as potent agonists for FPR2/ALX. Among them, the hexapeptide WKYMVM (Trp-Lys-Tyr-Met-Val-Met-NH2) is a selective agonist of FPR2/ALX, whereas WKYMVm (Trp-Lys-Tyr-Met-Val-d-Met-NH2) was found to be a potent agonist of FPR2/ALX and FPR1, and a weaker activator of FPR3 (3). So far, FPR3 has only one high-affinity endogenous ligand known as F2L that promotes calcium mobilization and chemotaxis (4). F2L is an acetylated amino-terminal peptide derived from the cleavage of the human heme-binding protein.
The sequence identity between the three receptors is high and the activation of all three receptors triggers similar cellular responses involved in bactericidal functions such as superoxide production and release of granule constituents that cause tissue damage and killing of bacteria. Because excessive cellular responses may have harmful effects, a fine regulation of receptor activation must be maintained. After stimulation with the agonist, the cellular responses rapidly decline in intensity and cells become refractory to subsequent stimulation with the same agonist. This loss of responsiveness termed receptor desensitization is a common feature of GPCRs, which is believed to be the conjunction of receptor-G protein uncoupling and receptor removal from the cell surface through agonist-mediated receptor internalization. It is well established that most, if not all, GPCRs have cytoplasmic loci of phosphorylation. Following ligand binding, GPCRs adopt a conformation that catalyzes the activation of the G protein and become susceptible to interact with, and be phosphorylated by one of the G protein-coupled receptor kinases on serine/threonine residues located in the intracellular loops and the cytoplasmic carboxyl tail. This phosphorylation process promotes the high-affinity binding of β-arrestins to the receptor, thereby preventing further coupling to G proteins (5). The β-arrestins target many GPCRs for internalization in clathrin-coated vesicles via a direct interaction of their carboxyl-terminal domain with both clathrin and the clathrin adaptor AP2 (6, 7). The very high degree of amino acid identity between the sequences of the carboxyl-terminal tail of the three N-formyl peptide receptors (presented in Fig. 1A) suggests that they may share common mechanisms of regulation. However, in the case of FPR1, it has been shown that β-arrestins are dispensable for receptor internalization but phosphorylation is a prerequisite (8). FPR1 phosphorylation is sufficient to inhibit interactions with the G-protein (9–11). In contrast, FPR2/ALX internalization was found to be a β-arrestin-dependent process (12). Upon agonist binding, FPR1 is phosphorylated on serines and threonines in the carboxyl tail (13). Eight of the 11 serines and threonines in the carboxyl tail of FPR1 have been found to be critical for FPR1 desensitization and internalization (14, 15). Likewise, FPR2/ALX is phosphorylated in an agonist-dependent manner but the sites that are phosphorylated or critical for phosphorylation have not yet been identified. Little is known as to the exact involvement of phosphorylation in FPR2/ALX desensitization and internalization. In contrast to FPR1 and FPR2/ALX, FPR3 displays a marked basal phosphorylation in the absence of agonist stimulation (3).
FIGURE 1.

Schematic representation of mutant and chimeric receptors. A, alignment of the primary sequence of the intracellular carboxyl terminus of human FPR1, FPR2/ALX, and FPR3. Residues that are identical in FPR2/ALX and FPR3 are linked by a vertical hyphen. B, amino acid sequence of the carboxyl terminus of wild type FPR2/ALX and location of point mutations in FPR2/ALX. Positions of putative phosphorylation sites in the primary sequence of the wild type FPR2/ALX C terminus are indicated in bold. Altered residues are shown for the different mutants. Residues corresponding to the anti-FPR2 epitope are underlined. C, schematic representation of wild type and chimeric receptors obtained by swapping C-terminal or N-terminal domains. The topology of FPR2/ALX (black lines) and FPR3 (gray lines) in chimeric receptors is illustrated.
In this study, we identified the phosphoacceptor sites that are critical for agonist-mediated phosphorylation of FPR2/ALX by testing a series of serine/threonine to alanine replacement mutants for their ability to be phosphorylated in response to agonist binding. In addition, we determined the role of FPR2/ALX phosphorylation in receptor desensitization. By exchanging domains between FPR2/ALX and FPR3, we uncovered the determinants involved in basal phosphorylation of FPR3. A pattern of receptor distribution in small vesicles with an intracellular localization was observed by immunofluorescence in unstimulated FPR3-transfected cells. Experiments aimed at monitoring receptor-bound antibody uptake showed that the intracellular distribution of FPR3 results from a constitutive internalization. The amino acid sequence encompassing the N-terminal region and the first transmembrane domain (amino acids 1 to 53) contains the sequence determinants that govern the constitutive internalization and vesicular localization of FPR3. A clathrin-independent, possibly caveolae-mediated, pathway seemed to be involved in the constitutive internalization of FPR3.
EXPERIMENTAL PROCEDURES
Construction of FPR2/ALX (FPR2) Phosphorylation Mutants
The serines and threonines present in the C-terminal cytoplasmic domain of FPR2/ALX were grouped in three clusters, referred to as clusters A, B, or C, as illustrated in Fig. 1B. Serines and threonines were replaced by alanines in each cluster. Clusters were mutated alone or in combination. Mutant receptors were created by using the PCR strategy described by Yon and Fried (16). Briefly, using FPR2/ALX in CDM8 as a template, two intermediary polymerase chain reaction fragments, PCR1 and PCR2, were generated with two couples of primers. PCR1 was produced with a sense primer located upstream from the start codon. The 5′-end of the primer contained an extension with an appropriate restriction site for further ligation in an expression vector, whereas the reverse primer carried the desired mutations. Likewise, PCR2 was created with a sense primer, which is complementary to the reverse primer used for the synthesis of PCR1, and a reverse primer, located downstream from the stop codon, with an appropriate restriction site. After purification, the two PCR fragments were mixed, denatured, and hybridized. The mixture was used to generate PCR3, which contains the entire sequence of the mutated FPR2/ALX. Ten cycles of amplification were first performed in the absence of primers to increase the number of copies of full-length cDNA carrying the mutations on both strands. Then, 35 cycles of amplification were carried out in the presence of the sense and reverse primers used to generate PCR1 and PCR2. PCR3 was cleaved with the appropriate restriction enzymes for ligation in pCDNA3.1 (Invitrogen) or pEF-neo (17). For each mutant receptor, plasmid cDNA was prepared and the open reading frame was entirely sequenced.
Construction of Chimeric Receptors
Chimeric receptors FPR3-R2 and FPR2-R3 were constructed by swapping the cytoplasmic C-terminal domain of FPR3 for that of FPR2/ALX and conversely. The construction of the chimeras was greatly facilitated by the presence of a conserved PvuII restriction site located in the nucleotide sequence coding for the end of the seventh transmembrane domain. A chimeric receptor, in which the C-terminal domain was replaced by FPR2/ALX with all serines and threonines mutated into alanines, was constructed and referred to as FPR3-R2-ABC. The wild type forms of the 3HA-tagged FPR2/ALX and FPR3 were purchased from the UMR cDNA Resource Center, University of Missouri-Rolla. Taking advantage of the presence of a unique BspEI restriction site, a chimeric receptor, named 3HA-FPR2(1–53)-R3, was engineered that combined the N-terminal 3HA-tagged domain (amino acids 1 to 53) of FPR2/ALX with amino acids 54 to 353 of FPR3. The converse chimeric receptor, named 3HA-FPR3(1–53)-R2, was also constructed that combined the N-terminal 3HA-tagged domain (amino acids 1 to 53) of FPR3 with amino acids 54 to 351 of FPR2/ALX.
Cell Culture and Transfection
HEK293 and HEK293T cells and HL60 cells were obtained from the American Type Culture Collection. Cells were cultured at 37 °C in a humidified atmosphere with 5% CO2 in Dulbecco's modified Eagle's/F-12/GlutaMAX I medium (HEK293 and HEK293T cells) or RPMI 1640 medium/GlutaMax I (HL60 cells) supplemented with 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 10% heat-inactivated fetal calf serum. The maximal density of HL60 cells was maintained below 2 × 106 cells/ml and cells were centrifuged at each passage. Culture medium, serum, and antibiotics were obtained from Invitrogen.
Transient Expression of Wild Type FPR2/ALX, Mutant FPR2/ALX, FPR3, and Receptor Chimeras in HEK293 Cells
Lipofectamine 2000® (Invitrogen) was used for transient DNA transfections. HEK293 and HEK293T cells were grown to reach ∼80% confluence and transfections were performed according to the manufacturer's recommendations.
Stable Expression of FPR2/ALX and Phosphorylation-deficient Mutant FPR2-ABC in HL60 Cells
Wild type FPR2/ALX and phosphorylation-deficient mutant FPR2-ABC were stably expressed in undifferentiated HL60 cells. Transfection of HL60 cells was performed by electroporation with a gene pulser apparatus (Bio-Rad) as previously described (18). Following electroporation, cells were allowed to recover in 20 ml of culture medium for 48 h prior to selection in medium containing 1 mg/ml of G418 (Geneticin, Invitrogen). G418-resistant clones were obtained by limited dilution into 24-well microtiter plates and receptor-expressing clones were identified for their ability to mobilize intracellular calcium upon addition of the WKYMVm peptide (100 nm final concentration). The hexapeptide WKYMVm was synthesized and HPLC purified by NeoSystem (Strasbourg, France).
Metabolic Labeling and Immunoprecipitation of the Phosphorylated Receptors
HEK293T cells were seeded in duplicate in Costar 6-well plates and transfected with wild type FPR2/ALX, mutated FPR2/ALX, FPR3, or chimeric receptors. One plate was used to determine the level of cell surface receptor expression, whereas the second plate was used to assess their ability to be phosphorylated after metabolic labeling with [32P]orthophosphoric acid (0.3–0.5 mCi/ml) as previously described (19). Receptor phosphorylation was initiated by the addition of WKYMVm (1 μm final concentration) or F2L peptide (1 μm final concentration). F2L peptide was obtained from AnyGen (Gwangju, Korea). After a 15-min stimulation, cells were lysed, and phosphorylated receptors were immunoprecipitated as previously described with affinity-purified rabbit IgGs directed against the last 10 residues (PPAETELQAM) of FPR2/ALX (3). These IgGs cross-react with the corresponding sequence (PPEETELQAM) in FPR3. 3HA-tagged receptors were immunoprecipited with a mouse monoclonal anti-HA antibody (12CA5, Roche Diagnostics). To analyze receptor phosphorylation, immunoprecipitates were treated with 2-fold Laemmli sample buffer under reducing conditions for 10 min at 50 °C and then subjected to SDS-PAGE and autoradiography.
Peptide Iodination and Binding Assays on Cells Expressing Wild Type and Mutant Forms of FPR2/ALX
The highly potent FPR2/ALX agonist WKYMVm was labeled with 125I as previously described (3). HEK293T cells expressing wild type or mutant forms of FPR2/ALX were incubated for 1 h at 4 °C with increasing concentrations of 125I-labeled WKYMVm peptide in RPMI medium and 0.5% bovine serum albumin (BSA) supplemented or not with an excess of nonradioactive peptide. Cells were washed three times with chilled phosphate-buffered saline, lysed in 2 n NaOH, and bound radioactivity was recorded in a γ-counter. The results were expressed as the percentage of 125I-labeled peptide bound on wild type FPR2/ALX expressing cells.
Intracellular Calcium Mobilization
The Fura 2-based calcium mobilization assay in transfected HL60 cells was performed as previously described (20). For desensitization of the calcium mobilization response, Fura 2-loaded cells were stimulated with or without 100 nm WKYMVm for 10 min at 37 °C. Cells were washed three times at room temperature to remove the peptide, and subsequently assayed for calcium mobilization in response to different concentrations of WKYMVm.
ERK1/2 Phosphorylation
HL60 cells that stably expressed the wild type or nonphosphorylated forms of FPR2/ALX were stimulated with the hexapeptide WKYMVm (100 nm final concentration) in RPMI 1640 for various periods of time at 37 °C. Cells were lysed in 2-fold Laemmli sample buffer and briefly sonicated. Proteins were resolved by electrophoresis on 10% SDS-PAGE and transferred to nitrocellulose membrane for Western blotting. Phosphorylated ERK1/2 were detected by immunoblotting with anti-phospho-ERK1/2 (E4) obtained from Santa Cruz Biotechnologies. Immunoblot analysis with polyclonal anti-ERK2 antibody (C14, Santa Cruz Biotechnology) was performed to confirm that the same amounts of proteins were loaded. Antigen-antibody complexes were visualized after incubating the membrane with goat anti-mouse IgG or goat anti-rabbit IgG antibody coupled to horseradish peroxidase using an enhanced chemiluminescence (ECL) detection system (Amersham Biosciences). Phosphorylated ERK2 signals were quantified by densitometry using ImageJ software (National Institute of Health).
Immunofluorescence
HEK293 cells were plated into poly-d-lysine-coated 8-chamber glass bottomed LAB-TEK® (Nalge Nunc International, Hereford, UK). HEK293 cells were transiently transfected to express FPR2/ALX, FPR3, or chimeric receptors with Lipofectamine 2000® according to the manufacturer's instructions (Invitrogen). In some experiments, the plasmid encoding 3HA-tagged FPR3 was co-transfected with a plasmid encoding β-arrestin 2 fused to EGFP, a plasmid encoding a dominant-negative mutant of dynamin (dynamin K44A), or a plasmid encoding the fragment 318–419 of β-arrestin 1 fused to EGFP. We described and used these plasmids in previous work (21). Two days after transfection, cells were fixed with 3% paraformaldehyde in PBS supplemented with 2% sucrose for 30 min at room temperature, quenched with 50 mm NH4Cl in RPMI 1640 for 15 min, and permeabilized with 0.1% Nonidet P-40 for 10 min. Fixed and permeabilized cells were incubated with blocking buffer (2% BSA in RPMI 1640) for 30 min at room temperature. The receptors were labeled for 1 h at room temperature with the polyclonal anti-FPR2 antibody or with the anti-HA monoclonal antibody (12CA5) at 1 μg/ml when 3HA-tagged receptors were expressed. Cells were washed three times and incubated with green fluorescent Alexa 488-conjugated goat anti-rabbit or anti-mouse antibody, or red fluorescent Alexa 568-conjugated goat anti-mouse antibody (Molecular Probes, Eugene, OR) for 30 min at room temperature. After washing, cells were embedded in anti-quenching medium DABCO (2.3% 1,4-diazabicyclo(2,2,2)octane), 50% glycerol, 10% 0.2 m Tris-HCl, pH 8.0, 0.02% NaN3. Fluorescence micrographs and layouts were performed using a Leica DMIRE2 inverse microscope equipped with a digital Leica DC350F camera using the QFluoro software (Leica Microsystems).
Constitutive Receptor Internalization Assay Monitored by Antibody Uptake
HEK293 cells were transiently transfected to express 3HA-tagged receptors. Two days after transfection, cells were incubated for 30 min at 37 °C, with the monoclonal mouse anti-HA antibody diluted at 1 μg/ml in pre-warmed RPMI, 2% BSA medium. As a control, cells were incubated for the same period of time in ice-cold RPMI, 2% BSA supplemented with monoclonal antibody. In some experiments, the transferrin receptor was tracked by adding transferrin Alexa Fluor 568 conjugate (15 μg/ml) 15 min after antibody uptake had been initiated. In another series of experiments, cells were simultaneously treated for 30 min at 37 °C with the monoclonal anti-HA antibody and filipin III (5 μg/ml) diluted in RPMI, 2% BSA medium. Cells were also co-transfected with the plasmid encoding 3HA-tagged FPR3 and a plasmid encoding a dominant-negative mutant of dynamin 1 (dynamin K44A) or a plasmid encoding the β-arrestin 1(318–418)-EGFP fusion. After being washed in RPMI medium, cells were immediately fixed, permeabilized, and treated for immunofluorescence as described in the previous section.
RESULTS
Agonist-induced Phosphorylation of FPR2/ALX and FPR3
Despite a high level of amino acid identity between FPR2/ALX and FPR3, especially in the transmembrane and cytoplasmic regions (Fig. 1A), FPR3 is phosphorylated in the absence of agonist binding, whereas FPR2/ALX is not (3). In this study, we compared the phosphorylation of each receptor induced by their respective high-affinity agonist. The hexapeptide WKYMVm displays a high affinity for FPR2/ALX and a low affinity for FPR3 (3). The acetylated peptide F2L, an amino-terminal cleavage product of the human heme-binding protein, is the sole high-affinity ligand identified for FPR3 (4). As illustrated in Fig. 2 (left panel), the incorporation of 32P in FPR2/ALX was strictly dependent on the binding of WKYMVm. Even at a high concentration, F2L was unable to induce phosphorylation of FPR2/ALX. Compared with FPR2/ALX, FPR3 displayed a significant level of phosphorylation even in the absence of stimulation (Fig. 2, right panel, first lane). We thus confirmed the basal phosphorylation of FPR3. The binding of WKYMVm to FPR3 barely increased the incorporation of 32P in FPR3 (Fig. 2, right panel, second lane) even though it was previously shown to induce a robust calcium mobilization in FPR3 expressing HL60 cells (3). Surprisingly, although F2L has been described as a high-affinity agonist for FPR3, it behaved as a weak inducer of FPR3 phosphorylation (Fig. 2, right panel, third lane).
FIGURE 2.

Agonist-induced phosphorylation of FPR2/ALX and FPR3. HEK293T cells expressing wild type FPR2/ALX (right panel) or FPR3 (left panel) were metabolically labeled with [32P]orthophosphoric acid and incubated for 15 min at 37 °C with no agonist (first lane), 1 μm WKYMVm (second lane), or 1 μm F2L (third lane). After cell lysis, receptors were immunoprecipitated with an antibody directed against the last 10 amino acids of FPR2/ALX that cross-reacts with FPR3. Immunoprecipitates were treated with 2-fold Laemmli sample buffer and subjected to SDS-PAGE and autoradiography. Data are representative of three independent experiments.
Identification of Phosphorylated Sites in the Agonist-dependent Phosphorylation of FPR2/ALX
In preliminary experiments, phosphoamino acid analysis was used to determine the nature of the phosphoacceptor sites of FPR2/ALX as described previously (22). The phosphoamino acids resulting from acid hydrolysis of the 32P-labeled FPR2/ALX were resolved on thin layer cellulose plates by two-dimensional electrophoresis and visualized by autoradiography. The 32P-labeled FPR2/ALX contained phosphoserines and to a lesser extent phosphothreonines (data not shown). This correlated with the amino acid sequence, and the extent of FPR2/ALX labeling was proportional to the number of serine and threonine residues present in the C-terminal tail.
To determine which serine and threonine residues were involved in agonist-mediated FPR2/ALX phosphorylation, a series of mutants were generated by changing one cluster of three Ser/Thr at a time (mutant A, B, and C in Fig. 1B). Four additional mutants were generated in which two of three clusters were mutated simultaneously (AB, AC, and BC mutants in Fig. 1B) or in which the three clusters have been mutated (mutant ABC in Fig. 1B). In this latter case, all serines and threonines were replaced by alanines, except Thr346, which is located in the antigenic epitope (PPAETELQAM) recognized by the rabbit polyclonal antibody. Judging from the binding of 125I-labeled WKYMVm (Fig. 3, A and B, left panel), wild type and mutant receptors exhibited similar levels of surface expression. The agonist-mediated phosphorylation of FPR2-B was markedly reduced, whereas the level of phosphorylation for both FPR2-A and FPR2-C remained comparable with that observed with the wild type receptor (Fig. 3A, right panel). Moreover, the alanine substitution in both clusters A and C yielded a mutant (FPR2-AC) that had conserved a significant capacity to incorporate 32P, indicating that the main phosphoacceptor sites are located in cluster B (Fig. 3B, right panel). As expected, mutants that associated mutations in cluster B and either cluster A or C (mutants FPR2-AB and FPR2-BC) exhibited a phosphorylation close to the background level (Fig. 3B, right panel). The replacement of all serines and threonines by alanines yielded a receptor (FPR2-ABC) unable to incorporate radioactive phosphate. The lack of phosphorylation of this mutant indicated that no other phosphorylation site within the intracellular loops was involved. Altogether, these results suggest that the phosphoacceptor sites within cluster B are essential for receptor phosphorylation, and that the agonist-mediated phosphorylation of FPR2/ALX proceeds in a hierarchical manner.
FIGURE 3.

Cell surface expression and phosphorylation of the wild type and mutant forms of FPR2/ALX. A, cell surface expression. HEK293T cells expressing wild type or mutant FPR2/ALX were incubated with increasing concentrations of 125I-labeled WKYMVm peptide for 1 h at 4 °C. Radioactivity specifically bound to cells was recorded with a γ-counter. The results are expressed as the percentage of 125I-labeled peptide bound on wild type FPR2/ALX expressing cells. B, receptor phosphorylation. HEK293T cells that expressed wild type or mutant forms of FPR2/ALX were metabolically labeled with [32P]orthophosphoric acid. Cells were stimulated with 100 nm WKYMVm for 15 min at 37 °C. Receptors were immunoprecipitated from cell lysates with an antibody directed against the last 10 amino acids of FPR2/ALX. Receptor immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography. Data are representative of three independent experiments.
Agonist-induced Calcium Mobilization and ERK1/2 Phosphorylation by Wild Type FPR2/ALX and Its Nonphosphorylated Mutant Form FPR2-ABC
Ligand binding to N-formyl peptide receptors induces the activation of a number of effectors triggering intracellular signaling pathways, thereby initiating cellular responses such as chemotaxis, superoxide production, and degranulation. Despite the persistent presence of chemoattractants, the intracellular signaling events elicited by chemoattractant receptors are transient. This attenuated responsiveness is thought to result from the desensitization of receptors through their phosphorylation and rapid sequestration. One would thus predict that the lack of phosphorylation of FPR2-ABC should result in prolonged intracellular signaling.
To compare the functionality of wild type and nonphosphorylated forms of FPR2/ALX, we stably expressed each receptor in HL60 cells, a myeloid cell line of physiological relevance that does not express FPR2/ALX unless differentiated in neutrophil-like cells. The ability of wild type and mutant expressing cells to mobilize intracellular calcium after agonist stimulation was determined by monitoring calcium fluxes with Fura 2. As shown in Fig. 4A, mutant receptor triggered a calcium response similar to that of the wild type receptor. Whether analysis was performed in the presence (Fig. 4A, left panel) or absence (Fig. 4A, right panel) of extracellular calcium, the extents of calcium responses as well as the kinetics of decay to basal calcium levels were similar. Wild type FPR2/ALX and mutant FPR2-ABC were further compared with respect to the activation of the MAP kinase pathway in HL60 cells (Fig. 4B). As previously observed for the calcium response, the absence of FPR2/ALX phosphorylation had no impact on the extent or the kinetics of ERK1/2 activation.
FIGURE 4.

Agonist-induced intracellular signaling by the wild type FPR2/ALX and mutant FPR2-ABC. A, agonist-induced calcium mobilization. HL60 cells expressing the wild type (solid line) and mutant form of FPR2/ALX (dashed line) were analyzed for agonist-induced desensitization of calcium mobilization. Following loading with Fura2, cells were stimulated with 100 nm WKYMVm and assayed for calcium mobilization in the presence (left panel) or absence (right panel) of extracellular calcium. Data are expressed as mean responses ± S.E. (n > 3). B, agonist-induced ERK1/2 phosphorylation. HL60 cells expressing the wild type FPR2/ALX and the mutant FPR2-ABC were stimulated with 100 nm WKYMVm for various periods of time. Phosphorylation of ERK1/2 was visualized by immunoblotting with anti-phospho-ERK1/2 antibody (upper panel). Blots are representative of three experiments. Western blot analyses with an anti-ERK2 antibody were performed to check that an equal amount of ERK2 was present in each sample (not shown). Phosphorylation of ERK2 was subsequently quantified by densitometry (lower panel). The results are expressed in percent of maximum phosphorylation in wild type expressing cells. Values in graphs represent the mean ± S.E. from three independent experiments.
Desensitization of Calcium Mobilization by Wild Type FPR2/ALX and Mutant FPR2-ABC
To investigate receptor desensitization, we compared the ability of wild type FPR2/ALX and FPR2-ABC to stimulate intracellular calcium movement following a previous exposure to agonist. Desensitization was accomplished by treating stably transfected HL60 cells with a saturating dose of WKYMVm for 10 min at 37 °C. Cells were washed extensively to remove the peptide, and assayed for calcium mobilization in response to various concentrations of WKYMVm. Wild type receptor expressing cells were desensitized as evidenced by the absence of response even at high concentrations of ligand (Fig. 5A). In contrast, the nonphosphorylated mutant FPR2-ABC was refractory to desensitization, exhibiting a similar dose-response curve after the second agonist stimulation as compared with cells that had not been desensitized (Fig. 5B). Thus, although the lack of phosphorylation of FPR2/ALX did not result in prolonged or enhanced cellular responses, phosphorylation of the C terminus of FPR2/ALX is essential for its desensitization.
FIGURE 5.

Desensitization of calcium mobilization by wild type FPR2/ALX and phosphorylation-deficient mutant FPR2-ABC. HL60 cells expressing FPR2/ALX (A) and FPR2-ABC (B) were analyzed for agonist-induced desensitization of calcium mobilization. Fura-2-loaded cells were incubated with either 100 nm WKYMVm (black symbols) or vehicle (clear symbols) for 10 min at 37 °C. Cells were then washed three times at room temperature and subsequently assayed for calcium mobilization in response to various doses of WKYMVm. Data expressed in percent of the maximum response are representative of three independent experiments.
FPR3 Constitutive Phosphorylation
To identify the determinant(s) responsible for the basal phosphorylation of FPR3, we first engineered two reciprocal chimeras, FPR2-R3 (FPR2/ALX with the C terminus of FPR3) and FPR3-R2 (FPR3 with the C terminus of FPR2/ALX) (see Fig. 1C). These chimeras were transiently expressed in HEK293 cells and their phosphorylation was compared with FPR2 and FPR3 in the absence of agonist stimulation. As shown in Fig. 6A, the C terminus of FPR3, in the context of the chimera FPR2-R3 was not phosphorylated, whereas radioactive phosphate was incorporated in the converse chimera FPR3-R2. Thus, in the absence of agonist, FPR3 incorporated 32P whether it bears its own C-terminal domain or that of FPR2/ALX. This suggests that it is the core of the receptor extending from the N terminus to the end of the seventh transmembrane domain rather than the cytoplasmic C-terminal amino acid sequence itself that confers to FPR3 the capacity to be phosphorylated in the absence of stimulation.
FIGURE 6.
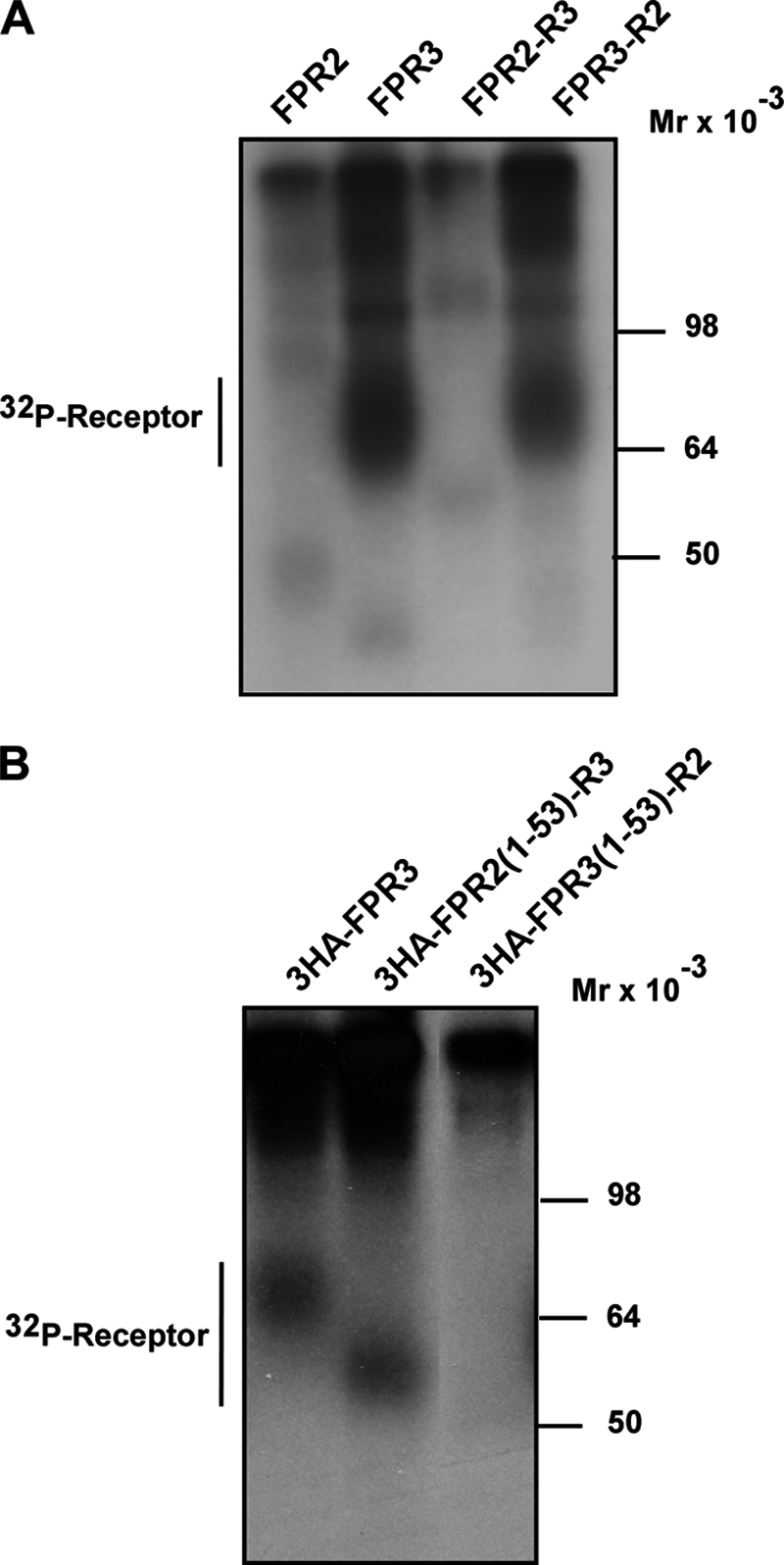
Constitutive phosphorylation of FPR3 and chimeric receptors obtained by swapping C-terminal or N-terminal domains. HEK293 cells expressing either FPR3 or the indicated chimeric receptors were metabolically labeled with [32P]orthophosphoric acid. Cells were not challenged with agonist. Receptors were immunoprecipitated from cell lysates with the anti-FPR2/ALX antibody (A) or with an anti-HA antibody when cells expressed 3HA-tagged receptors (B). Receptor immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography. Data are representative of three independent experiments.
To gain further insight on the molecular determinants responsible for basal phosphorylation of FPR3, additional chimeric receptors were engineered by swapping N-terminal domains and transiently expressed in HEK293 cells. These chimeras were constructed from receptors tagged with a triple HA sequence at the N terminus. Ligand binding or signaling profiles of wild type FPR2/ALX and FPR3 receptors were not affected by insertion of the 3HA sequence (data not shown). The chimera named 3HA-FPR2(1–53)-R3 combined the N-terminal 3HA-tagged domain (amino acids 1 to 53) of FPR2/ALX with amino acids 54 to 353 of FPR3. The converse chimeric receptor was named 3HA-FPR3(1–53)-R2 (see Fig. 1C). The domain 1–53 encompasses the extracellular N terminus and the first transmembrane domain of the receptors. As shown in Fig. 6B, the chimeric receptor 3HA-FPR2(1–53)-R3 exhibited a level of basal phosphorylation similar to that observed with 3HA-FPR3. The faster electrophoretic mobility results from the difference in the number of glycosylation sites in the N-terminal domain. No basal phosphorylation could be detected with the reciprocal chimera 3HA-FPR3(1–53)-R2. Thus, N-terminal domain 1–53 does not contain the determinants that drive the specific conformation responsible for basal phosphorylation of FPR3.
Constitutive Internalization of FPR3
Phosphorylation of the receptor C-terminal domain is a key step for agonist-mediated receptor internalization. We have previously shown by immunofluorescence experiments that upon binding of the agonist peptide WKYMVm, both FPR2/ALX and FPR3 expressed in RINm5F cells accumulated in large intracellular vesicles (3). However, in the absence of stimulation, distribution of the two receptors was different. FPR2/ALX was evenly distributed at the cell surface as evidenced by fluorescent immunostaining of the plasma membrane, whereas FPR3 was mainly detected as a punctuate intracellular fluorescence.
To further investigate the peculiar behavior of FPR3, FPR2/ALX and FPR3 were transiently expressed in HEK293 cells. As shown in Fig. 7A, FPR2/ALX was evenly distributed at the cell surface, as illustrated by the bright fluorescent staining of the plasma membrane (Fig. 7A, left panels). In contrast, in cells expressing FPR3, fluorescent vesicles were seen throughout the cytoplasm (Fig. 7A, right panels). The distribution pattern of constitutively phosphorylated chimera FPR3-R2 was also examined. As shown in Fig. 7B, the fluorescence pattern of this chimera was similar to that of FPR3, suggesting that the constitutive phosphorylation of the receptor may be associated with a constitutive internalization of the receptor in the absence of agonist stimulation. However, the chimeric receptor (FPR3-R2-ABC), which comprises the nonphosphorylated C terminus of FPR2-ABC exhibited also a punctuate distribution throughout the cytoplasm similar to that observed with FPR3 and FPR3-R2.
FIGURE 7.
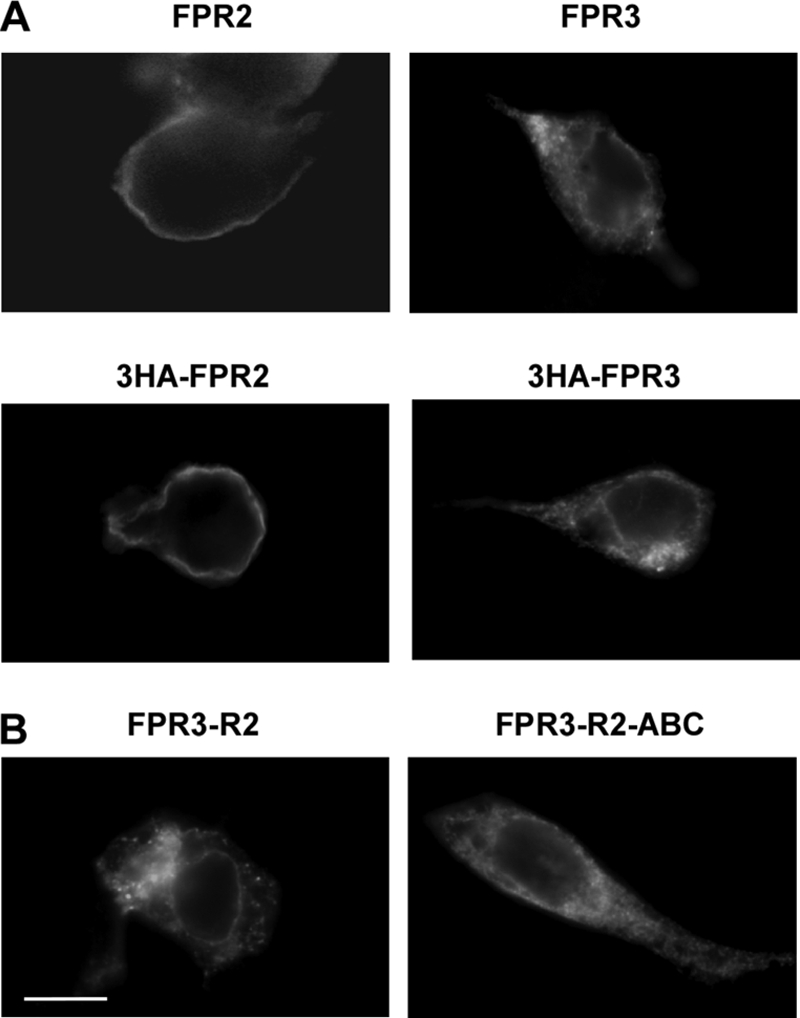
Cellular distribution of FPR2, FPR3, and chimeric receptors FPR3-R2 and FPR3-R2-ABC. HEK293 cells were transiently transfected with FPR2/ALX or FPR3 (tagged or not with 3HA) (A), or the chimeric receptors obtained by swapping C-terminal domains (B). Cells were fixed and permeabilized. The cellular distribution of receptors was visualized by labeling with either a polyclonal anti-FPR2/ALX antibody, which cross-react with FPR3, or a monoclonal anti-HA antibody followed by immunofluorescence staining with Alexa Fluor 488-conjugated secondary antibodies. Scale bar, 10 μm.
To directly examine whether wild type and mutant FPR3 reached the cell surface and rapidly accumulate in endocytic vesicles or were retained in the endoplasmic reticulum due to improper protein folding, we performed antibody uptake experiments using receptors tagged at the N terminus with a triple HA epitope. As shown in Fig. 7A, the presence of the 3HA tag did not modify the distribution pattern of the receptors. This experimental setup allowed the labeling of receptors that were accessible to the anti-HA antibody, i.e. at the cell surface, during a 30-min incubation period. After this period of time, cells were washed and immediately fixed and permeabilized. As a control, cells were fixed and permeabilized, and then incubated with the anti-HA antibody. Subsequently, antibody-fed and control cells were visualized with an Alexa Fluor 488-conjugated secondary antibody (Fig. 8, right and left panels, respectively). When the anti-HA uptake was performed at 37 °C, the 3HA-FPR3 receptor was mostly visualized inside the cells (Fig. 8A, right panel). This indicates that the receptor had first reached the cell surface, because it had bound the anti-HA antibody, and had been subsequently internalized in the absence of agonist. When the antibody uptake experiment was conducted at 4 °C (a temperature that inhibits internalization), a very faint labeling of the membrane was detected. This indicates that only a small amount of 3HA-FPR3 was present on the cell surface. Consistent with plasma membrane distribution and agonist-dependent internalization of FPR2/ALX (12), the antibody uptake experiments performed with 3HA-FPR2 indicated that the receptor was essentially present on the cell surface and was not internalized in the absence of agonist (Fig. 8B). However, it was visualized as a line of dots, probably due to an anti-HA-mediated receptor clustering. The nonphosphorylated chimera 3HA-FPR3-R2-ABC behaved as 3HA-FPR3 and was constitutively internalized (Fig. 8C). The constitutive internalization of FPR3 can thus be considered as independent of the phosphorylation status of the C terminus. Unexpectedly, in fixed and permeabilized cells, the chimera 3HA-FPR2(1–53)-R3 was present on the cell surface and not intracellularly as in the case of the wild type receptor (Fig. 8D, left panel). In the anti-HA uptake experiments (Fig. 8D, right panel), this chimera was mainly visualized as a series of dots at the cell periphery, indicating that it was not constitutively internalized. The converse chimera 3HA-FPR3(1–53)-R2 behaved as FPR3 and was localized in small cytoplasmic vesicles (Fig. 8E). Thus, amino acid sequence 1–53 of FPR3 drives its constitutive internalization most probably by imposing to the core of the receptor a particular conformation.
FIGURE 8.

Constitutive internalization of FPR3. Antibody uptake (right panels) was performed on living cells. Cells expressing the indicated HA-tagged receptors were incubated with a monoclonal anti-HA antibody for 30 min at 37 °C (or at 4 °C to inhibit internalization) to label the receptors present on the cell surface. Subsequently, cells were fixed, permeabilized, incubated with anti-mouse Alexa Fluor 488-conjugated antibody, and processed for fluorescence microscopy. In parallel (left panels), the distribution of the receptors was visualized in cells fixed and permeabilized prior to incubation with monoclonal anti-HA antibody and immunofluorescence staining. Scale bar, 10 μm.
Because these two chimeric receptors presented a different cellular distribution, we examined the effect of the two FPR3 ligands, F2L and WKYMVm, regarding their capacity to induce the phosphorylation of these chimeras. The F2L-induced incorporation of 32P was markedly increased in 3HA-FPR2(1–53)-R3 as compared with 3HA-FPR3 (Fig. 9A, right and left panels, respectively). This observation is consistent with the massive cell surface expression of 3HA-FPR2(1–53)-R3 (see Fig. 8E). Unexpectedly, this cell surface-expressed chimera was not phosphorylated upon WKYMVm stimulation (Fig. 9A, left panel). The robust F2L-induced phosphorylation of the chimeric receptor, 3HA-FPR2(1–53)-R3, prompted us to investigate whether, in the presence of F2L, the chimera was endocytosed through the classical β-arrestin-dependent pathway. Cells were cotransfected with the chimera and β-arrestin1-EGFP, which is known to interact with phosphorylated chemoattractant receptors, upon agonist binding (12). In the absence of F2L, the receptor was localized at the cell surface, whereas β-arrestin1-EGFP was evenly distributed in the cytoplasm (Fig. 9B). Upon addition of F2L, both the chimeric receptor and β-arrestin1-EGFP accumulated and colocalized in a perinuclear compartment.
FIGURE 9.

Agonist-induced phosphorylation and internalization of chimeric receptors. A, HEK293 cells expressing 3HA-tagged FPR3 or the chimeric receptor in which the N-terminal domain of FPR2/ALX and FPR3 has been exchanged (3HA-FPR2(1–53)-R3), were metabolically labeled with [32P]orthophosphoric acid. Cells were not stimulated or stimulated for 15 min at 37 °C with 1 μm WKYMVm or 1 μm F2L as indicated. After cell lysis, receptors were immunoprecipitated with an anti-HA antibody. Receptor immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography. Data are representative of two independent experiments. B, HEK293 cells were cotransfected with the chimera 3HA-FPR2(1–53)-R3 and β-arrestin-1 in fusion with EGFP. Cells were not stimulated or stimulated with 1 μm F2L for 30 min, at 37 °C. Cells were fixed and permeabilized. The 3HA-FPR2(1–53)-R3 receptor was visualized by incubation with monoclonal anti-HA antibody and staining with a red fluorescent Alexa Fluor 568-conjugated anti-mouse antibody. Scale bar, 10 μm.
Endocytic Pathway Involved in the Constitutive Internalization of FPR3
We next examined which internalization pathway could be involved in the constitutive internalization of FPR3. Beside macropinocytosis, three basic mechanisms are involved in macromolecule endocytosis: clathrin-mediated endocytosis, caveolae-mediated endocytosis, and a number of clathrin- and caveolae-independent internalization pathways. GPCR internalization is, in many cases, a ligand-mediated phenomenon that occurs through clathrin-coated pits. The β-arrestins are thought to act as scaffolding proteins in coupling GPCRs to clathrin-coated vesicles (6, 23, 24). Agonist stimulation of GPCRs promotes the formation of receptor-containing vesicles, which are pinched off from the plasma membrane and translocated into endocytic compartments.
To determine whether clathrin is required for constitutive endocytosis of FPR3 in the absence of agonist stimulation, 3HA-FPR3 was coexpressed in HEK293 cells with a fragment of β-arrestin 1 (amino acids 318–419) in fusion with the enhanced green fluorescent protein (β-Arr(318–419)-EGFP). This fragment, which constitutively binds to clathrin is unable to interact with phosphorylated GPCRs. Consequently, it acts as a dominant-negative mutant that inhibits agonist-stimulated endocytosis of GPCRs via the classical clathrin- and β-arrestin-dependent internalization pathway (25). As previously observed in RINm5F cells (21), the β-Arr(318–419)-EGFP was distributed throughout the cell in small intracellular vesicles as well as in large perinuclear vesicles (Fig. 10A). The 3HA-FPR3 was detected with a red fluorescent Alexa 568-conjugated goat anti-mouse antibody. As shown in Fig. 10A, the distribution of 3HA-FPR3 was not affected by the presence of the β-arrestin fragment. Additional experiments were performed that combined the use of anti-HA uptake to track HA-FPR3 and the capture of transferrin-Alexa Fluor 568 conjugate by the transferrin receptor, a marker of the clathrin endocytic pathway. As seen in Fig. 10B, the punctuate distribution of the green fluorescence of 3HA-FPR3 showed minimal colocalization with the red fluorescence of the transferrin-labeled receptor. Thus, 3HA-FPR3 and the transferrin receptor seemed to be located in distinct endocytic vesicles. Altogether, the results strongly suggest that a clathrin-independent pathway is involved in the constitutive internalization of FPR3.
FIGURE 10.

Endocytosis pathway involved in FPR3 constitutive internalization. A, the 3HA-FPR3 receptor was coexpressed in HEK293 cells with a fragment of β-arrestin 1 (amino acids 318–419) in fusion with EGFP (β-Arr(318–419)-EGFP). The 3HA-FPR3 was labeled with the monoclonal anti-HA and a red fluorescent Alexa 568-conjugated anti-mouse antibody. B, antibody uptake experiments were performed on living cells expressing 3HA-FPR3. The cells were incubated with anti-HA antibody for 30 min at 37 °C. Internalization of the transferrin receptor was visualized by adding transferrin-Alexa Fluor 568 conjugate during the last 15 min of incubation. Cells were fixed, permeabilized, and incubated with an Alexa Fluor 488-conjugated anti-mouse antibody to label the anti-HA antibody bound to the internalized 3HA-FPR3. C, antibody uptake was performed in HEK293 cells cotransfected with 3HA-FPR3 and the dominant-negative mutant of dynamin (dynamin K44A). D, antibody uptake was performed in HEK293 cells expressing the 3HA-FPR3 receptor in the presence of filipin III. Scale bar, 10 μm.
Several distinct endocytic pathways are regulated by dynamin, a multidomain GTPase, involved in the scission of newly formed vesicles from the membrane (25, 26). Dynamin is required in clathrin- and caveolae-mediated endocytosis, as well as in some clathrin- and caveolae-independent endocytic pathways (27). The 3HA-FPR3 was coexpressed with dynamin K44A, a dominant-negative mutant of dynamin previously shown to inhibit endocytosis via clathrin-coated pits (26). Judging from the staining of 3HA-FPR3 as bright dots at the periphery of the cells in anti-HA uptake experiments (Fig. 10C), dynamin is required for the constitutive internalization of FPR3.
To better characterize the clathrin-independent pathway for the constitutive internalization of FPR3, we examined the effects induced by disruption of lipid rafts and caveolae function. Caveolae are a subset of lipid rafts that constitute specialized cholesterol-rich invaginations of the plasma membrane thought to participate in clathrin-independent endocytosis (28). Filipin III is a sterol-binding agent that binds to cholesterol and disrupts caveolae structure and function. Sensitivity to filipin is thus a general indicator that endocytosis of particular cargos is mediated by caveolae. When the anti-HA uptake was performed in the presence of filipin III, 3HA-FPR3 was visualized as a faint dotted line at the plasma membrane (Fig. 10D), indicating that filipin III inhibited the constitutive internalization of the receptor. The concentration of filipin III (5 μg/ml) used in this study was similar to that previously shown in other cell types to effectively reduce plasma membrane cholesterol levels with minimal cell damage. We did not detect significant changes in cell morphology, and cell viability assessed by trypan blue was not affected. Taken together our results suggest that the constitutive internalization of FPR3 occurs through a clathrin-independent but dynamin- and caveolae-dependent mechanism.
DISCUSSION
The N-formyl peptide receptors FPR1, FPR2/ALX, and FPR3 belong to the chemoattractant receptor family. They are expressed mainly in phagocytic leukocytes and thought to be important in host defense and inflammation. Following an initial exposure to ligand, phagocytic leukocytes rapidly but reversibly become unresponsive to subsequent stimulation with chemoattractants. Multiple mechanisms contribute to this desensitization, including receptor phosphorylation and endocytosis. Under ligand stimulation, FPR1 and FPR2/ALX are rapidly phosphorylated in a dose-dependent manner (3, 29, 30). The present study emphasizes the similarities between FPR1 and FPR2/ALX regarding the agonist-dependent phosphorylation process. Similarly to FPR1, the phospho-acceptor sites of FPR2/ALX appear to be restricted to serines and threonines located in the carboxyl-terminal region and the phosphorylation process to occur through a hierarchical mechanism. The phosphorylation of Ser/Thr in the sequence 325SEDSAPT331 (termed cluster B in the present study) was essential for the subsequent phosphorylation of other Ser/Thr. The sequence 325SEDSAPT331 is in a similar position to that of cluster D328STQTS332 in FPR1, of which phosphorylation of Ser/Thr is required to allow the phosphorylation of other phospho-acceptor sites (13). Replacement of all Ser/Thr in the C-terminal domain of FPR2/ALX (mutant termed FPR2-ABC) did not alter agonist binding but resulted in the lack of phosphorylation. In the case of the C5a chemoattractant receptor, the lack of phosphorylation has been previously shown to result in sustained calcium mobilization and extracellular signal-regulated kinase (ERK1/2) activation (31). Therefore, one would predict that the phosphorylation-deficient mutant FPR2-ABC should exhibit prolonged intracellular signaling. Surprisingly, the stimulation of wild type receptor and phosphorylation-deficient mutant triggered intracellular calcium mobilization and ERK1/2 activation with similar magnitude and duration as if the absence of phosphorylation had no impact on the cellular responses. Nevertheless, the phosphorylation-deficient receptor FPR2-ABC was refractory to desensitization. After being challenged with a saturating concentration of agonist, FPR2-ABC was still able to respond to a second stimulation with the same agonist, whereas wild type FPR2/ALX was refractory to subsequent ligand stimulation. Therefore, as observed for FPR1 (32), phosphorylation of FPR2/ALX is a necessary and sufficient step in cellular desensitization.
Despite a high level of amino acid identity in the C-terminal domain of the three formyl peptide receptors, only FPR3 displayed a significant basal level of phosphorylation in the absence of ligand binding. It is worth noting that the C-terminal domain of FPR3 contains two additional amino acids (Val-Pro329). The proline residue may bring about a conformation favoring the phosphorylation of FPR3 in a resting state. After stimulation by the high-affinity ligand F2L, the incorporation of radioactive phosphate in FPR3 was modestly increased, suggesting that only a few additional phosphoacceptor sites are prone to incorporate phosphate upon agonist-induced conformational changes. Domain swapping between FPR2/ALX and FPR3 indicated that the C terminus sequence of each receptor can undergo a constitutive phosphorylation only when linked to the core of FPR3. Thus, it is the global conformation of the receptor rather than the sequence of its C terminus that directs the constitutive phosphorylation of FPR3. Of particular interest is the observation that the exchange of the N-terminal domain (1–53) of FPR3 for that of FPR2/ALX confers to the resulting chimera the capacity to be highly phosphorylated upon F2L binding. The chimeric receptor 3HA-FPR2(1–53)-R3 could thus provide a valuable tool to identify the phosphoacceptor sites of FPR3.
Another peculiarity of FPR3 is its cellular distribution. Corroborating our previous observation (3), we found that FPR3 was expressed primarily in small intracellular vesicles throughout the cell, as monitored by immunofluorescence staining. This cellular distribution in the absence of agonist stimulation suggested that FPR3 was undergoing constitutive endocytosis. However, staining permeabilized cells for receptor does not definitively distinguish the potentially misfolded receptor retained in the secretory pathway from the receptor that had been internalized in the absence of agonist. An experimental design, monitoring receptor-bound antibody uptake allowed us to show that FPR3 reached the cell surface and was endocytosed in the absence of agonist. To further assess the determinants that underlie this phenomenon, we examined the behavior of chimeric receptors in which domains have been exchanged between FPR3 and FPR2/ALX. The latter is not internalized in the absence of ligand. C-terminal domain swapping showed that the core of FPR3 dictates both basal phosphorylation and constitutive internalization of the receptor. However, the observation that the phosphorylation deficient 3HA-FPR3-R2-ABC chimera was constitutively internalized suggests that basal phosphorylation and constitutive internalization are likely to be independent events. This view is also supported by the fact that N-terminal domain swapping did not interfere with receptor constitutive phosphorylation, whereas it had an impact on receptor localization.
Growing evidence indicates that GPCRs are allosteric proteins that can adopt many distinct conformations, some of which may promote continuous endocytosis in the absence of stimulation and more attention is dedicated now to what appears to be an important aspect of receptor regulation. In many cases, GPCRs are constitutively internalized via a β-arrestin-dependent pathway and undergo a clathrin-dependent constitutive internalization (33). However, other mechanisms seem to be involved. For example, the constitutive internalization of a mutant of the protease-activated receptor 1 is not affected in β-arrestin knockout mouse embryonic fibroblast cells (34). The constitutive β-arrestin-independent internalization of protease-activated receptor 1 is regulated by the clathrin adapter AP2 (35). In general, cargo proteins at the plasma membrane enter the cell through a variety of endocytic mechanisms that can be divided into two main groups: clathrin-dependent and -independent endocytosis. A number of GPCRs undergo a clathrin-independent constitutive internalization occurring through lipid rafts. Caveolae, a distinct subset of lipid rafts, are enriched in the structural scaffolding protein caveolin, which is thought to sequester membrane proteins, including GPCRs. For example, it has been shown that a phosphorylation-deficient mutant of PAR2 (protease activated receptor 2) is constitutively internalized through a dynamin-dependent but clathrin- and β-arrestin-independent pathway (36). A caveolin-binding motif is present within the carboxyl-terminal portion of the seventh transmembrane domain of PAR2 (YYFVSHDF, ΦXΦXXXXΦ), which may interact with caveolin, thereby facilitating GPCRs recruitment into caveolae and endocytosis. In the present study, results indicate that FPR3 is constitutively internalized through a clathrin- and β-arrestin-independent pathway as suggested by the absence of vesicular colocalization of FPR3 with the transferrin receptor and the lack of effect of the clathrin binding domain of β-arrestin. The inhibitory effect of fillipin III on the constitutive internalization of FPR3 suggests a caveolae-mediated mechanism. Interestingly, we identified a putative caveolin-binding motif YVFMGRNF matching the canonical ΦXΦXXXXΦ caveolin binding motif at the end of the seventh transmembrane domain of FPR3. Although caveolin expression is controversial in neutrophils, caveolae and caveolin have been identified in monocytes, macrophages, and dendritic cells (37–39). The expression and properties of caveolae and caveolin in immune cells are likely to depend on several factors, including cell type and their states of activation and/or maturation. Caveolae may thus play an important role in specific immune-cell functions.
GPCR constitutive internalization has been proposed as a means to maintain a quiescent intracellular pool of receptor protected from desensitization and allow the delivery of functional receptor to the cell surface when required. The significance of the high basal internalization and large intracellular pool of FPR3 is presently unclear. Phosphorylation and internalization appeared to be independent events, suggesting that constitutive endocytosis may not be the consequence of a basal activity of the receptor. Furthermore, we were unable to detect G-protein activation in FPR3 expressing cells. FPR3 internalization may also serve an alternate, ligand scavenging function similar to chemokine receptors D6 and CXCR7 (40, 41) and the chemoattractant C5a receptor-like 2 (C5L2) (42). Such decoy receptors that do not transduce signal have been shown to exhibit minimal plasma membrane expression but undergo rapid constitutive recycling to bind extracellular ligand and internalize it for degradation. A recent study concerning C5L2 (43) offers another new interesting prospect for constitutively internalized receptors. C5L2 is mainly localized in intracellular vesicles and functions as an intracellular receptor that forms heterodimers with C5a-activated C5aR. C5L2 is thought to act as a negative modulator of C5aR signal transduction. This represents a novel regulatory mechanism in the activation of myeloid cells and fine-tuning of host defense. Constitutive internalization of FPR3 may similarly serve to regulate the function of one or two other formyl peptide receptors. The differential expression of the three formyl peptide receptors, FPR1, FPR2/ALX, and FPR3, in human myeloid cells (4, 44–46) may reflect their respective functions in the activation of various types of immune cells. Unveiling the function of FPR3 will provide an additional clue to the complex regulation of innate immunity.
Footnotes
- FPR
- formyl peptide receptors
- GPCR
- G protein-coupled receptors
- EGFP
- enhanced green fluorescent protein
- C5L2
- C5a receptor-like 2.
REFERENCES
- 1. Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., Serhan C. N., Murphy P. M. (2009) Pharmacol. Rev. 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabiet M. J., Huet E., Boulay F. (2005) Eur. J. Immunol. 35, 2486–2495 [DOI] [PubMed] [Google Scholar]
- 3. Christophe T., Karlsson A., Dugave C., Rabiet M. J., Boulay F., Dahlgren C. (2001) J. Biol. Chem. 276, 21585–21593 [DOI] [PubMed] [Google Scholar]
- 4. Migeotte I., Riboldi E., Franssen J. D., Grégoire F., Loison C., Wittamer V., Detheux M., Robberecht P., Costagliola S., Vassart G., Sozzani S., Parmentier M., Communi D. (2005) J. Exp. Med. 201, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McDonald P. H., Lefkowitz R. J. (2001) Cell. Signal. 13, 683–689 [DOI] [PubMed] [Google Scholar]
- 6. Goodman O. B., Jr., Krupnick J. G., Santini F., Gurevich V. V., Penn R. B., Gagnon A. W., Keen J. H., Benovic J. L. (1996) Nature 383, 447–450 [DOI] [PubMed] [Google Scholar]
- 7. Laporte S. A., Oakley R. H., Holt J. A., Barak L. S., Caron M. G. (2000) J. Biol. Chem. 275, 23120–23126 [DOI] [PubMed] [Google Scholar]
- 8. Vines C. M., Revankar C. M., Maestas D. C., LaRusch L. L., Cimino D. F., Kohout T. A., Lefkowitz R. J., Prossnitz E. R. (2003) J. Biol. Chem. 278, 41581–41584 [DOI] [PubMed] [Google Scholar]
- 9. Bennett T. A., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2001) J. Biol. Chem. 276, 49195–49203 [DOI] [PubMed] [Google Scholar]
- 10. Gilbert T. L., Bennett T. A., Maestas D. C., Cimino D. F., Prossnitz E. R. (2001) Biochemistry 40, 3467–3475 [DOI] [PubMed] [Google Scholar]
- 11. Key T. A., Bennett T. A., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2001) J. Biol. Chem. 276, 49204–49212 [DOI] [PubMed] [Google Scholar]
- 12. Huet E., Boulay F., Barral S., Rabiet M. J. (2007) Cell. Signal. 19, 1939–1948 [DOI] [PubMed] [Google Scholar]
- 13. Prossnitz E. R., Kim C. M., Benovic J. L., Ye R. D. (1995) J. Biol. Chem. 270, 1130–1137 [DOI] [PubMed] [Google Scholar]
- 14. Maestes D. C., Potter R. M., Prossnitz E. R. (1999) J. Biol. Chem. 274, 29791–29795 [DOI] [PubMed] [Google Scholar]
- 15. Potter R. M., Key T. A., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2002) J. Biol. Chem. 277, 8970–8978 [DOI] [PubMed] [Google Scholar]
- 16. Yon J., Fried M. (1989) Nucleic Acids Res. 17, 4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhen L., King A. A., Xiao Y., Chanock S. J., Orkin S. H., Dinauer M. C. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 9832–9836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tardif M., Rabiet M. J., Christophe T., Milcent M. D., Boulay F. (1998) J. Immunol. 161, 6885–6895 [PubMed] [Google Scholar]
- 19. Naik N., Giannini E., Brouchon L., Boulay F. (1997) J. Cell Sci. 110, 2381–2390 [DOI] [PubMed] [Google Scholar]
- 20. Dahlgren C., Christophe T., Boulay F., Madianos P. N., Rabiet M. J., Karlsson A. (2000) Blood 95, 1810–1818 [PubMed] [Google Scholar]
- 21. Braun L., Christophe T., Boulay F. (2003) J. Biol. Chem. 278, 4277–4285 [DOI] [PubMed] [Google Scholar]
- 22. Giannini E., Brouchon L., Boulay F. (1995) J. Biol. Chem. 270, 19166–19172 [DOI] [PubMed] [Google Scholar]
- 23. Barak L. S., Ferguson S. S., Zhang J., Caron M. G. (1997) J. Biol. Chem. 272, 27497–27500 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Barak L. S., Winkler K. E., Caron M. G., Ferguson S. S. (1997) J. Biol. Chem. 272, 27005–27014 [DOI] [PubMed] [Google Scholar]
- 25. Krupnick J. G., Santini F., Gagnon A. W., Keen J. H., Benovic J. L. (1997) J. Biol. Chem. 272, 32507–32512 [DOI] [PubMed] [Google Scholar]
- 26. Damke H., Baba T., Warnock D. E., Schmid S. L. (1994) J. Cell Biol. 127, 915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hinshaw J. E. (2000) Annu. Rev. Cell Dev. Biol. 16, 483–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurzchalia T. V., Parton R. G. (1999) Curr. Opin. Cell Biol. 11, 424–431 [DOI] [PubMed] [Google Scholar]
- 29. Tardif M., Mery L., Brouchon L., Boulay F. (1993) J. Immunol. 150, 3534–3545 [PubMed] [Google Scholar]
- 30. Ali H., Richardson R. M., Tomhave E. D., Didsbury J. R., Snyderman R. (1993) J. Biol. Chem. 268, 24247–24254 [PubMed] [Google Scholar]
- 31. Christophe T., Rabiet M. J., Tardif M., Milcent M. D., Boulay F. (2000) J. Biol. Chem. 275, 1656–1664 [DOI] [PubMed] [Google Scholar]
- 32. Prossnitz E. R. (1997) J. Biol. Chem. 272, 15213–15219 [DOI] [PubMed] [Google Scholar]
- 33. Xu Z. Q., Zhang X., Scott L. (2007) Acta Physiol. 190, 39–45 [DOI] [PubMed] [Google Scholar]
- 34. Paing M. M., Stutts A. B., Kohout T. A., Lefkowitz R. J., Trejo J. (2002) J. Biol. Chem. 277, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 35. Paing M. M., Johnston C. A., Siderovski D. P., Trejo J. (2006) Mol. Cell. Biol. 26, 3231–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ricks T. K., Trejo J. (2009) J. Biol. Chem. 284, 34444–34457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kiss A. L., Kittel A. (1995) Cell Biol. Int. 19, 527–538 [DOI] [PubMed] [Google Scholar]
- 38. Kiss A. L., Geuze H. J. (1997) Eur. J. Cell Biol. 73, 19–27 [PubMed] [Google Scholar]
- 39. Werling D., Hope J. C., Chaplin P., Collins R. A., Taylor G., Howard C. J. (1999) J. Leukocyte Biol. 66, 50–58 [DOI] [PubMed] [Google Scholar]
- 40. Graham G. J., McKimmie C. S. (2006) Trends Immunol. 27, 381–386 [DOI] [PubMed] [Google Scholar]
- 41. Naumann U., Cameroni E., Pruenster M., Mahabaleshwar H., Raz E., Zerwes H. G., Rot A., Thelen M. (2010) PLoS One 5, e9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scola A. M., Johswich K. O., Morgan B. P., Klos A., Monk P. N. (2009) Mol. Immunol. 46, 1149–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bamberg C. E., Mackay C. R., Lee H., Zahra D., Jackson J., Lim Y. S., Whitfeld P. L., Craig S., Corsini E., Lu B., Gerard C., Gerard N. P. (2010) J. Biol. Chem. 285, 7633–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang D., Chen Q., Le Y., Wang J. M., Oppenheim J. J. (2001) J. Immunol. 166, 4092–4098 [DOI] [PubMed] [Google Scholar]
- 45. Yang D., Chen Q., Gertz B., He R., Phulsuksombati M., Ye R. D., Oppenheim J. J. (2002) J. Leukocyte Biol. 72, 598–607 [PubMed] [Google Scholar]
- 46. Devosse T., Guillabert A., D'Haene N., Berton A., De Nadai P., Noel S., Brait M., Franssen J. D., Sozzani S., Salmon I., Parmentier M. (2009) J. Immunol. 182, 4974–4984 [DOI] [PubMed] [Google Scholar]