

Abstract
Many medications in common clinical use consist of “mirror image” isomers that differ only in the direction in which they rotate plane-polarized light. These stereoisomers exist as mixtures of “right” and “left” handed molecules that are the product of chemical syntheses. However, the biochemical milieu of the human body is a highly stereospecific environment where the fit of medication and receptor may depend on the shape of the molecule in 3-dimensional space. Recent advances in chemistry have allowed the more ready preparation of single isomers of various drugs that were previously available only as racemic mixtures. For those compounds in which the isomers differ in stereospecificity, this separation into single isomers can result in significant changes in potency, tolerability, and efficacy. This article reviews some basic information about stereochemistry and describes the development of a new single isomer antidepressant, escitalopram, which is one of the components of the widely used selective serotonin uptake inhibitor citalopram.
Among Louis Pasteur's many accomplishments was his discovery that some organic molecules exist as mirror images. Pasteur made this discovery in 1848 while experimenting with tartaric acid, a by-product of wine-making. He was able to resolve 2 crystal forms of this compound and then manually separate them on the basis of their visibly different structures. The separation was conducted with the aid of a microscope and a small set of forceps. He proceeded to make solutions of the 2 crystal forms and found they were identical in physicochemical properties with the exception of their ability to rotate plane-polarized light (Figure 1).1
Figure 1.

Louis Pasteur, Discoverer of Stereoisomersa
In 1874, Jacobus H. van't Hoff provided the explanation for why these crystals rotated light differently. He hypothesized that the carbon atom had a tetrahedral structure. When 1 atom of carbon binds to 4 different substituents in a tetrahedral structure, 2 configurations are possible that are mirror images of each other. Such compounds are said to be “chiral,” and the carbon is considered a chiral center (Figure 2).1
Figure 2.
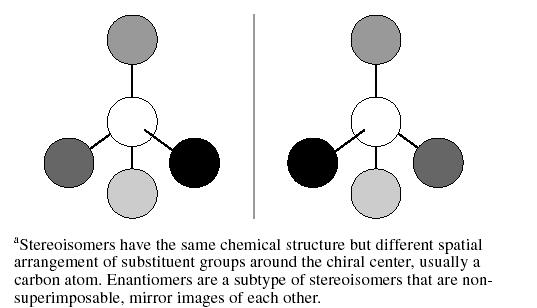
Basic Structure of Stereoisomersa
The word chiral is derived from the Greek word cheir, which means hand. Just like a hand or glove, chiral molecules are non-superimposable mirror images. Because of the orientation of the 4 groups around the carbon, these “stereoisomers” interact with the surrounding environment in unique ways. This stereospecificity can result in important stereoselectivity of the individual molecules at receptors. Enantiomers is the term for stereoisomers that are non-superimposable mirror images of one another. The presence of 1 chiral center in a molecule gives rise to a pair of enantiomers. If there is more than 1 chiral center, the yield is 2n stereoisomers, where “n” is the number of chiral centers, and half as many pairs of enantiomers. Isomers that are not enantiomers are called “diastereomers.”3 Since enantiomers differ in their ability to rotate plane-polarized light, they are also referred to as optical isomers.3
TERMINOLOGY
The proper designation of enantiomers can be confusing since there are at least 3 different systems to classify them. The first is the “d/l” or “(+)/(−)” system that relies on the direction that the compound rotates plane-polarized light. Enantiomers that rotate plane-polarized light to the right are termed dextrorotatory (indicated by a [+] or “d” before the name of the compound), and those that rotate light to the left are termed levorotatory (indicated by a [−] or “l” prefix).3 A racemic mixture is indicated by either a “(+)/(−)” or a “d/l” prefix. Interestingly, rotation of light is not an absolute property of a compound but can vary in different settings. For example, the active stereoisomer of chloramphenicol is dextrorotatory in alcohol and levorotatory in ethyl acetate.3 Likewise, ibuprofen and naproxen are dextrorotatory as their free acids but levorotatory as their sodium salts.3 Most drugs with a chiral center produced by chemical synthesis are available only as racemates since chemical synthesis usually leads to a racemic mixture.4
The other 2 classification schemes for designating enantiomers can be used once the structure has been determined. The older “D/L” system (no relationship to the “d/l” system noted above) assigns a label based on comparison to a standard reference compound, either the carbohydrate D-glyceraldehyde or the amino acid L-serine. This designation, however, now is used only for carbohydrates and amino acids.
The current system assigns a prefix of either an “R” (rectus) or an “S” (sinister) on the basis of assigning priority to the atoms attached to the chiral center. The atoms attached to the chiral center are ranked by atomic number, with the highest atomic number given the highest priority. The molecule is then viewed from the side opposite the group with the lowest priority, and a prefix is assigned according to convention. The absolute configuration nomenclature “R/S” has no relationship to the rotational prefixes “d” and “l” obtained by measurement of the rotation of light.
It is not unusual for stereoisomers to differ significantly in their activity for a particular function or to have a high degree of stereoselectivity for one action, but no stereoselectivity for another action.4 The term eutomer is used to denote the more active enantiomer, and distomer, the less active. The “eudismic ratio” is the potency ratio between the eutomer and distomer.
An important concept for medications is that although enantiomers differ only in how they rotate plane-polarized light, they may have very different biological properties in the human body. For example, the R-enantiomer of the chemical carvone has the odor of spearmint, while S-carvone smells like caraway. Enantiomers with stereospecific indications are dextropropoxyphene and levopropoxyphene. Dextropropoxyphene (Darvon) is marketed as an analgesic, while levopropoxyphene (once marketed as Novrad—Darvon spelled backward) is an antitussive.5 Similarly, the toxicity of a compound may be selective for an enantiomer. The S-enantiomer of ketamine produces an anesthetic effect, while the R-enantiomer can cause hallucinations. These diverse effects reflect the fact that most biological systems consist of chiral environments, thus the orientation of a molecule in space may be crucial. A right-handed molecule may fit perfectly in a chiral protein receptor, while the enantiomer may exhibit much less activity because of its inability to fit in that receptor. This is expressed as “Pfeiffer's rule,” which states that the more potent a drug is, the more likely it is to show stereoselectivity of action as a consequence of the greater steric demand for tight receptor binding.6
POTENTIAL ADVANTAGES OF ENANTIOMERS
It has been argued that giving drugs as racemates is a form of polypharmacy driven by chemical rather than therapeutic considerations. Racemic mixtures can also be considered compounds that contain a 50% impurity.3 Using a single enantiomer can result in an improved therapeutic index resulting from presumed higher potency and selectivity while removing those side effects that may be due to the less active enantiomer. Use of a single enantiomer thus can result in an improved onset of action and duration of action and a decrease in the propensity for drug-drug interactions.7 Importantly, single stereoisomers have less complex pharmacokinetic profiles and less complex plasma concentration–effect relationships.3 When a racemate is given, the patient is really being given 2 different drugs with different pharmacokinetic and pharmacodynamic profiles. Other advantages of stereochemically pure drugs are reducing the total dose given and removing a source of intersubject variability.6
Caldwell has made the point that the “pharmacokinetic importance of drug stereochemistry depends on the mechanism of the process under consideration: passive processes such as diffusion across membranes do not involve macromolecular interactions and stereochemistry has little influence, but when the drug interacts with an enzyme or a transporter system then discrimination may be seen.”6(p40) These are precisely the types of interactions seen in the use of psychotropic medications in which the drug needs to interact with a receptor to have its effect. This effect may entirely depend on the spatial configuration of the molecule that confronts the receptor.
SELECTIVE SEROTONIN REUPTAKE INHIBITORS
Of the currently available selective serotonin reuptake inhibitors (SSRIs), fluvoxamine lacks a chiral center, whereas paroxetine and sertraline have always been available as single enantiomers.8 Sertraline, which has 2 chiral centers (and thus 4 different enantiomers), is marketed as the 1S(+)-,4S(+)-enantiomer.8 Citalopram and fluoxetine are marketed as racemates, although the potential for reintroducing each of these drugs as one of their respective stereoisomers has been investigated.7,8 Fluoxetine and its main metabolite, norfluoxetine, are present as enantiomers. The R(−)- and S(+)-enantiomers of fluoxetine are roughly equivalent inhibitors of serotonin uptake (Table 1).8 On the other hand, S(+)-norfluoxetine is a more potent serotonin reuptake inhibitor than the R(−)-enantiomer. When patients are treated with racemic fluoxetine, the ratio of S(+)-fluoxetine/R(−)-fluoxetine has been reported to vary from 1 to 3.5.8 R(−)-fluoxetine tends to be eliminated more rapidly, but in the presence of a cytochrome P450 2D6 enzyme (CYP2D6) inhibitor the ratio decreases. Furthermore, the R(−)-enantiomers of fluoxetine and norfluoxetine are weaker inhibitors of CYP2D6 than the corresponding S(+)-enantiomers.9 The more rapid elimination of R(−)-fluoxetine and its decreased potential for drug-drug interactions led to efforts to develop this compound as a specific enantiomer for the treatment of depression. This effort was abandoned due to concerns about the possibility of corrected QT interval prolongation with R(−)-fluoxetine when administered as a single isomer.
Table 1.
Pharmacologic Properties of the Enantiomers of Fluoxetine and Citaloprama
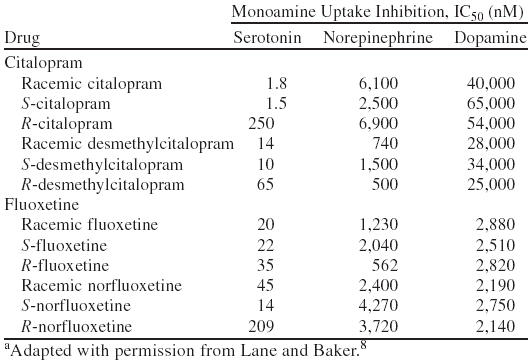
In the case of citalopram (Figure 3), the S(+)-enantiomer (whose generic name is escitalopram) is greater than 2 orders of magnitude more potent than R(−)-citalopram in vitro as an inhibitor of serotonin (5-HT) uptake.10,11 The eudismic ratio for citalopram has been calculated to be 167.11,12 Furthermore, escitalopram has very little effect on other receptors, making it the most selective SSRI.10,13 The plasma concentration of the eutomer of citalopram (escitalopram) is usually one third of the total citalopram concentration, with the implication being that the other two thirds of the total citalopram concentration is drug that is mainly inactive as an antidepressant.12
Figure 3.
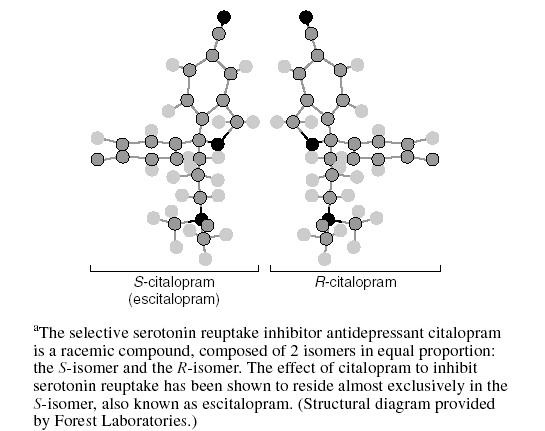
Structure of R- and S-Citaloprama
In a study of 29 depressive patients treated with various doses (20–80 mg/day) of citalopram,12 the concentrations of the distomer were higher than those of the eutomer with a mean S/R ratio of 0.56 and a range of 0.32 to 0.97. In those same subjects, the mean S/R ratio of desmethylcitalopram (the main metabolite of citalopram) was 0.72. This study noted that escitalopram and R(−)-citalopram levels correlated strongly (r = 0.866, p < .0001), as did those of the desmethyl metabolites (r = 0.932, p < .0001). The implication is that enantiomers of citalopram may be metabolized by the same enzymes but at different rates.12 This difference in the rate of metabolism would presumably be due to stereoselective metabolism by the cytochrome P450 enzyme system. Levels of desmethylcitalopram observed in steady-state conditions generally reach less than 50% of those of the parent compound, leading to the suggestion that the role of S(+)-desmethylcitalopram in the overall activities of escitalopram can probably be ignored.9
Unlike the situation with fluoxetine, for which both enantiomers appear to have significant effects as antidepressants, a wealth of information from animal models suggests that the psychotropic properties of citalopram reside almost exclusively in the S(+)-enantiomer. For example, the effect of acute intravenous administration of citalopram and its enantiomers on neuronal activity of 5-HT cells in the rat dorsal raphe nucleus (DRN) was measured using extracellular single unit recording.14 Escitalopram and citalopram, but not R(−)-citalopram, reduced the firing activity of 5-HT cells in the DRN in a dose-dependent fashion. Escitalopram was twice as potent as citalopram in this model. In the forced swim test, one of the most widely used animal models for depression, both citalopram and escitalopram demonstrated a dose-dependent reduction in immobility in mice, while R(−)-citalopram was inactive in this model.15 In the rat “resident-intruder” paradigm, escitalopram was 2 to 4 times more potent than racemic citalopram.16
The lack of antidepressant activity of R(−)-citalopram in these models is certainly consistent with the IC50 data presented in Table 1. It is also consistent with Pfeiffer's rule, noted above, which suggests that the more potent the drug, the more likely it is to be stereoselective. In the chiral environment of the 5-HT receptor, it can be pictured that the R-enantiomer binds much less readily to the receptor than escitalopram. The “key” simply does not fit the “lock.”
Clinical studies with escitalopram for the treatment of major depression have been conducted and provide further evidence of the potency and efficacy of escitalopram. A fixed-dose, placebo-controlled, randomized clinical trial of 10 and 20 mg of escitalopram, 40 mg of citalopram, and placebo was conducted.17 This trial showed efficacy for all active drugs compared with placebo, but additionally that the 10-mg dose of escitalopram was at least as efficacious as 40 mg of citalopram while having an overall rate of side effects comparable to placebo. Additionally, there was a trend for the 20-mg dose of escitalopram to have greater efficacy than 40 mg of citalopram.
One of the potential benefits of using a eutomer mentioned above is that a more rapid onset of action may be realized. For antidepressants, a lag time of 3 to 4 weeks is considered standard. It is therefore of interest that escitalopram, at both doses, was statistically superior to placebo early in the study, and this superiority was reflected in multiple measures of depression.17 For example, scores on the depressed mood item of the Hamilton Rating Scale for Depression (HAM-D) significantly decreased by the end of week 1 compared with placebo. Additionally, the overall scores on the HAM-D and Montgomery-Asberg Depression Rating Scale (MADRS) separated by the end of week 2. This finding has been further explored in a report that pooled data from 3 placebo-controlled, randomized clinical trials comparing escitalopram and citalopram with placebo in depressed outpatients.18 The pooled data, which include data from the study by Burke and colleagues17 mentioned above, demonstrated that the mean change from baseline in MADRS scores was significantly greater at week 1 with escitalopram, while citalopram did not significantly separate from placebo until week 6. Similar changes were seen in the mean change from baseline in the Clinical Global Impressions-Improvement (CGI-I) scale. Escitalopram treatment significantly decreased CGI-I scores within 1 week, whereas citalopram treatment significantly decreased CGI-I scores at week 4.
CONCLUSIONS
The Nobel Prize for Chemistry in 2001 was awarded to the scientists who created catalysts that could produce 1 stereoisomer without creating the mirror-image compound. The ability to synthesize a single isomer has opened the way to produce drugs that are more selective and “pure.” Chemical synthesis is no longer limited to producing racemic mixtures that contain unwanted stereoisomers. In the case of citalopram, this has allowed the division of the stereoisomer that contains all of the desired activity of the racemic mixture, escitalopram, from its much less potent counterpart, R(−)-citalopram.
Additional studies will need to be done to elucidate the ultimate place of escitalopram in the armamentarium of medications used to treat depression, but at present it appears to hold promise as a potent, effective, and well-tolerated antidepressant that may offer a more rapid onset of action than other antidepressants for some patients.
Drug names: citalopram (Celexa), fluoxetine (Prozac and others), fluvoxamine (Luvox and others), paroxetine (Paxil), propoxyphene (Darvon and others), sertraline (Zoloft).
Footnotes
Grant/research support provided by Forest Laboratories.
Dr. Burke has received honoraria from and is a speakers/advisory board member for Forest Laboratories.
References
- Brocks DR, Jamali F. Stereochemical aspects of pharmacotherapy. Pharmacotherapy. 1995;15:551–564. doi: 10.1002/j.1875-9114.1995.tb02863.x. [DOI] [PubMed] [Google Scholar]
- Pennisi E. Parasitology. Close encounters: good, bad, and ugly. Science. 2000 290:1491–1493. [DOI] [PubMed] [Google Scholar]
- Hutt AJ, Tan SC. Drug chirality and its clinical significance. Drugs. 1996;52(suppl 5):1–12. doi: 10.2165/00003495-199600525-00003. [DOI] [PubMed] [Google Scholar]
- Eichelbaum M. Enantiomers: implications and complications in developmental pharmacology. Dev Pharmacol Ther. 1992;18:131–134. [PubMed] [Google Scholar]
- Wainer IW. Three-dimensional view of pharmacology. Am J Hosp Pharm. 1992;49(9, suppl 1):S4–S8. [PubMed] [Google Scholar]
- Caldwell J. Stereochemical determinants of the nature and consequences of drug metabolism. J Chromatogr A. 1995;694:39–48. doi: 10.1016/0021-9673(94)00812-n. [DOI] [PubMed] [Google Scholar]
- Tucker GT. Chiral switches. Lancet. 2000;355:1085–1087. doi: 10.1016/S0140-6736(00)02047-X. [DOI] [PubMed] [Google Scholar]
- Lane RM, Baker GB. Chirality and drugs used in psychiatry: nice to know or need to know? Cell Mol Neurobiol. 1999;19:355–372. doi: 10.1023/A:1006997731966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Rochat B. Comparative pharmacokinetics of selective serotonin reuptake inhibitors: a look behind the mirror. Int Clin Psychopharmacol. 1995;10(suppl 1):15–21. doi: 10.1097/00004850-199503001-00004. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Knight DL, Nemeroff CB. Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry. 2001;50:345–350. doi: 10.1016/s0006-3223(01)01145-3. [DOI] [PubMed] [Google Scholar]
- Hyttel J, Bøges KP, Perregaard J, et al. The pharmacological effect of citalopram resides in the (S)-(+)-enantiomer. J Neural Transm Gen Sect. 1992;88:157–160. doi: 10.1007/BF01244820. [DOI] [PubMed] [Google Scholar]
- Rochat B, Amey M, Baumann P. Analysis of enantiomers of citalopram and its demethylated metabolites in plasma of depressive patients using chiral reverse-phase liquid chromatography. Ther Drug Monit. 1995;17:273–279. doi: 10.1097/00007691-199506000-00011. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467–489. doi: 10.1023/A:1006986824213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PJ, Hogg S. Beavioural effects of escitalopram predict potent antidepressant activity. Presented at the 7th World Congress of Biological Psychiatry. 1–6July2001 Berlin, Germany. [Google Scholar]
- Sanchez C, Hogg S. The antidepressant effect of citalopram resides in the S-enantiomer (Lu 26-054). Presented at the 55th annual meeting of the Society of Biological Psychiatry. 11–13May2000 Chicago, Ill. [Google Scholar]
- Mitchell P, Hogg S. Escitalopram: rapid antidepressant activity in rats. Presented at the 7th World Congress of Biological Psychiatry. 1–6July2001 Berlin, Germany. [Google Scholar]
- Burke WJ, Gergel I, Bose A. Fixed-dose trial of the single isomer SSRI escitalopram in depressed outpatients. J Clin Psychiatry. 2002;63:331–336. doi: 10.4088/jcp.v63n0410. [DOI] [PubMed] [Google Scholar]
- Gorman JM, Korotzer A, Su G. Efficacy comparison of escitalopram and citalopram in the treatment of major depressive disorder: pooled analysis of placebo-controlled trials. CNS Spectrums. 2002;7(suppl 1):40–44. doi: 10.1017/s1092852900028595. [DOI] [PubMed] [Google Scholar]