

Abstract
Botulinum Neurotoxins (BoNTs) are organized into seven serotypes, A-G. Although several BoNT serotypes enter neurons through synaptic vesicle cycling utilizing dual receptors (a ganglioside and a synaptic vesicle-associated protein), the entry pathway of BoNT/D is less well understood. Although BoNT/D entry is ganglioside-dependent, alignment and structural studies show that BoNT/D lacks key residues within a conserved ganglioside binding pocket that are present in BoNT serotypes A, B, E, F, and G, which indicate that BoNT/D-ganglioside interactions may be unique. In this study BoNT/D is shown to have a unique association with ganglioside relative to the other BoNT serotypes, utilizing a ganglioside binding loop (GBL, residues Tyr-1235—Ala-1245) within the receptor binding domain of BoNT/D (HCR/D) via b-series gangliosides, including GT1b, GD1b, and GD2. HCR/D bound gangliosides and entered neurons dependent upon the aromatic ring of Phe-1240 within the GBL. This is the first BoNT-ganglioside interaction that is mediated by a phenylalanine. In contrast, Trp-1238, located near the N terminus of the ganglioside binding loop, was mostly solvent-inaccessible and appeared to contribute to maintaining the loop structure. BoNT/D entry and intoxication were enhanced by membrane depolarization via synaptic vesicle cycling, where HCR/D colocalized with synaptophysin, a synaptic vesicle marker, but immunoprecipitation experiments did not detect direct association with synaptic vesicle protein 2. Thus, BoNT/D utilizes unique associations with gangliosides and synaptic vesicles to enter neurons, which may facilitate new neurotoxin therapies.
Keywords: Bacterial Toxins, Botulinum Toxin, Cell Surface Receptor, Ganglioside, Glycolipid Structure, Membrane Trafficking, Neurons, Neurotoxin, Receptors
Introduction
Botulinum neurotoxins (BoNTs)3 are the most potent protein toxins for humans (1) and are categorized as Category A agents (2). Consumption of less than 1 ng/kg of body weight, often through contaminated food, may elicit sustained flaccid paralysis (1). In localized use, BoNT serotypes A and B are therapies for dystonias (2), whereas BoNT serotypes C-G are being evaluated for clinical applications (3). Botulinum neurotoxin serotype D (BoNT/D) was identified in 1929 as the etiological agent in outbreaks of cattle botulism (4). Human intoxication by BoNT/D has not been reported, and early studies reported that human neuromuscular junctions were resistant to BoNT/D intoxication (5). However, BoNT/D efficiently intoxicates rats and mice along with various ruminants (6), which suggests that the natural species preference of BoNT/D may be complex.
BoNTs enter α-motor neurons and inhibit acetylcholine signaling at neuromuscular junctions. BoNTs are single chain AB toxins that comprise an N-terminal 50-kDa light chain (LC, the enzymatic A subunit) and a C-terminal 100-kDa heavy chain (HC) that is cleaved between a disulfide bridge to yield a di-chain toxin. The HC (B subunit) comprises a translocation domain and a receptor binding domain (HCR) (7). The HCR domain of BoNT binds neurons via dual receptors: that is, a ganglioside and a synaptic vesicle (SV) protein (8–13). Cycling of the SV from the nerve terminus facilitates endocytosis of the BoNT-receptor complex. Acidification of the SV luminal compartment triggers LC translocation across the vesicle membrane and into the cytoplasm (14) where LC, a zinc metalloprotease, cleaves target SNARE (soluble N-ethylmaleimide factor attachment protein receptor) proteins, blocking subsequent SV fusion to the plasma membrane (15). BoNT/D as well as serotypes B, G, and F and tetanus toxin (TeNT) cleaves synaptobrevin/vesicle-associated membrane protein 2 (VAMP2) (16, 17). The intoxications are long-lived; acetylcholine blockage can last multiple months depending on the serotype (4).
Clostridium botulinum strains carrying BoNT/D cluster with C. botulinum strains carrying BoNT/C by 16 S RNA analysis (18). Thus far, clustered serotypes have been shown to recognize similar host receptors and use the same entry pathway (19). The A-E-F serotype cluster binds synaptic vesicle protein 2 (SV2) as a receptor, whereas the B-G serotype cluster binds synaptotagmin 1 and 2 (8, 11–13, 20). Host receptors for the C-D serotype cluster are the least well understood among the BoNTs. The association of BoNT/D with gangliosides has been unclear. Early studies reported that BoNT/D bound derivatives of phosphatidylethanolamine and not gangliosides in vitro, although exogenous gangliosides were observed to inhibit toxicity in mouse hemidiaphragm preparations (21, 22). BoNT/D lacks the H…SXWY ganglioside binding motif present in other BoNT serotypes (21, 23, 24). Strotmeier et al. (25) implicated the presence of two carbohydrate binding pockets within BoNT/D; that is, a site for sialic acid and a site for a yet unidentified secondary carbohydrate receptor (indicated by a glycerol molecule resolved in the crystal structure) as dual receptors for BoNT/D. Karalewitz et al. (23) showed that BoNT/D lacked conserved residues that are utilized by other BoNT serotypes to bind gangliosides, although the tertiary structure in the binding pocket was conserved. Recently, Peng et al. (26) reported the entry of BoNT/D into neurons via the SV through the direct binding to SV2, which was not mediated by glycosylation. Strotmeier et al. (25) proposed that BoNT/D uses dual carbohydrates as receptors, analogous to tetanus toxin, which binds sialic acid and lactose, whereas Peng et al. (26) propose that BoNT/D enters like BoNT/A by binding SV2.
To date, BoNT serotypes have been shown to utilize a Trp to coordinate ganglioside interactions. In this study ganglioside binding is shown to be facilitated by a Phe located within the ganglioside binding loop (GBL) and that SV uptake was abolished after disruption of ganglioside binding, implicating the GBL as a primary interaction for target cell recognition.
EXPERIMENTAL PROCEDURES
Expression and Purification of BoNT/HCR in Escherichia coli
Codon-optimized DNA encoding the HCR domain of BoNT/D-1873 (residues 861–1276) was synthesized by EZBiolab (Westfield, IN). DNA encoding HCR/D was subcloned into pET-28a (Novagen), which introduced His6 and 3-FLAG epitopes N-terminal to the HCR. pET28-HCR/D was transformed into E. coli BL-21(DE3) (Stratagene) for protein expression (27). Mutated forms of HCR/D were prepared using a QuikChange site-directed mutagenesis kit (Stratagene). Point mutations were confirmed by DNA sequencing. Mutated HCRs were purified from clarified lysates by Ni2+-nitrilotriacetic acid spin-column chromatography (Qiagen).
Cell Culture
Rat E-18 primary cortical neurons (BrainBits LLC) were cultured for 10–14 days in neurobasal medium (Invitrogen) plus B27 (Invitrogen), Glutamax (Invitrogen), and Primocin (InvivoGen) on glass-bottom 24-well culture dishes (MatTek) coated with poly-d-lysine (Sigma) and laminin (Invitrogen). Neurons were washed twice in DPBS (Invitrogen) and incubated with 40 nm HCRs in high potassium plus calcium (56 mm KCl, 2.2 mm CaCl2, 15 mm HEPES, 145 mm NaCl, 0.5 mm MgCl2, pH 7.4), high potassium minus calcium (56 mm KCl, 15 mm HEPES, 145 mm NaCl, 0.5 mm MgCl2, pH 7.4), low potassium plus calcium (5.6 mm KCl, 2.2 mm CaCl2, 15 mm HEPES, 145 mm NaCl, 0.5 mm MgCl2, pH 7.4), or low potassium without calcium (5.6 mm KCl,15 mm HEPES, 145 mm NaCl, 0.5 mm MgCl2, pH 7.4) for 5 min at 37 °C. Alternatively, cells were cooled to 4 °C and incubated with 40 nm HCRs for 30 min. Cells were washed twice and fixed in 4% (w/v) paraformaldehyde in DPBS for 15 min. For external labeling, cholera toxin B-subunit Alexa488 (Invitrogen) was incubated for 10 min followed by a second paraformaldehyde fix. For internal staining, cells were permeabilized with 0.1% Triton X-100 and 4% formaldehyde in DPBS for 15 min, washed twice, incubated with 150 mm glycine for 15 min, washed twice, and incubated in blocking solution (10% (v/v) fetal bovine serum, 2.5% (w/v) fish skin gelatin (Sigma), 0.1% Triton X-100, 0.05% Tween 20) for 1 h at room temperature. Primary antibodies were incubated overnight at 4 °C in antibody solution (5% (v/v) FBS, 2.5% (w/v) cold fish skin gelatin, 0.1%Triton X-100, 0.05% Tween 20), mouse-anti-FLAG (Sigma, 1:10,000), rat-anti-HA (Roche Applied Science, 1:2000) rabbit-anti-Rab5 (Abcam, 1:200), and guinea pig-anti-synaptophysin (Synaptic Systems, 1:2000). Secondary antibodies (goat-anti-mouse Alexa561, goat-anti-rat Alexa488, goat-anti-rabbit Alexa488, and goat-anti-guinea pig Alexa633 (Invitrogen) at 1:1000 were incubated for 1 h at room temperature. Cells were fixed in 4% paraformaldehyde in DPBS for 20 min and mounted in 200 μl of Citifluor AF-3 mounting media (Electron Microscopy Sciences).
Toxicity Experiments
Rat E-18 primary cortical neurons were cultured as described. BoNT/D and tetanus toxin (10 nm) were incubated with neurons in high potassium buffer or low potassium buffer for 20 min at 37 °C. Cells were washed and incubated further for 4 h in conditioned media before fixation. Cells were stained with VAMP2 (synaptobrevin 2) antibody (Synaptic Systems, 1:2000) and synaptophysin1. VAMP2 signal intensity was normalized to synaptophysin1. Twenty independent fields were imaged, and data represent averages and S.E.
Cell Imaging and Colocalization
Total internal reflection fluorescence (TIRF) microscopy images were captured on an Eclipse TE2000 inverted microscope equipped with a CFI Apo TIRF 100×/1.49 oil objective (Nikon Instruments Inc.) and Cool Snap HQ2 camera (Photometrics). Fields were selected to exclude cell bodies in the HCR analysis because HCR binding was primarily observed in the axonal appendages, not within the cell bodies. Relative intensities of bound HCR or VAMP2 were normalized to the average intensity of the image/the average intensity of the synaptophysin signal in the same field. Colocalization was analyzed with ImageJ (NIH) using the Manders Coefficients plugin (Tony Collins, Wright Cell Imaging Facility, Toronto, Canada) (28, 29). Values are reported as Pearson's correlation coefficients calculated individually from two corresponding channels from the same field. The correlation coefficients from individual fields were averaged and presented as a value ranging from 0 to 1 with S.E.
Binding Assays
Gangliosides (Matreya) in methanol (1 μg/well) were immobilized onto a 96-well non-binding plate (Corning) and blocked in 1% BSA in 50 mm sodium carbonate buffer, pH 9.6. Alternatively, synaptic vesicle lysate (lacking detergent, described below) was coated on a 96-well high binding plate (Corning) in 50 mm sodium carbonate buffer, pH 9.6, overnight, washed, and blocked with 1% w/v cold fish skin gelatin (Sigma). HCRs were bound for 2 h at 4 °C in PBS. Bound HCR/D was detected using an anti-FLAG antibody (Sigma, 1:10,000), and horseradish peroxidase (HRP)-conjugated secondary mAb. Bound antibody was measured with Ultra-TMB substrate (Pierce). The reaction was stopped with 1 m sulfuric acid, and absorbance was read at 450 nm on a plate reader (Victor 3 V, PerkinElmer Life Sciences). Data were generated in triplicate, and the absorbance detected in wells lacking ganglioside or synaptic vesicles was determined for each concentration and subtracted from the average to account for nonspecific binding. Data were analyzed with GraphPad Prism 5.
FLAG Immunoprecipitation
SV lysates were prepared as described previously with alterations (30, 31). All steps were carried out at 4 °C. Twenty rat cerebral cortices (PelFreeze Bio) were thawed and placed in buffered sucrose solution (5 mm HEPES, pH 7.4, 320 mm sucrose, 1 mm EDTA) and homogenized using a Potter-Elvehjem Tissue Homogenizer. Insoluble material and nuclei were removed by centrifugation at 800 × g for 10 min. Post-nuclear supernatant was separated into cytosolic soluble fraction and membrane pellet by centrifugation at 45,000 × g for 60 min. The pellet was suspended in hypotonic lysis buffer (5 mm HEPES, pH 7.4, 1 mm EDTA) using tissue homogenizer and stirred for 45 min. Large membranes were removed by centrifugation for 60 min at 45,000 × g. Vesicles in supernatant were precipitated using ammonium sulfate (55% w/v) while stirring. Synaptic vesicles were recovered by centrifugation at 10,000 × g for 30 min. Vesicles were dialyzed into 10 mm HEPES, 300 mm glycine pH 7.4 overnight, and centrifuged at 10,000 × g for 30 min to remove large aggregates. Vesicle protein concentration was measured using a BCA assay (Pierce) and determined to be ∼1 mg/ml. Vesicle purity was assayed by Western blot for vesicle markers (SV2, synaptophysin) and plasma membrane markers (SNAP25, sodium-potassium ATPase). Compared with membrane fractions, SVs were enriched for vesicle marker proteins but not enriched for membrane marker proteins. Lysates were diluted to 0.1 mg/ml, and proteins were extracted in 0.5% CHAPS or Triton X-100 and used immediately or stored at −80 °C.
5 μg of HCR was added to 1 ml of synaptic vesicle lysate (containing ∼0.1 mg of total protein) and incubated with rotation for 30 min at 4 °C. Anti-FLAG-agarose beads (Sigma) were blocked in 1% BSA, and 40 μl of bead slurry was added to each immunoprecipitation and incubated for 2 h. Beads were recovered by centrifugation and washed three times. FLAG interactions were eluted specifically with 150 μg of FLAG peptide (Sigma) for 30 min, and soluble material was analyzed by SDS-PAGE and Western blot. Antibodies used were SV2a, SV2b, SV2c, synaptotagmin 2, vATPase, and synaptophysin 1 (Synaptic Systems).
Crystallization and Data Collection
Purified HCR proteins were dialyzed and concentrated to 5 mg/ml in buffer containing 20 mm Tris-HCl at pH 7.6 and 100 mm NaCl. Crystals were produced by vapor diffusion using the hanging drop method (32) containing 2 μl of protein solution mixed with 2 μl of a well solution consisting of 0.1 m Pipes, pH 6.5, 1.0 m sodium malonate, and 2.5% 2-methyl-2,4-pentanediol (MPD) for HCR/D-W1238A, and 0.1 m MOPS, pH 7.0, 10% PEG4000, 12.5% MPD, and 100 mm NaCl for HCR/D-F1240A. Diffraction data for HCR/D-F1240A were collected at 100 K at the LS-CAT 21-ID-F beamline, Advanced Photon Source, Argonne National Laboratory, and data for HCR/D-W1238A were collected using an in-house R-AXIS IV2+ with a MicroMax 007 generator. HKL2000 (33) was used for data processing. Data collection and processing statistics for all crystals are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics
r.m.s.d., root mean square deviation.
| Crystal | HCR/D-W1238A | HCR/D-F1240A |
|---|---|---|
| Diffraction data | ||
| Resolution range (Å) | 50–2.30/2.38–2.30 | 50–2.75/2.80–2.75 |
| No. total reflections | 357,783 | 236,387 |
| No. unique reflections | 102,248/10,205 | 59,660/2,676 |
| Completeness (%) | 99.9/99.9 | 99.0/90.2 |
| Redundancy | 3.5/3.4 | 4.0/3.0 |
| I/σ(I) | 32.1/2.6 | 14.7/2.7 |
| Unit cell | ||
| a, b, c (Å) | 94.5, 115.6, 107.2 | 94.6, 115.6, 107.2 |
| β (°) | 91.9 | 91.9 |
| Space group | P21 | P21 |
| Rsymm | 0.036/0.444 | 0.082/0.330 |
| Vm (Å3/Da)/solvent content (%) | 2.9/56 | 2.9/56 |
| Monomers in an asymmetric unit | 4 | 4 |
| Refinement | ||
| Rcrystal/Rfree | 0.231/0.269 | 0.236/0.301 |
| r.m.s.d. bond length/bond angle | 0.007/1.25 | 0.008/1.38 |
| No. protein atoms | 13,125 | 13,293 |
| No. water molecules | 207 | 73 |
| No. glycerol atoms | 18 | |
| Average B factor | ||
| Main-Chain atoms (Å2) | 60.6 | 64.2 |
| Side-chain atoms (Å2) | 61.5 | 64.4 |
| Water molecules (Å2) | 41.3 | 37.5 |
| glycerol (Å2) | 77.7 | |
Structure Determination and Refinement
Both structures of HCR/D-F1240A and HCR/D-W1238A were solved by the molecular replacement method using MOLREP within the CCP4 (34) program suite and the structure of the HCR/D (residues 875–1295, PDB code 3N7J (23)) as the probe. Initial structures obtained from the molecular replacement trials were refined using the program CNS (35). The refinement procedure consisted of rigid body and positional refinements followed by a simulated-annealing protocol. Iterative rounds of positional and temperature factor refinement followed by manual fitting and rebuilding using the graphics program (36) with 2Fo − Fc and Fo − Fc difference Fourier maps. At later stages of refinement, water molecules were assigned where electron densities were greater than 3 σ in the Fo − Fc map and situated within 3.3 Å of a potential hydrogen bonding partner. The final models were completed with Rcrystal/Rfree of 0.231/0.269 for HCR/D-W1238A and 0.239/0.301 for HCR/D-F1240A (Table 1).
RESULTS
HCR/D-Ganglioside Interactions
Trp-1238 is important for BoNT/D toxicity (25). Space-filling models showed that Trp-1238 was partially buried with only one surface of the indole ring accessible to solvent (Fig. 1, A and B). Alignment analysis showed that HCR/D contained a loop that was similar to the GBL of BoNT/C, although BoNT/C has an exposed Trp that interacts with gangliosides. Instead, Phe-1240 was positioned at the tip of the loop in HCR/D (23, 37) (Fig. 1C). Trp-1238 is, therefore, less likely to contribute to ganglioside binding in a manner similar to the Trp-1258 within the GBL of BoNT/C.
FIGURE 1.

HCR/D structure and ganglioside specificity. A, the crystal structure of HCR/D (PDB code 3OBT) is shown with bound sialic acid and glycerol molecules (red). The ganglioside binding loop is colored blue. Trp-1238, Arg-1239, and Phe-1240 are shown in orange. Hydrophobic residues in the vicinity of Trp-1238 are colored green. Ala-1245 and Pro-1237 are on the GBL. Val-1117 and Val-1119 map to a β-hairpin loop adjacent to the GBL. The structure was rotated 90° and shown with van der Waals radii (right). The boxed area was enlarged (bottom). B, organization of the GBL is shown as in A (right, boxed area) but with the surface omitted. Only one face of the W1238 indole ring is exposed to solvent. The other face contacts hydrophobic residues present on the same or adjacent β-hairpin loops. C, alignment of the crystal structures of HCR/D (PDB code 3OBT, gray) and HCR/C (PDB 3N7K, green) is shown as in A, left. The GBLs from HCR/D and HCR/C are colored blue and magenta, respectively. Phe-1240 of HCR/D and Trp-1258 of HCR/C map to the apex of the respective GBLs. D, HCR/D and HCR/D-W1238A ganglioside binding preferences were assessed with a solid-phase binding assay featuring purified gangliosides immobilized on a 96-well microtiter plate. Bound HCR/D was detected using anti-FLAG antibody and HRP-conjugated secondary mAb and detected using Ultra-TMB. The reaction was stopped with sulfuric acid, and absorbance was read at 450 nm. Data were generated in triplicate and are shown as described in the “Experimental Procedures.”
Recent studies with ganglioside-deficient mice (25) and glycan array screens conducted by the Consortium for Functional Glycomics (Protein-Glycan Interaction Core (Core H)) support functional, direct binding of HCR/D to b-series ganglioside. To obtain a global assessment for the capacity to bind complex gangliosides, HCR/D was bound to a matrix of a- and b-series gangliosides. Under these conditions, HCR/D bound only the b-series gangliosides GD2, GT1b, and GD1b. This indicated that HCR/D recognized the 6,7-disialic acid unique to b-series gangliosides and the Gal-Nac-3 common to complex gangliosides (Fig. 1D). HCR/D bound GD2 with ∼4-fold higher affinity than GT1b at 50% binding, ∼6 nm for GD2 compared with ∼25 nm for GT1b (data not shown). The higher binding to GD2, relative to GD1b and GT1b, may represent steric hindrance by the terminal #4 Gal and #5 sialic acid. HCR/D-W1238A did not bind to gangliosides in the complex ganglioside panel, indicating the requirement for Trp-1238 in this interaction. Cell binding studies showed that HCR/D-W1238A did not bind neurons, indicating the interaction facilitated by Trp-1238 is necessary for HCR/D entry (Fig. 2).
FIGURE 2.
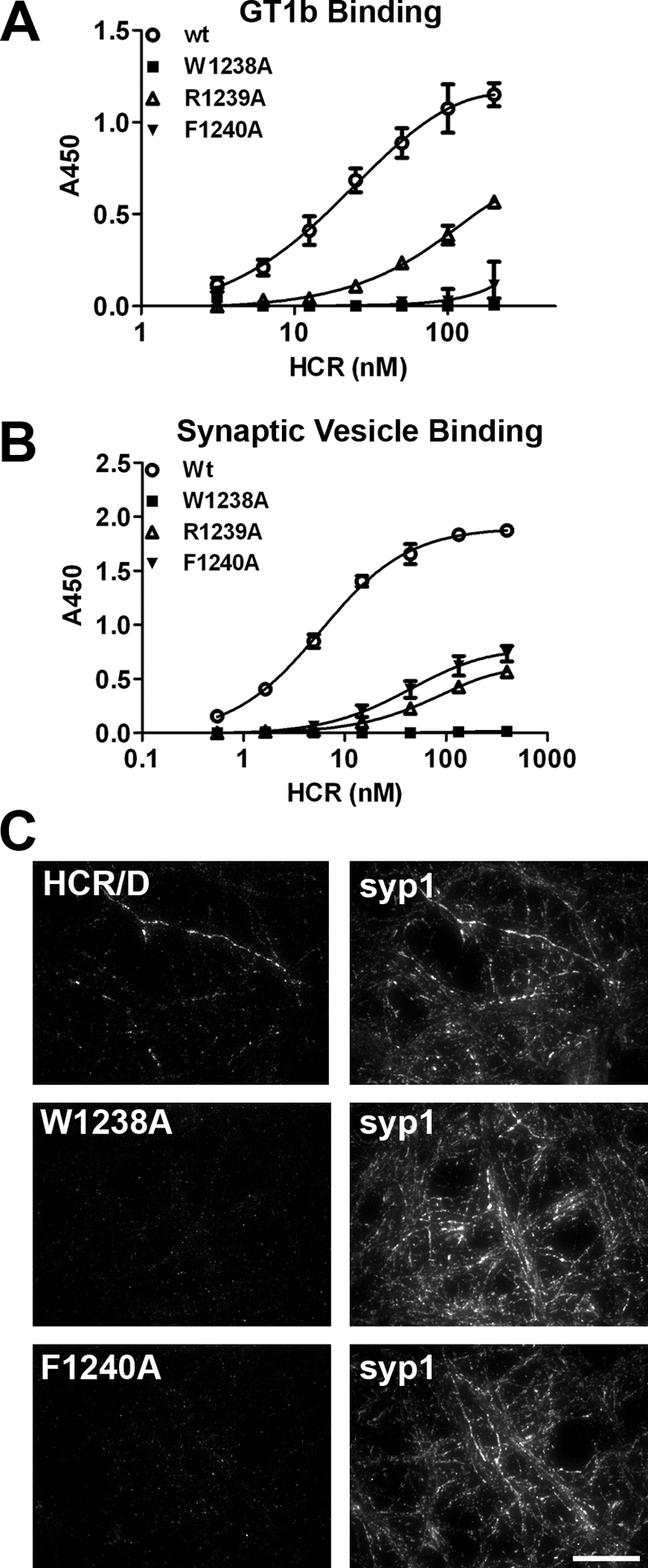
Role of the ganglioside binding loop in receptor recognition. A, GT1b (1 μg/well) was immobilized on a 96-well microtiter plate. HCR/D in indicated concentrations was bound for 1 h at 4 °C and detected as described in Fig. 1. HCR/D achieved 50% binding at ∼27 nm. B, isolated SVs were immobilized on a 96-well microtiter plate, and HCR affinity was determined as described under “Experimental Procedures.” HCR/D achieved 50% binding at ∼6 nm. C, HCRs were incubated with rat E18 primary cortical neurons for 5 min in depolarizing (56 mm potassium) or control (5.6 mm potassium) media. Cells were fixed and permeabilized with 0.1% Triton X-100 in 4% formaldehyde, blocked, and incubated with primary antibody overnight at 4 °C, and cells were then incubated with secondary antibody for 1 h at room temperature, fixed, and imaged. Representative images are shown; scale bar = 20 μm.
Mutations within the Ganglioside Binding Loop of HCR/D Reduce Ganglioside Binding and Synaptic Vesicle Binding
Although several BoNT serotypes utilize a conserved structural pocket to bind gangliosides, HCR/D lacks several key residues within this binding pocket that contribute to ganglioside binding (23, 25). Trp-1238, although critical for binding gangliosides, is largely inaccessible to solvent, suggesting that Trp-1238 may stabilize the GBL rather than providing direct contacts for ganglioside binding. In contrast, Phe-1240 is located at an analogous position to Trp-1258 in BoNT/C, at the apex of the GBL (Fig. 1, A and B). HCR/D-F1240A showed reduced GT1b binding, whereas a second adjacent mutation, HCR/D-R1239A, also showed reduced GT1b binding, with an ∼8-fold decrease in GT1b binding affinity (Fig. 2A). HCR/D-F1240W was generated to test the role of the aromatic residue in ganglioside binding, which has been shown to rescue neurotoxicity (supplemental Fig. 1). HCR/D-F1240W bound gangliosides similarly to HCR/D. Isolated synaptic vesicles were treated with hypotonic lysis to generate a population of inside-out vesicles (31) and used in a solid phase assay as described for the ganglioside binding assay. Binding of wild type HCR/D exhibited dose-dependent binding that reached saturation. HCR/D-R1239A or HCR/D-F1240A showed ∼30-fold reductions in binding affinity, whereas HCR/D-W1238A did not bind above background (Fig. 2B). Cell binding studies utilizing depolarizing conditions in high potassium showed that wild type HCR/D, but not HCR/D-W1238A or HCR/D-F1240A, bound neurons. This indicated that ganglioside binding was necessary for HCR/D entry (Fig. 2C). Thus, the loop that comprises Tyr-1235—Ala-1245 has been designated the GBL of BoNT/D.
Trp-1238 Contributes to the Structural Integrity of the HCR/D GBL
The crystal structures of HCR/D-F1240A and HCR/D-W1238A were determined to assess the effect of removing the R-group for each amino acid on protein structure. Both mutated proteins crystallized in the space group P21 with four molecules in an asymmetric unit. The overall structure of the HCR domain of HCR/D-W1238A was essentially the same as that of wild type HCR/D, except that, unlike the wild type GBL structure, which was well defined, the corresponding region in all four molecules in the W1238A structure was disordered to a varying degree; that is, from completely disordered in two molecules to barely traceable in the other two molecules. One of the HCR/D-W1238A molecules with a traceable GBL is shown (Fig. 3A). In contrast, the structure of HCR/D-F1240A was essentially identical to wild type HCR/D with well defined GBL except for the absence of the phenyl ring at the equivalent Phe-1240 residue (Fig. 3B). These data indicate the indole ring of Trp-1238 serves a structural role to stabilize the GBL. Because HCR/D-F1240A is significantly reduced for ganglioside binding and the GBL structure is the same as wild type HCR/D, we conclude that Phe-1240 is directly involved in ganglioside binding.
FIGURE 3.

Alanine substitution at Trp-1238, but not Phe-1240, disrupts GBL structural integrity for HCR/D. A, shown is the crystal structure of HCR/D-W1238Ala (orange, left) and HCR/D-W1240A (green, right) aligned with HCR/D (gray, PDB ID code 3N7J). The boxed area represents the GBL. B, enlargement of boxed area from A is shown. The GBL in two molecules in the asymmetric unit are completely disordered, and the other two are barely traceable. The GBL conformation shown in W1238A is taken from the molecule that has the best ordered GBL among the four molecules.
HCR/D Binding and Entry into Neurons
Previous studies observed that HCR/D did not bind to resting primary cortical neurons (23). To achieve greater sensitivity and resolution, TIRF microscopy was used to assay HCR/A and HCR/D association with E18 rat primary cortical neurons. TIRF has low background fluorescence and resolves synaptic vesicle interactions that are in close proximity to the plasma membrane (38). Although epifluorescence allows detection of staining, the greater depth of field complicates resolution of individual axons for measurement of colocalization.
In a 4 °C binding experiment, cholera toxin B-subunit-Alexa488 was used to label the plasma membrane and the HCR of tetanus toxin (HCR/TeNT) as a control that binds primary cortical neurons utilizing gangliosides as dual receptors (39). HCR/TeNT-bound neurons, but neither HCR/D nor HCR/A, showed detectable binding (Fig. 4).
FIGURE 4.
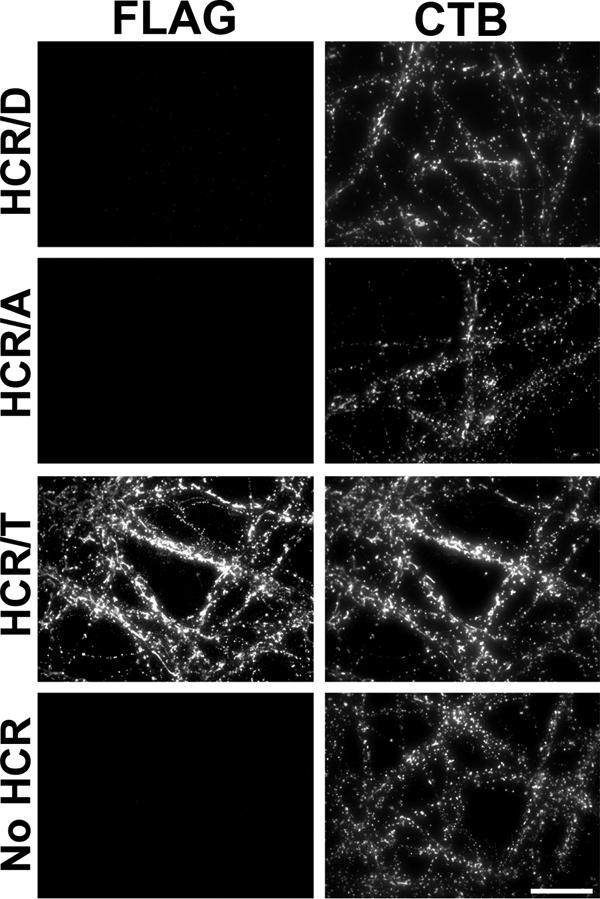
Binding of HCRs to neuronal plasma membranes. Rat E18 primary cortical neurons were incubated with 40 nm HCRs for 1 h at 4 °C in DPBS. Cells were fixed and stained for bound HCR as described in Fig. 1D. CTB, surface bound cholera toxin B. Representative immunofluorescence images are shown; scale bar = 20 μm.
Neurons in culture can be depolarized and synaptic vesicle exocytosis induced by a high concentration of potassium in the media. An enrichment of antibody that bound the luminal domain of synaptotagmin 1 was observed upon incubation of neurons in high K+ buffer (56 mm KCl) versus unstimulated conditions (5.6 mm KCl) (supplemental Fig. 2). At 37 °C, HCR/A and HCR/D again did not show detectable binding to unstimulated neurons; however, upon membrane depolarization by incubation in (+K+ + Ca2+) buffer, HCR/D and HCR/A association was detected (Fig. 5), with ∼3- and 7-fold increases in bound HCR/D and HCR/A, respectively, and HCR/A achieving a 2-fold higher intensity than HCR/D. In contrast, HCR/TeNT-bound unstimulated cortical neurons, as previously reported (39), with HCR/TeNT exhibiting an ∼30% increase in binding upon membrane depolarization, consistent with HCR/TeNT entering cortical neurons by both endosome- and SV-mediated pathways (40).4
FIGURE 5.

Membrane depolarization stimulates binding of HCR/D to neurons. HCRs were incubated with neurons for 5 min in depolarizing (56 mm K+) or unstimulated conditions (5.6 mm K+). Cells were fixed, permeabilized, and stained for FLAG-HCRs and synaptophysin. Quantification of FLAG-HCR signal was normalized to synaptophysin signal. Data are presented as the average of 5 fields with S.D. normalized to HCR/A in high K+. Scale bar = 20 μm. **, p < 0.01; ***, p < 0.001.
HCR/D Enters Neurons through Calcium-dependent SV Cycling
SV fusion is triggered by Ca2+ influx into depolarized neurons (41), and depletion of extracellular Ca2+ prevents SV cycling despite membrane depolarization. To determine if HCR/D uses Ca2+-dependent entry, HCR binding to neurons was measured in buffer containing high potassium plus calcium (+K+ + Ca2+), high potassium without calcium (+K+ − Ca2+), low potassium plus calcium (−K+ + Ca2+), and low potassium without calcium (−K+ − Ca2+) (supplemental Fig. 3). Maximum HCR/D binding was detected in +K++Ca2+ buffer, whereas in the absence of Ca2+ (+K+ − Ca2+ buffer), which decouples SV cycling with membrane depolarization, HCR/D binding was not detected. The assay would not resolve internalized from surface-bound HCR/D; however, endocytosis of SVs is rapid and should be complete by 5 min. The colocalization with synaptophysin, which served as a synaptic vesicle marker and was not enriched on the plasma membrane, supported HCR/D internalization. The observation that HCR/D did not bind to cells chilled to 4 °C (23) also supported HCR/D entry into SVs. These data were consistent with HCR/D entering neurons upon membrane depolarization and Ca2+-dependent SV cycling.
To test if the activity of the HCRs is relevant to toxicity, toxins were incubated with neurons in either unstimulated conditions (low potassium) or membrane-depolarizing conditions (high potassium) and after 4 h incubation stained for VAMP2. Although unstimulated cells incubated in BoNT/D showed reduced levels of VAMP2 to ∼60% of mock-treated cells, incubation of BoNT/D under membrane-depolarizing conditions reduced VAMP-2 to ∼20% of mock-treated cells, which was significant relative to unstimulated cells (Fig. 6). This was consistent with an increased fraction of BoNT/D entering neurons via membrane depolarizing pathways. Cleavage of VAMP2 by BoNT/D in unstimulated neurons may result from BoNT/D entry by spontaneously firing SVs or by a secondary entry mechanism (39, 40, 42). VAMP2 cleavage by TeNT in unstimulated and membrane-depolarized neurons was similar.
FIGURE 6.
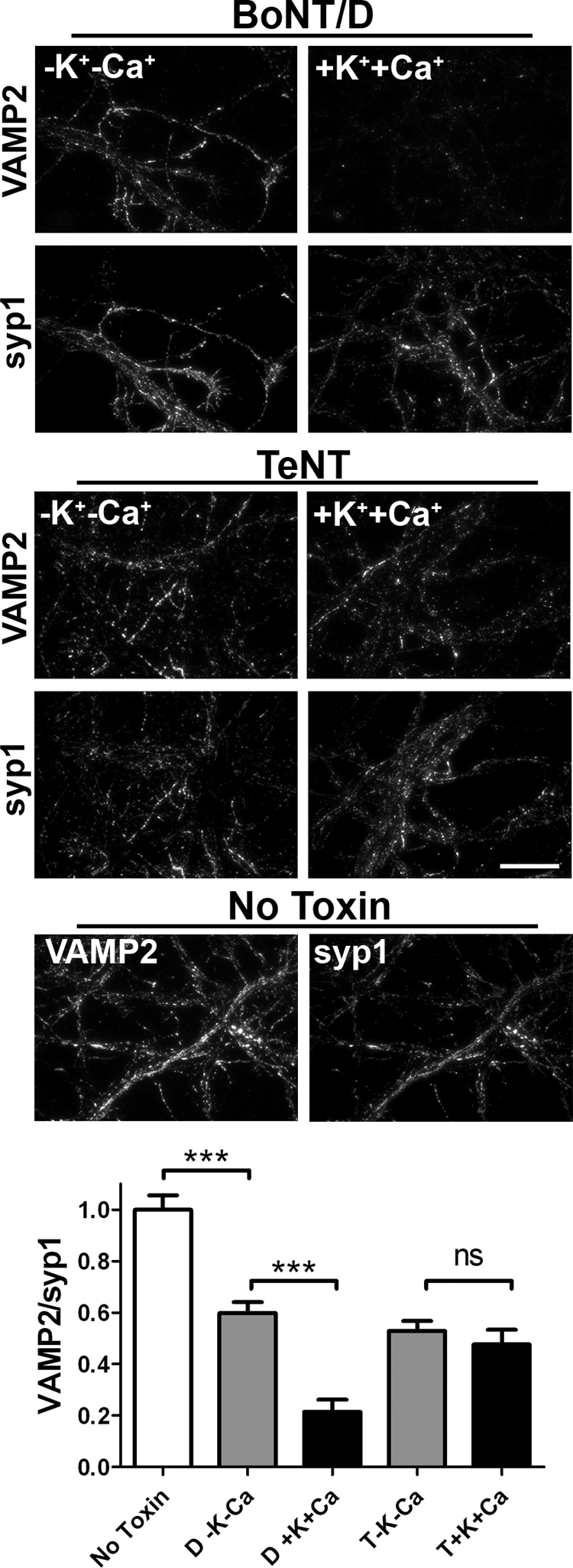
BoNT/D toxicity is enhanced by membrane depolarization. 10 nm BoNT/D and tetanus toxin were incubated with rat E18 primary cortical neurons for 20 min followed by a 4-h chase. Neurons were fixed and subjected to immunostaining for intact VAMP2 and synaptophysin1. Average VAMP2 intensity was normalized to synaptophysin1 and averaged from 20 fields. Data are presented normalized to intact VAMP in mock intoxication. Scale bar = 20 μm. ***, p < 0.001; ns, not significant.
Colocalization of HCR/D with Synaptophysin 1
HCR/D entry appeared to localize with intracellular synaptophysin. To quantify this observation, colocalization of HCRs with marker proteins, synaptophysin1 (SVs) and Rab5 (endosomes), was measured upon membrane depolarization (Fig. 7). Colocalization was analyzed by plotting the intensity of individual pixels representing bound HCR on one axis versus marker protein intensity on the other axis and determining the Pearson correlation coefficients for each data set. Controls showed that a protein with two bound marker proteins yielded a correlation that approached 1, whereas the correlation for two proteins that are known to segregate in the cells yielded correlation coefficients that were near 0.2 (data not shown) (29). HCR/A and HCR/D colocalized well to synaptophysin, achieving correlations of ∼0.63 and 0.70, respectively (Fig. 7, black bars). In contrast, colocalization of HCR/A and HCR/D with Rab5 (white bars) was lower, averaging ∼0.25 and 0.17, respectively. A fractional population of synaptophysin1 and Rab5 double-positive vesicles was also observed, and rarely observed areas of colocalization between HCRs and Rab5 were nearly always triple positive for synaptophysin1. Together, these data show that HCR/D and HCR/A associate with SVs (colocalized with synaptophysin) and that a fraction of HCR may enter neurons via endosome compartments colocalized with Rab5+ and synaptophysin1, possibly via rapid SV protein sorting endosomes (42).
FIGURE 7.

Colocalization of HCR/D to synaptophysin. Rat E18 primary cortical neurons were incubated with 40 nm HCRs for 5 min at 37 °C in depolarizing (56 mm K+) buffer. Cells were fixed, permeabilized with TritonX-100, and stained for FLAG-HCRs, synaptophysin, and Rab5 as described in Fig. 2. Overlays were generated between HCR (red) and either Rab5 or syp1 (green), with areas of positive colocalization rendered yellow. Colocalization was evaluated by measuring the correlation coefficients (28) between HCR and either syp1 or Rab5 for eight independent fields; graphs represent the average with S.E. Scale bar = 20 μm. ***, p < 0.001.
Limited Association of HCR/D with SV Resident Proteins
Potential protein receptors for BoNT/D among SV proteins were assayed using a CHAPS- or Triton-extracted SV lysate isolated from rat brain (30, 43) (Fig. 8). SV2 proteins form unique complexes upon extraction with CHAPS and Triton X-100 (43). HCR/A was previously shown to bind SV2 in CHAPS and Triton X-100-extracted vesicles; whether detergents perturb unique HCR/D interactions synaptic vesicle interactions is unknown (30). In CHAPS-extracted SVs, HCR/A bound several SV proteins, whereas HCR/D showed limited binding to SV2a, SV2b, and Syt2. In contrast, in Triton X-100-extracted SVs, HCR/A bound SV2 and synaptotagmin, which form a complex upon Triton extraction (44). HCR/D did not bind detectable SV2 or synaptotagmin above background controls. This indicates that HCR/D does not directly bind SV2 or synaptotagmin or that the affinity of HCR/D for SV2 or synaptotagmin was below the limit of detection of the assay.
FIGURE 8.
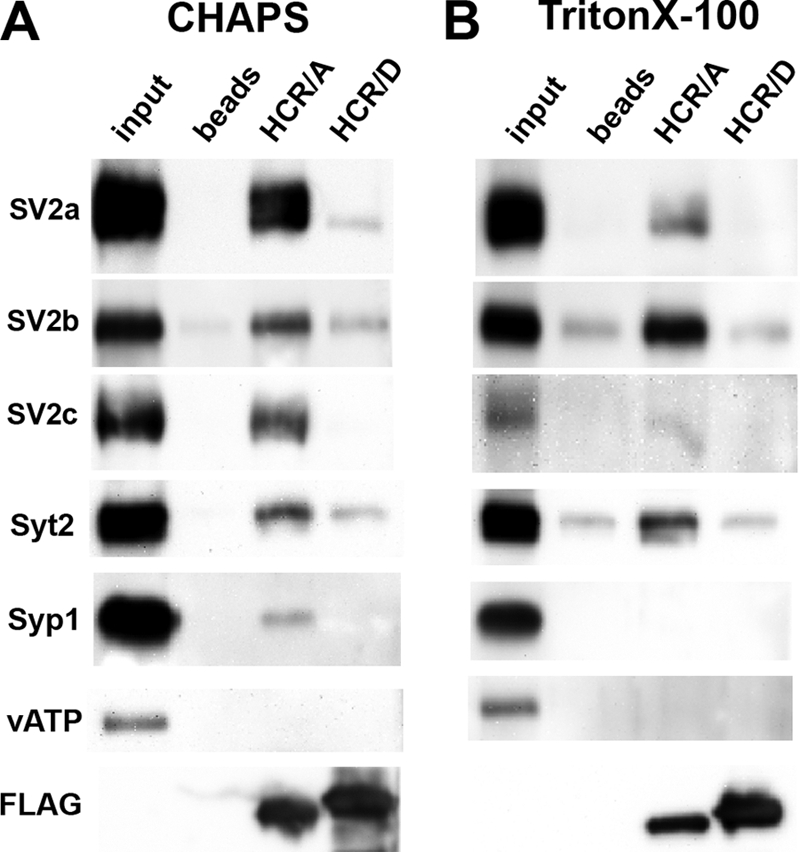
HCR/D shows limited interaction with SV proteins. FLAG-HCR (5 μg) was incubated with ∼100 μg of total SV protein lysate extracted with either 0.5% CHAPS (A) or 0.5% Triton X-100 (B). HCRs were precipitated with anti-FLAG-agarose beads. Bead bound HCRs were eluted with FLAG peptide and analyzed by Western blot, which is shown. Quantification showed that with CHAPS-extracted SVs, HCR/D bound SV2a at 0.7%, SV2b at 11%, and Syt2 at 15% of the relative binding observed for HCR/A. In Triton X-100-extracted SVs, HCR/D did not bind detectable amounts of SV proteins above background.
DISCUSSION
Recently, the structure of HCR/D was solved, completing the structural determination for all seven BoNT HCR serotypes (23, 25, 45). The overall secondary and tertiary structure of HCR/D was similar to the HCRs of the other BoNT serotypes, where HCRs consists of an N-terminal subdomain conforming to a jellyroll motif and a C-terminal subdomain folded into a β-trefoil motif. The C-terminal subdomain contains the known receptor binding sites for each serotype and also possesses the greatest divergence of structure and primary amino acid homology (18, 46). In BoNT/A,/B,/E-G, a short α-helix near to the C terminus contains an Ser-X-Trp-Tyr motif that forms a portion of the ganglioside binding pocket (47). HCR/C and HCR/D lack these residues, although the helical structure of the pocket is retained (23, 25). Whether this analogous pocket of HCR/D and HCR/C contain receptor binding activity is unclear, but these pockets within HCR/C or HCR/D do not support ganglioside binding. Recently, Strotmeier et al. (25) showed that this region within HCR/D can support glycerol binding and thus may represent a binding site for other classes of macromolecules.
SVs contain specialized proteins for uptake of neurotransmitters and Ca2+-regulated exocytosis. HCR/D binds SVs, indicating a high affinity binding where gangliosides present in SVs may provide an optimal binding site over plasma membrane ganglioside populations. Gangliosides are critical for nervous system development and organizing neurotransmitter receptor in lipid rafts and myelin; mice lacking components in the ganglioside synthesis pathway display numerous defects (48–50). Although the specific roles of gangliosides in SV cycling have not been elucidated, the requirement for complex gangliosides in normal synaptic development and function has been shown (51), where vacuolar ATPase activation and intensity of synaptic response are modulated by gangliosides (52, 53). Several groups estimate that gangliosides comprise only a minor component of the SV lipidome; likewise, SVs contain a limited number of molecules of SV2 (1–5 molecules) and synaptotagmin (7–15 molecules) that serve as protein receptors for several BoNT serotypes (54–56). Thus, gangliosides presented within SV components may provide a high affinity binding site for HCR/D. Recently, Peng et al. (26) reported that HCR/D entered gangliosides via SVs mediated through the direct binding to SV2; our analysis did not detect a direct interaction between HCR/D and SV2 under conditions where HCR/A and SV2 association were detected. Of note, the earlier study used ∼100-fold more HCR to detect HCR/D-SV2 interactions than needed to detect the association of HCR/A and SV2 in the current study. The lack of, or low affinity, interaction of HCR/D to SV proteins suggests the use of a unique SV specific interaction along with the observed unique ganglioside preference to accomplish neuronal entry comparable with BoNT/A.
Despite detectable binding of HCR/D to gangliosides in vitro, a similar level of binding was not observed in neurons. This effect may be related to the unique ganglioside binding specificities of HCR/D, which shows an affinity for GD2. In contrast, HCR/A forms contacts with the terminal galactose and sialic acid (4Gal-5Sia) of GT1b and GD1a, which are absent from GD2 (47). Because HCR/D can bind GD1b and GT1b on a solid phase, but apparently not on a neuronal membrane, the terminal 4Gal-5Sia may inhibit HCR/D interaction, reducing the affinity below levels detectable by TIRF microscopy. The disialic acid motif may assume a more favorable conformation for HCR/D upon presentation within an SV receptor component. The recent observation showing the resistance of ganglioside knock-out mice to BoNT/D intoxication supports a role for gangliosides in the intoxication process (25). Future studies will determine the role of membrane-bound BoNT/D in toxin entry, which has been implicated as contributing to the productive entry of BoNT/A into neurons (57).
HCR/C and HCR/D contain a prominent hydrophobic loop (the GBL) (58). In HCR/A, HCR/E, and HCR/F, the equivalent region representing the GBL loop is reduced in size and is less hydrophobic in composition (7, 13, 59). In HCR/C, the GBL contains a Trp that was required for ganglioside binding (23). Although the GBL of HCR/D resembles the GBL of HCR/C, the residues implicated in ganglioside binding appear to differ. Mutation of Trp-1238 reduced the capacity of HCR/D to bind gangliosides (this study) and synaptosomes (25). Structural studies showed that the indole ring of Trp-1238 is partially solvent-exposed and does not contact sialic acid in the structure of the HCR/D-sialic acid complex (25). In addition, the GBL loop in the HCR/D-W1238A structure was disordered and thus Trp-1238 appeared to serve a structural role to stabilize the GBL and was not directly involved in binding of gangliosides. Strotmeier et al. (25) showed that substitution of Phe for Trp-1238 could only partially rescue synaptosome binding or neuron toxicity, indicating a required role for the indole ring. The current study showed that mutation of Phe 1240 to Ala, located on the GBL of HCR/D, reduced the ganglioside binding capacity, but the GBL structure was conserved. We propose that Phe-1240 in HCR/D is analogous to Trp1358 in the GBL of HCR/C in the coordination of ganglioside binding, likely by a ring-stacking mechanism, which is supported by the F1240W substitution. The data are consistent with BoNT/D being the first serotype to utilize a Phe to support ganglioside binding. Of note, the two known BoNT/D subtypes, excluding the D/C mosaic toxins, are nearly identical; thus, Phe-1240 and the ganglioside interaction are most likely conserved (18). The recent observation that Phe-1240 contributes to BoNT/D toxicity supports this conclusion (25). Continued studies may determine how the unique interactions between HCR/D and neurons can be utilized for the development of novel clinical therapies.
Supplementary Material
Acknowledgments
We acknowledge the assistance of Amanda Przedpelski for HCR production. Glycan microarray data were obtained through the Protein-Glycan Interaction Core (Core H) of the Consortium for Functional Glycomics supported by National Institutes of Health Grant GM62116.
This work was supported, in whole or in part, by the National Institutes of Health Regional Center of Excellence for Bio-defense and Emerging Infectious Diseases Research Program and the NIAID (to J. T. B. and J. J. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
F. C. Blum and J. T. Barbieri unpublished data.
- BoNT
- botulinum neurotoxin
- BoNT/D
- BoNT serotype D
- LC
- light chain
- HC
- heavy chain
- HCR
- heavy chain receptor binding domain
- SV
- synaptic vesicle
- GBL
- ganglioside binding loop
- DPBS
- Dulbecco's PBS
- TIRF
- total internal reflection fluorescence
- TeNT
- tetanus toxin
- VAMP2
- vesicle-associated membrane protein 2.
REFERENCES
- 1. Gill D. M. (1982) Microbiol. Rev. 46, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnon S. S., Schechter R., Inglesby T. V., Henderson D. A., Bartlett J. G., Ascher M. S., Eitzen E., Fine A. D., Hauer J., Layton M., Lillibridge S., Osterholm M. T., O'Toole T., Parker G., Perl T. M., Russell P. K., Swerdlow D. L., Tonat K. (2001) JAMA 285, 1059–1070 [DOI] [PubMed] [Google Scholar]
- 3. Eleopra R., Tugnoli V., Rossetto O., Montecucco C., De Grandis D. (1997) Neurosci. Lett. 224, 91–94 [DOI] [PubMed] [Google Scholar]
- 4. Davletov B., Bajohrs M., Binz T. (2005) Trends Neurosci. 28, 446–452 [DOI] [PubMed] [Google Scholar]
- 5. Coffield J. A., Bakry N., Zhang R. D., Carlson J., Gomella L. G., Simpson L. L. (1997) J. Pharmacol. Exp. Ther. 280, 1489–1498 [PubMed] [Google Scholar]
- 6. Woodward L. A., Arimitsu H., Hirst R., Oguma K. (2003) Infect. Immun. 71, 2941–2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lacy D. B., Tepp W., Cohen A. C., DasGupta B. R., Stevens R. C. (1998) Nat. Struct. Biol. 5, 898–902 [DOI] [PubMed] [Google Scholar]
- 8. Dong M., Liu H., Tepp W. H., Johnson E. A., Janz R., Chapman E. R. (2008) Mol. Biol. Cell 19, 5226–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ochanda J. O., Syuto B., Ohishi I., Naiki M., Kubo S. (1986) J. Biochem. 100, 27–33 [DOI] [PubMed] [Google Scholar]
- 10. Rummel A., Mahrhold S., Bigalke H., Binz T. (2004) Mol. Microbiol. 51, 631–643 [DOI] [PubMed] [Google Scholar]
- 11. Dong M., Yeh F., Tepp W. H., Dean C., Johnson E. A., Janz R., Chapman E. R. (2006) Science 312, 592–596 [DOI] [PubMed] [Google Scholar]
- 12. Dong M., Richards D. A., Goodnough M. C., Tepp W. H., Johnson E. A., Chapman E. R. (2003) J. Cell Biol. 162, 1293–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu Z., Chen C., Barbieri J. T., Kim J. J., Baldwin M. R. (2009) Biochemistry 48, 5631–5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koriazova L. K., Montal M. (2003) Nat. Struct. Biol. 10, 13–18 [DOI] [PubMed] [Google Scholar]
- 15. Montecucco C., Schiavo G. (1993) Trends Biochem. Sci. 18, 324–327 [DOI] [PubMed] [Google Scholar]
- 16. Schiavo G., Benfenati F., Poulain B., Rossetto O., Polverino, de Laureto P., DasGupta B. R., Montecucco C. (1992) Nature 359, 832–835 [DOI] [PubMed] [Google Scholar]
- 17. Yamasaki S., Baumeister A., Binz T., Blasi J., Link E., Cornille F., Roques B., Fykse E. M., Südhof T. C., Jahn R. (1994) J. Biol. Chem. 269, 12764–12772 [PubMed] [Google Scholar]
- 18. Hill K. K., Smith T. J., Helma C. H., Ticknor L. O., Foley B. T., Svensson R. T., Brown J. L., Johnson E. A., Smith L. A., Okinaka R. T., Jackson P. J., Marks J. D. (2007) J. Bacteriol. 189, 818–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rummel A., Häfner K., Mahrhold S., Darashchonak N., Holt M., Jahn R., Beermann S., Karnath T., Bigalke H., Binz T. (2009) J. Neurochem. 110, 1942–1954 [DOI] [PubMed] [Google Scholar]
- 20. Dong M., Tepp W. H., Liu H., Johnson E. A., Chapman E. R. (2007) J. Cell Biol. 179, 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsukamoto K., Kohda T., Mukamoto M., Takeuchi K., Ihara H., Saito M., Kozaki S. (2005) J. Biol. Chem. 280, 35164–35171 [DOI] [PubMed] [Google Scholar]
- 22. Simpson L., Rapport M. (1971) J. Neurochem. 18, 1341–1343 [DOI] [PubMed] [Google Scholar]
- 23. Karalewitz A. P., Kroken A. R., Fu Z., Baldwin M. R., Kim J. J., Barbieri J. T. (2010) Biochemistry 49, 8117–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsukamoto K., Kozai Y., Ihara H., Kohda T., Mukamoto M., Tsuji T., Kozaki S. (2008) Microb. Pathog. 44, 484–493 [DOI] [PubMed] [Google Scholar]
- 25. Strotmeier J., Lee K., Völker A. K., Mahrhold S., Zong Y., Zeiser J., Zhou J., Pich A., Bigalke H., Binz T., Rummel A., Jin R. (2010) Biochem. J. 431, 207–216 [DOI] [PubMed] [Google Scholar]
- 26. Peng L., Tepp W. H., Johnson E. A., Dong M. (2011) PLoS Pathog. 7, e1002008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baldwin M. R., Tepp W. H., Przedpelski A., Pier C. L., Bradshaw M., Johnson E. A., Barbieri J. T. (2008) Infect. Immun. 76, 1314–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manders E. E. M., Verbeek F. J., Aten J. A. (1993) J. Microsc. 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 29. Li Q., Lau A., Morris T. J., Guo L., Fordyce C. B., Stanley E. F. (2004) J. Neurosci. 24, 4070–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baldwin M. R., Barbieri J. T. (2007) Biochemistry 46, 3200–3210 [DOI] [PubMed] [Google Scholar]
- 31. Huttner W. B., Schiebler W., Greengard P., De Camilli P. (1983) J. Cell Biol. 96, 1374–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McPherson A. (1999) Crystallization of Biological Macromolecules, pp. 188–192, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 33. Minor W., Tomchick D., Otwinowski Z. (2000) Structure 8, R105–R110 [DOI] [PubMed] [Google Scholar]
- 34. CCP4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 35. Brunger A. T. (2007) Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 36. Roussel A., Inisan A. G., Knoop-Mouthuy A., Cambillau C. (1999) Turbo-Frodo, Version OpenGL:1, CNRS/Universitite, Marseille, France [Google Scholar]
- 37. Chai Q., Arndt J. W., Dong M., Tepp W. H., Johnson E. A., Chapman E. R., Stevens R. C. (2006) Nature 444, 1096–1100 [DOI] [PubMed] [Google Scholar]
- 38. Zenisek D., Steyer J. A., Almers W. (2000) Nature 406, 849–854 [DOI] [PubMed] [Google Scholar]
- 39. Chen C., Fu Z., Kim J. J., Barbieri J. T., Baldwin M. R. (2009) J. Biol. Chem. 284, 26569–26577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yeh F. L., Dong M., Yao J., Tepp W. H., Lin G., Johnson E. A., Chapman E. R. (2010) PLoS Pathog. 6, e1001207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pang Z. P., Südhof T. C. (2010) Curr. Opin. Cell Biol. 22, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoopmann P., Punge A., Barysch S. V., Westphal V., Bückers J., Opazo F., Bethani I., Lauterbach M. A., Hell S. W., Rizzoli S. O. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 19055–19060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bennett M. K., Calakos N., Kreiner T., Scheller R. H. (1992) J. Cell Biol. 116, 761–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lazzell D. R., Belizaire R., Thakur P., Sherry D. M., Janz R. (2004) J. Biol. Chem. 279, 52124–52131 [DOI] [PubMed] [Google Scholar]
- 45. Zhang Y., Buchko G. W., Qin L., Robinson H., Varnum S. M. (2010) Biochem. Biophys. Res. Commun. 401, 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lacy D. B., Stevens R. C. (1999) J. Mol. Biol. 291, 1091–1104 [DOI] [PubMed] [Google Scholar]
- 47. Stenmark P., Dupuy J., Imamura A., Kiso M., Stevens R. C. (2008) PLoS Pathog. 4, e1000129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cole A. A., Dosemeci A., Reese T. S. (2010) Biochem. J. 427, 535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheikh K. A., Sun J., Liu Y., Kawai H., Crawford T. O., Proia R. L., Griffin J. W., Schnaar R. L. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 7532–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takamiya K., Yamamoto A., Furukawa K., Yamashiro S., Shin M., Okada M., Fukumoto S., Haraguchi M., Takeda N., Fujimura K., Sakae M., Kishikawa M., Shiku H., Furukawa K., Aizawa S. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10662–10667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sugiura Y., Furukawa K., Tajima O., Mii S., Honda T., Furukawa K. (2005) Neuroscience 135, 1167–1178 [DOI] [PubMed] [Google Scholar]
- 52. Yoshinaka K., Kumanogoh H., Nakamura S., Maekawa S. (2004) Neurosci. Lett. 363, 168–172 [DOI] [PubMed] [Google Scholar]
- 53. Ando S., Tanaka Y., Waki H., Kon K., Iwamoto M., Fukui F. (1998) Ann. N.Y. Acad. Sci. 845, 232–239 [DOI] [PubMed] [Google Scholar]
- 54. Mutch S. A., Kensel-Hammes P., Gadd J. C., Fujimoto B. S., Allen R. W., Schiro P. G., Lorenz R. M., Kuyper C. L., Kuo J. S., Bajjalieh S. M., Chiu D. T. (2011) J. Neurosci. 31, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Takamori S., Holt M., Stenius K., Lemke E. A., Grønborg M., Riedel D., Urlaub H., Schenck S., Brügger B., Ringler P., Müller S. A., Rammner B., Gräter F., Hub J. S., De Groot B. L., Mieskes G., Moriyama Y., Klingauf J., Grubmüller H., Heuser J., Wieland F., Jahn R. (2006) Cell 127, 831–846 [DOI] [PubMed] [Google Scholar]
- 56. Bonanomi D., Benfenati F., Valtorta F. (2006) Prog. Neurobiol. 80, 177–217 [DOI] [PubMed] [Google Scholar]
- 57. Keller J. E., Cai F., Neale E. A. (2004) Biochemistry 43, 526–532 [DOI] [PubMed] [Google Scholar]
- 58. Swaminathan S., Eswaramoorthy S. (2000) Nat. Struct. Biol. 7, 693–699 [DOI] [PubMed] [Google Scholar]
- 59. Kumaran D., Eswaramoorthy S., Furey W., Navaza J., Sax M., Swaminathan S. (2009) J. Mol. Biol. 386, 233–245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.