

Abstract
Background
Relatives of breast cancer cases have an increased risk of the disease. The risk increases with increasing numbers and decreasing age of onset of affected relatives. In families with a BRCA1 or a BRCA2 mutation, individual carrier status predicts the risk of breast cancer. In relatives of cases where both BRCA1 and BRCA2 mutations are excluded, the risk remains undetermined.
Methods
Standardized incidence ratios (SIRs) and cumulative cancer incidences were calculated for relatives of a population-based set of early-onset breast cancer index cases (younger than age 41 years) with a defined BRCA mutation status (n = 203).
Results
In first-degree relatives (FDRs) of mutation-negative cases, breast cancer incidences (SIR = 2.3), prostate cancer incidences (SIR = 1.7), cervix cancer incidences (SIR = 3.3) and nonmelanoma skin cancer incidences (SIR = 2.8) were increased. The risks of breast cancer, prostate cancer and nonmelanoma skin cancer were further increased in FDRs of breast cancer cases younger than 36 years of age. In high-risk individuals with at least one relative with breast cancer apart from the index case, but no BRCA mutation in the family, breast cancer incidence was increased (SIR = 5.3); again the prostate cancer incidence was elevated (SIR = 2.5). The cumulative incidence of breast cancer at ages 50 and 70 years for FDRs of index cases without a BRCA mutation was 3.6% and 12.8%, respectively. Similarly, the cumulative incidence of breast cancer for high-risk women was 6.3% and 21.1% at ages 50 and 70 years, and that for FDRs of BRCA mutation carriers was 17.2% and 27.7% at the same ages.
Conclusion
The incidence of breast cancer is increased for FDRs of women with early-onset breast cancer irrespective of the BRCA status in the family. Risk increases with decreasing age and with increasing number of affected relatives. The incidences of prostate cancer, cervix cancer and nonmelanoma skin cancer are elevated for FDRs of early-onset breast cancer cases without a BRCA mutation, indicating a possible association between these cancers and early-onset breast cancer.
Keywords: breast cancer, BRCA1, BRCA2, familial risk, prostate cancer.
Introduction
The influence of hereditary factors over breast cancer risk in women is well established. In a review and meta-analysis of previously published case–control and cohort studies, Pharoah and colleagues concluded in 1997 that a woman with a relative with breast cancer has an approximately doubled breast cancer risk compared with controls. The risk of breast cancer tended to be higher in first-degree relatives (FDRs) of affected individuals than in second-degree relatives, tended to be higher if there was more than one close relative with breast cancer, and tended to be higher if the affected woman was younger than 50 years at the time of diagnosis compared with if she was older than 50 years [1]. Similar data have been reported from a Swedish population [2].
Epidemiologically based data from different cohort and case–control studies of different populations have been used to create risk models that are applied clinically in oncogenetic counselling. The two most used models are the so-called Claus model [3] and the Gail model [4]. The Claus model provides cumulative breast cancer risks for women with different numbers of relatives with breast cancer or ovarian cancer at different ages. In addition to the number of FDRs with breast cancer, the Gail model also takes nonhereditary risk factors into account.
The identification of the inherited breast cancer susceptibility genes, most notably BRCA1 and BRCA2 [5-7], has greatly improved the possibility of providing distinct estimates of future breast cancer risk in mutation-positive families, and above all has opened up the possibility of differentiating between carriers with a markedly increased risk and noncarriers with a population-like risk in these families. However, the introduction of BRCA1 and BRCA2 mutation testing in clinical oncogenetic counselling will inevitably lead to bias in the risk estimates if the same epidemiologically based risk-calculation models are used in families where BRCA1 and BRCA2 have been excluded as the causative factor of inherited cancer.
In families with an identified BRCA1 or BRCA2 mutation, the risk estimate of breast cancer in mutation carriers will depend on the population on which the calculation is based and on the way family members were ascertained (discussed by Dörum and colleagues [8]). Generally speaking, penetrance estimates of mutation carriers tend to indicate a higher cumulative risk of breast cancer if based on a set of high-risk families [9,10] than if based on a population-based set of mutation carriers not recruited due to the occurrence of multiple cases of breast cancer in their families [11-15]. Families identified in oncogenetic counselling have penetrance estimates of breast cancer between these two extremes [16].
Despite intense research efforts and the publication of several potential chromosomal loci [17-22] since and before the identifications of BRCA1 and BRCA2, no generally accepted high-penetrant 'breast cancer gene 3' has been identified. A recent study of FDRs of incident breast cancer cases where BRCA1 and BRCA2 were excluded indicates that obtaining a complete picture of the remaining molecular genetic factors involved in hereditary breast cancer may be difficult given the fact that several common alleles may be involved in a polygenic model, and that recessive genes are as likely as dominant ones to be involved in 'unexplained' hereditary breast cancer families [23]. For those families where BRCA1 and BRCA2 mutations are not identified, therefore, data from epidemiological studies have, and probably will in the future continue to have, a strong impact on clinical oncogenetic counselling.
The main purpose of the present study is to estimate relative and absolute cancer risks in FDRs (i.e. siblings and parents) of a population-based set of index individuals with early-onset breast cancer (onset younger than age 41 years), a known family history of cancer, and a known BRCA1 and BRCA2 mutation status. These estimates are made to provide data that can be used in the counselling of individuals in a BRCA1-screened and BRCA2-screened population.
Materials and methods
During the period 1 January–31 December 1995, a total of 262 women aged 40 years or younger were diagnosed with breast cancer in the South Swedish Health Care Region. After approval was obtained from the ethical committee at Lund University Hospital, these women were invited to a population-based screening of BRCA1 and BRCA2 germline mutations and a survey of their family history of cancer. Among a total of 234 women screened for germline mutations, 16 mutation carriers (6.8%) were initially detected for BRCA1 and five mutation carriers (2.1%) for BRCA2 [24]. After the publication of the study, however, an additional mutation was detected in one of the families: the sister of an index case with a medullary breast cancer contracted breast cancer and sought oncogenetic counselling. In this situation, mutation screening was reiterated in the index individual using denaturating high-performance liquid chromatography, a more sensitive method than that previously used (single-stranded conformal polymorphism). A 300T → G germline mutation in BRCA1 that had escaped previous detection was identified. The rates of mutation of BRCA1 was thus modified to 17/234 (7.3%), and the total rate of BRCA mutation carriers was set to 22/234 (9.4%).
All data regarding cancer diagnoses in cases and relatives in the study were obtained from independent sources. Children, siblings, parents and their siblings, as well as grandparents of the index women were identified for this study by contacting the local revenue authorities that keep the local census registers. Questionnaire data from the index individuals were used only to make sure no relatives were overseen when identifying family members. Malignant diagnoses of these individuals were obtained from the regional and national cancer registries. Only study subjects where diagnoses in FDRs could be checked-up in the local or national cancer register were included in the statistical analyses (n = 203). Dropouts were either a result of the index individual not participating in the mutation analysis or due to the fact that data on family history could not be traced in Swedish registers, most frequently because the family was of foreign ancestry. The material has been described in greater detail in a previous publication [24].
Statistical risk calculations were performed separately on three sets of FDRs. Index individuals were excluded from all analyses. The first set of individuals included all FDRs of identified index individuals without a BRCA1 or BRCA2 germline mutation (n = 646). The second analysis group consisted of a selected group of high-risk individuals with at least one FDR or second-degree relative with breast cancer in the same branch of the family, other than relative to the index woman (n = 180). The families of these individuals were families that would have been considered for mutation screening of BRCA1 and BRCA2 if they had been identified through the ordinary oncogenetic reception, a clinical unit for oncogenetic counselling jointly run by the departments of oncology and clinical genetics at Lund University Hospital. The third set of individuals included all FDRs of index cases with an identified BRCA1 or BRCA2 germline mutation (n = 82).
Individual family members of index individuals that were mutation carriers were not uniformly tested for both BRCA1 and BCRA2, and thus data on the individual carrier status were not included in the analyses. Data on cancer incidence not verified in the national or regional cancer register were accepted for the purpose of identifying individuals with at least one FDR or second-degree relative with breast cancer other than the index individual, but not for inclusion in the cohort analysis itself. For example, if the index individual reported that the maternal grandmother of an index individual had died from breast cancer and this could not be verified in the cancer register, the mother and all siblings of the index individual were included in the analysis of high-risk women with one FDR or second-degree relative with breast cancer other than the index woman (Fig. 1a). On the contrary, if a paternal aunt and a sister of the index case were recorded to have breast cancer, then the father, siblings and children of the index case were included, but not the mother (Fig. 1b).
Figure 1.
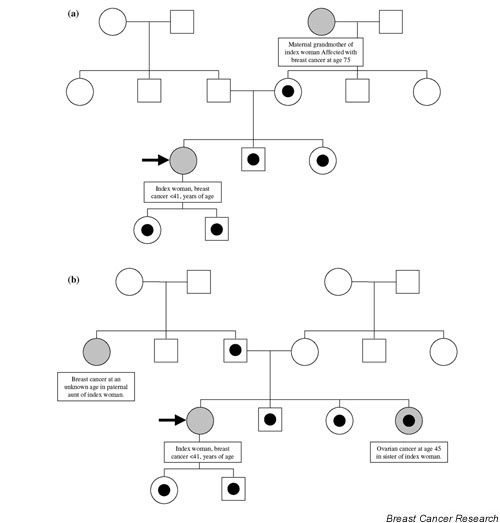
Pedigrees indicating which individuals were included in the BRCA nonassociated high-risk group. The cohort of high-risk individuals was selected based on the prevalence of at least one additional case of breast cancer apart from the index woman in those families. In these two hypothetical families, (a) and (b), individuals affected with breast cancer appear in greyish colour. Individuals included in the analyses are marked with a black dot.
Because of the small number of mutation-positive index cases in the study (n = 22), calculations of the dominantly inherited breast cancer susceptibility genes in BRCA1-positive and BRCA2-positive families (n = 82 FDRs) were not carried out separately, but were pooled in one group.
Calculation of standard incidence ratios
Cancer incidence in relatives of the index cases was analysed by calculating standardized incidence ratios (SIRs) with stratification by sex, by calendar year, and by 5-year age groups. Incidence rates from the southern region of Sweden were used as references. The different study cohorts were stratified by the index cases' age of onset. Individuals in the cohorts were followed from date of birth until the earliest event of death, of a second primary cancer diagnosis, of emigration, or of end of study follow-up (i.e. 31 December 2000).
In standard cohort analyses, the statistical inference is based on the assumption that the observations are independent and Poisson distributed. The Poisson assumption is not correct if there is a family factor (hereditary or environmental), since this means that there is a dependency between the individuals of a family. The estimate of the SIR from a standard cohort analysis will be correct even when a dependency is present between data, but the estimate of its standard error may be too small if the dependency between family members is not taken into account. Therefore, we have used a sampling-based standard error when calculating approximate confidence intervals based on the normal distribution [2].
Calculation of cumulative breast cancer risks
To be able to calculate cumulative breast cancer incidence in mutation carriers in the families where individual testing had not been performed, we calculated the cumulative breast cancer risk using the following method. Let SMRE denote the standardized morbidity ratio of individuals with exposure E (E can, for instance, indicate the prevalence of a FDR with breast cancer). The probability of an individual with exposure E developing breast cancer before the age of t is then:
![]()
where  is the age-specific and calendar year-specific breast cancer incidence in the reference population, and Δtij is the time in age interval i and calendar year j.
is the age-specific and calendar year-specific breast cancer incidence in the reference population, and Δtij is the time in age interval i and calendar year j.
The cumulative risk for a BRCA1 or BRCA2 mutation carrier was calculated using the estimate of the cumulative risk for FDRs of the index women with a BRCA1 or BRCA2 mutation:
P(brca <t | FDR of index women) = 0.5 [P(brca <t | no mutation)] + 0.5 [P(brca <t | BRCA1 or BRCA2 mutation)]
⇔
P(brca <t | BRCA1 or BRCA2 mutation) = {P(brca <t | FDR of index women) - 0.5 [P(brca <t | no mutation)]} / 0.5
The estimation of the standard errors of the cumulative risks was calculated using Gauss approximation. Cumulative incidences and confidence intervals were calculated in 5-year intervals.
Stata 6.0 statistical software was used for all statistical analyses [25].
Results
The mutation status and the family history according to the criteria for inclusion in the study (i.e. confirmed data on cancer incidence among siblings and parents of the index individual) were known for 203 out of 262 index individuals (77.5%). The families of these index individuals included 728 FDRs. In addition, 804 female second-degree relatives (paternal and maternal aunts and grandmothers) of the index individuals were identified. In total, 22 families where BRCA1 or BRCA2 mutations were identified in index individuals (17 families with BRCA1 mutations and five families with BRCA2 mutations) and 181 families without mutations were included in the study. Out of the families with no BRCA1 or BRCA2 mutation, 70 had a family history with at least one additional case of breast cancer in a FDR or a second-degree relative of the index individual; by definition, a woman younger than the age of 41 years with breast cancer (Fig. 1a,1b).
Relative cancer incidence in FDRs of BRCA nonmutation carriers (Table 1)
Table 1.
Cancer incidences in first-degree relatives of BRCA1/BRCA2 noncarriers
| ICD-7 | Site | Observed (n) | Expected (n) | Standardized incidence ratio | 95% confidence interval |
| 140–209 | All malignant tumours | 120 | 82.3 | 1.5 | 1.2–1.7 |
| Non-170 | All malignant tumours except breast | 91 | 69.9 | 1.3 | 1.1–1.6 |
| 140 | Lip | 1 | 0.50 | 2.0 | 0.05–11.1 |
| 141 + 143–144 | Cavum oris | 0 | 0.56 | 0 | 0–6.6 |
| 142 | Salivary glands | 1 | 0.31 | 3.2 | 0.08–17.8 |
| 145–148 | Throat | 0 | 0.40 | 0 | 0–9.1 |
| 150 | Oesophagus | 0 | 0.53 | 0 | 0–6.9 |
| 151 | Stomach | 2 | 2.8 | 0.71 | 0.09–2.6 |
| 153 | Large bowel | 5 | 5.6 | 0.89 | 0.29–2.1 |
| 154 | Rectum | 6 | 3.8 | 1.6 | 0.58–3.5 |
| 1550 | Liver | 1 | 0.61 | 1.6 | 0.04–9.1 |
| 1551–1559 | Gall tract | 1 | 1.2 | 0.85 | 0.02–4.8 |
| 157 | Pancreas | 0 | 2.3 | 0 | 0–1.6 |
| 160 | Nose and sinuses | 0 | 0.18 | 0 | 0–20.4 |
| 161 | Larynx | 0 | 0.50 | 0 | 0–7.3 |
| 1620–1621 | Trachea and lung | 8 | 6.6 | 1.2 | 0.53–2.4 |
| 1622 | Pleura | 0 | 0.21 | 0 | 0–17.8 |
| 170 | Breast | 29 | 12.4 | 2.3 | 1.6–3.4 |
| 171 | Cervix uteri | 5 | 1.5 | 3.3 | 1.1–7.6 |
| 172 + 174 | Uterine corpus | 2 | 3.0 | 0.68 | 0.80–2.4 |
| 175 | Ovaries | 3 | 2.5 | 1.2 | 0.24–3.5 |
| 176 | Vulva, vagina | 0 | 0.32 | 0.0 | 0–11.7 |
| 177 | Prostate | 17 | 9.9 | 1.7 | 1.0–2.8 |
| 178 | Testes | 0 | 0.61 | 0 | 0–6.0 |
| 180 | Kidneys | 3 | 3.2 | 0.93 | 0.19–2.7 |
| 181 | Bladder and urinary tract | 3 | 3.9 | 0.76 | 0.16–2.2 |
| 190 | Melanoma | 5 | 3.2 | 1.5 | 0.4–93.5 |
| 191 | Skin, nonmelanoma | 6 | 2.1 | 2.8 | 1.0–6.1 |
| 193 | Central nervous system | 6 | 3.7 | 1.6 | 0.60–3.6 |
| 194 | Thyroid | 0 | 1.53 | 0 | 0–2.4 |
| 196–197 | Bone, connective tissue | 0 | 0.79 | 0 | 0–4.7 |
| 200 + 202 | Non-Hodgkin's lymphoma | 4 | 3.0 | 1.4 | 0.37–3.4 |
| 201 | Hodgkin's disease | 2 | 0.59 | 3.4 | 0.41–12.2 |
| 203 | Myeloma | 0 | 1.2 | 0 | 0–2.9 |
| Acute leukaemia | 3 | 0.76 | 4.0 | 0.82–11.6 | |
| 2041 | Chronic lymphocytic leukaemia | 1 | 0.84 | 1.2 | 0.03–6.6 |
| 2051 | Chronic myelogenous leukaemia | 0 | 0.25 | 0 | 0–14.9 |
| 208 | Polycytaemia vera | 0 | 0.34 | 0 | 0–10.8 |
Age 0–79 years; n = 646; 25,244.0 person-years; 1958–2000.
The incidence of all malignant tumours was increased among FDRs of women without a BRCA1 or BRCA2 mutation (n = 646) (observed cases = 120 versus expected cases = 82.3, SIR = 1.5, 95% confidence interval [CI] = 1.2–1.7). The incidence of all malignant tumours excluding breast cancer was also significantly elevated (observed cases = 91 versus expected cases = 69.9, SIR = 1.3, 95% CI = 1.1–1.6). The risk of breast cancer among FDRs of non-BRCA1/non-BRCA2 mutation carriers was more than doubled (observed cases = 29 versus expected cases = 12.4, SIR = 2.3, 95% CI = 1.6–3.4). In addition, the SIR of prostate cancer was increased (observed cases = 17 versus expected cases = 9.9, SIR = 1.7, 95% CI = 1.0–2.8), as was the SIR of cervix cancer (observed cases = 5 versus expected cases = 1.5, SIR = 3.3, 95% CI = 1.1–7.6) and of nonmelanoma skin cancer (observed cases = 6 versus expected cases = 2.1, SIR = 2.8, 95% CI = 1.0–6.1).
The FDRs of non-BRCA1/non-BRCA2 mutation carriers were subdivided into groups on the basis of the age of the index case. In FDRs of index women aged younger than 36 years (n = 184) breast cancer was increased more than threefold (observed cases = 9 versus expected cases = 2.8, SIR = 3.2, 95% CI = 1.5–6.1) compared with FDRs of index women aged 36 years and older (n = 462), among which the increase was only twofold (observed cases = 20 versus expected cases = 9.6, SIR = 2.1, 95% CI = 1.3–3.2). Furthermore, the increase of prostate cancer appeared to be confined to the FDRs of index women younger than the age of 36 years (observed cases = 9 versus expected cases = 2.0, SIR = 4.4, 95% CI = 2.0–8.4). Among FDRs of index women aged 36 years or older, no elevation in prostate cancer risk was observed (observed cases = 8 versus expected cases = 7.8, SIR = 1.0, 95% CI = 0.44–2.0). In female second-degree relatives of BRCA nonmutation carriers (n = 746) the breast cancer risk was not significantly elevated compared with what could be expected for the population (observed cases = 48 versus expected cases = 39.4, SIR = 1.2, 95% CI = 0.90–1.6).
Individuals with a positive family history of breast cancer (Table 2)
Table 2.
Cancer incidence in first-degree relatives of index cases with breast cancer younger than age 41 but noBRCA1/BRCA2 germline mutation, and at least one additional case of breast cancer in a first-degree or second-degree relative
| ICD-7 | Site | Observed (n) | Expected (n) | Standardized incidence ratio | 95% confidence interval |
| 140–209 | All malignant tumours | 43 | 18.5 | 2.3 | 1.6–3.1 |
| Non-170 | All malignant tumours except breast | 29 | 15.9 | 1.8 | 1.2–2.6 |
| 153 | Large bowel | 1 | 1.2 | 0.82 | 0.02–4.6 |
| 154 | Rectum | 2 | 0.82 | 2.4 | 0.30–8.8 |
| 162 | Trachea and lung | 3 | 1.5 | 2.0 | 0.42–5.9 |
| 170 | Breast | 14 | 2.6 | 5.3 | 2.9–9.0 |
| 171 | Cervix uteri | 2 | 0.39 | 5.1 | 0.62–18.5 |
| 175 | Ovaries | 1 | 0.50 | 2.0 | 0.05–11.0 |
| 177 | Prostate | 6 | 2.4 | 2.5 | 0.91–5.4 |
| 180 | Kidney | 2 | 0.74 | 2.7 | 0.33–9.8 |
| 181 | Bladder and urinary tract | 2 | 0.93 | 2.1 | 0.26–7.8 |
| 191 | Skin nonmelanoma | 1 | 0.50 | 2.0 | 0.05–11.2 |
| 193 | Central nervous system | 2 | 0.86 | 2.3 | 0.28–8.4 |
| 200 + 202 | Non-Hodgkin's lymphoma | 3 | 0.66 | 4.5 | 0.94–13.3 |
Age 0–79 years; n = 180; 6885.2 person-years; 1958–2000.
Among individuals without a BRCA1 or BRCA2 mutation in the family but with at least one case of breast cancer in a FDR or a second-degree relative other than the index case (n = 180, related to 61 index individuals), the SIR of all malignant tumours was elevated (observed cases = 43 versus expected cases = 18.5, SIR = 2.3, 95% CI = 1.6–3.1). When breast cancer incidence was excluded, the risk of cancer was still increased in this group (observed cases = 29 versus expected cases = 15.9, SIR = 1.8, 95% CI = 1.2–2.6). In this group, breast cancer (observed cases = 14 versus expected cases = 2.6, SIR = 5.3, 95% CI = 2.9–9.0) was elevated, and there were also nonsignificant increases of prostate cancer (observed cases = 6 versus expected cases = 2.4, SIR = 2.5, 95% CI = 0.91–5.4) and non-Hodgkin's lymphoma (NHL) (observed cases = 3 versus expected cases = 0.66, SIR = 4.5, 95% CI = 0.94–13.3).
FDRs of BRCA mutation carriers (Table 3)
Table 3.
Cancer incidence in first-degree relatives ofBRCA1/BRCA2 carriers
| ICD-7 | Site | Observed (n) | Expected (n) | Standardized incidence ratio | 95% confidence interval |
| 140–209 | All malignant tumours | 20 | 7.3 | 2.7 | 1.7–4.2 |
| Non-170 | All malignant tumours except breast | 12 | 6.2 | 1.9 | 1.0–1.6 |
| 151 | Stomach | 0 | 0.21 | 0 | 0–17.3 |
| 153 | Large bowel | 0 | 0.44 | 0 | 0–8.3 |
| 157 | Pancreas | 1 | 0.17 | 5.8 | 0.15–32.2 |
| 170 | Breast | 8 | 1.1 | 7.2 | 3.1–14.3 |
| 171 | Cervix uteri | 0 | 0.17 | 0 | 0–21.5 |
| 175 | Ovaries | 3 | 0.20 | 14.9 | 3.1–43.7 |
| 176 | Vulva, vagina | 1 | 0.02 | 45.6 | 1.1–254 |
| 177 | Prostate | 2 | 0.82 | 2.4 | 0.30–8.8 |
| 190 | Melanoma | 1 | 0.36 | 2.8 | 0.70–15.6 |
| 191 | Skin, nonmelanoma | 0 | 0.19 | 0 | 0–19.8 |
| 193 | Central nervous system | 1 | 0.40 | 2.5 | 0.06–13.9 |
| 194 | Thyroid | 1 | 0.17 | 5.9 | 0.15–33.0 |
Age 0–79 years; n = 82; 3106.4 person-years; 1958–2000.
Among FDRs of BRCA1 and BRCA2 mutation carriers (n = 82), the SIR of all malignant tumours was increased (observed cases = 20 versus expected cases = 7.3, SIR = 2.7, 95% CI = 1.7–4.2). When breast cancer cases were excluded, the increase diminished but was still statistically significant (observed cases = 12 versus expected cases = 6.2, SIR = 1.9, 95% CI = 1.0–1.6). When it came to individual diagnoses, breast cancer (observed cases = 8 versus expected cases = 1.1, SIR = 7.2, 95% CI = 3.1–14.3) and ovarian cancer (observed cases = 3 versus expected cases = 0.20, SIR = 14.9, 95% CI = 3.1–43.7) were observed more frequently than expected. In female second-degree relatives of BRCA1 and BRCA2 mutation carriers (n = 61), breast cancer (observed cases = 8 versus expected cases = 2.8, SIR = 2.8, 95% CI = 1.2–5.6) and ovarian cancer (observed cases = 4 versus expected cases = 0.64, SIR = 6.2, 95% CI = 1.7–16.0) were also elevated.
Cumulative breast cancer risks
The cumulative incidence of breast cancer at ages 50 and 70 years among FDRs of index women without an identified BRCA1 or BRCA2 germline mutation was 3.6% (95% CI = 1.3–5.7%) and 12.8% (95% CI = 6.5–15.7%), respectively (Fig. 2a). The cumulative incidence of breast cancer in the group of individuals without a BRCA1 mutation or a BRCA2 mutation but with at least one FDR or second-degree relative with breast cancer other than the index case was 6.3% (95% CI = 3.2–10.9%) at age 50 years and 21.1% (95% CI = 11.1–34.7%) at age 70 years (Fig. 2b). The cumulative breast cancer incidence in the female population of the South Swedish Health Care Region was 1.5% at age 50 years and 5.3% at age 70 years, significantly lower compared with all study groups (Figs 2a,2b,2c,2d). The cumulative incidence of breast cancer at ages 50 and 70 years among FDRs of index individuals with an identifiable BRCA1 or BRCA2 mutation was 17.2% (95% CI = 6.7–33.7%) and 27.7% (95% CI = 8.2–54.4%), respectively (Fig. 2c), translating to a 32.9% (95% CI = 8.2–62.1%) and 50.2% (95% CI = 14.9–100%) risk of breast cancer among BRCA1 or BRCA2 mutation carriers (Fig. 2d).
Figure 2.
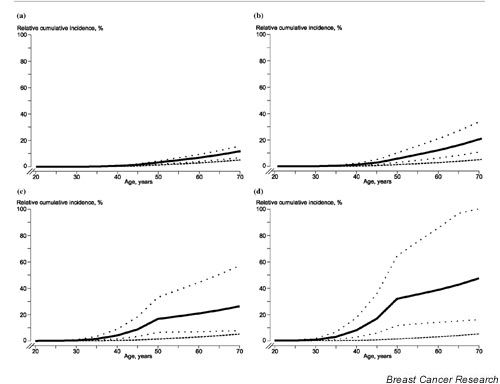
Cumulative breast cancer incidences up to the age of 70 years of the different study cohorts. (a) Cumulative incidence of breast cancer (solid line) (dotted lines, 95% confidence interval) for first-degree relatives of breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2) mutation noncarriers younger than age 41 years. A general female South Swedish population is added for comparison (dashed line). (b) Cumulative incidence of breast cancer (solid line) (dotted lines, 95% confidence interval) for women with at least two first-degree relatives with breast cancer, of which one is an index individual diagnosed younger than age 41 years and is without an identified BRCA1/BRCA2 germline mutation. A general female South Swedish population is added for comparison (dashed line). (c) Cumulative incidence of breast cancer (solid line) (dotted lines, 95% confidence interval) for first-degree relatives of BRCA1 and BRCA2 mutation carriers younger than age 41 years. A general female South Swedish population is added for comparison (dashed line). (d) Calculated cumulative incidence of breast cancer (solid line) (dotted lines, 95% confidence interval) for BRCA1 and BRCA2 mutation carriers younger than age 41 years. A general female South Swedish population is added for comparison (dashed line).
Discussion
In the present paper we present a population-based overview of the hereditary and familial risks of cancer in relatives of early-onset breast cancer cases from southern Sweden. All individuals and all diagnoses included in the statistical estimations were verified from independent and reliable data sources. In several previous studies of familial risk of cancer that are frequently used in oncogenetic counselling of cancer risk in breast cancer families, unknown numbers of unidentified families with BRCA1 and BRCA2 mutations are hidden among other families. This will introduce bias in the counselling situation if data from such studies are utilized to estimate breast cancer risk in individuals from families where BRCA1 and BRCA2 have been excluded as the disease-causing factor, a commonplace procedure nowadays in oncogenetic clinics worldwide. Providing clinically useful data for the large group of BRCA1-negative and BRCA2-negative families seeking oncogenetic counselling has been the main objective of the present study. Although the size of study sample may seem limited, the quality of data, the high inclusion rate and the thorough BRCA screening that was performed justifies certain conclusions to be drawn from the dataset.
One BRCA1 mutation carrier was detected due to reanalysis of that particular case with a more sensitive technique. Since one of the main aims of the study was to analyse cancer incidence in BRCA1 and BRCA2 noncarriers, we were not willing to include a known mutation carrier in that study group even if the detection of this case was not strictly population based. Second, denaturating high-performance liquid chromatography was introduced to replace the single-stranded conformal polymorphism technique in our laboratory when about one-half of the cases were analysed. The intention was to increase the sensitivity of the mutation screening. It was therefore reasonable to accept its use in an additional case.
Regarding FDRs of non-BRCA1/non-BRCA2-mutation carriers, the main findings were increased risks of breast cancer, of prostate cancer, of cervix cancer and of nonmelanoma skin cancer. The risks seemed to depend on the age of the index individual at the time of diagnosis, and were higher for breast cancer in FDRs of index individuals diagnosed with breast cancer younger than the age of 36 years compared with FDRs of index individuals diagnosed between the ages of 36 and 41 years. The risks of prostate cancer and of skin cancer were only elevated in FDRs of index individuals younger than the age of 36 years. The increase in cervix cancer risk was not significant in any of the subgroups of FDRs of index cases younger or older than the age of 36 years (data not shown). This finding might indicate an association between very early-onset breast cancer and one or several of prostate cancer, of nonmelanoma skin cancer and, possibly, of cervix cancer.
In the literature, partly conflicting data regarding the association between breast cancer and prostate cancer have been presented. In two studies of relatives of prostate cancer patients, no excess risks of breast cancer or any other nonprostate cancers were identified [26,27]. Other studies have indicated an association. In an American case-cohort study of 981 cases with prostate cancer and 1315 controls, an association between prostate cancer and breast cancer in sisters was identified, but this was not the case in mothers of prostate cancer cases [28]. In a study of families from Utah, a small but statistically significant increase in prostate cancer was observed in relatives of breast cancer patients [29]. Finally, in Iceland, relatives of breast cancer patients were subject to a 1.5-fold increase in prostate cancer risk [30], an increase most probably attributable to carriers of the Icelandic BRCA2 founder mutation.
Since BRCA1 and BRCA2 were characterized, prostate cancer has been linked to breast cancer through these genes. In some studies on mutation carriers, BRCA1 and BRCA2 have been associated with increased risk of prostate cancer [9,31]. It has been claimed that BRCA2 is associated with a sevenfold increase in prostate cancer risk in mutation carriers compared with that in the general population [31]. The increased risk of prostate cancer in relatives of BRCA2 mutation carriers has also been seen in a previous study from our institute [32].
Several of the aforementioned studies suggest an association between breast cancer and prostate cancer that could well be mediated through an increased risk of both malignancies in BRCA2 mutation carriers. The present study suggests that there may be a residual association not mediated through this gene in families with very early-onset breast cancer. A possible explanation could be that we have overlooked a number of mutation carriers during mutation screening. Sensitivity of mutation screening is not 100%; it is thus probable that not all mutation carriers in the material were identified, and that the over-risk of prostate cancer could be due to undetected BRCA2 families in the dataset. It should be noted, however, that if a significant number of BRCA1 mutation carriers have been overlooked during mutation screening, then one would expect to observe an excess of ovarian cancer cases in the remaining families; this was not the case. The observation concerning the increased risk of prostate cancer in FDRs of breast cancer cases younger than the age of 36 years needs further study to be confirmed.
An association between breast cancer and nonmelanoma skin cancer was previously reported in a study on BRCA1-associated families from Southern Sweden [32]. To our knowledge, such an association has not been reported elsewhere.
BRCA mutations are generally detected in only a fraction of the families that are offered mutation screening. It is of great clinical importance to be able to provide reliable estimates of cancer risk among individuals of those mutation-negative families for which mutation status will provide no further information; at our institute, almost three-quarters of the analysed families (unpublished observation). To explore this group further, we selected a group of FDRs of nonmutation carriers who had at least one additional FDR or second-degree relative in the same side of the pedigree with breast cancer. In this selected subgroup of BRCA1 and BRCA2 mutation-negative families, which corresponds to families offered clinical mutation screening according to the aforementioned criteria, we observed a significantly increased greater than twofold risk for all malignant tumours and a fivefold increased risk of breast cancer. Prostate cancer and NHL were frequent, but were not statistically significantly increased. A similar observation regarding NHL was made previously in a large family material from Utah, where the risk of NHL was increased in relatives of cases of early-onset breast cancer (onset younger than 50 years of age) [29].
We noted that germline mutations in BRCA1 and BRCA2 do not appear to be associated with any major risk increase in any malignant diagnoses apart from breast cancer and ovarian cancer. Furthermore, all observed cases of ovarian cancer were seen in first-degree relatives of BRCA1 mutation carriers (n = 63); that is, no cases of ovarian cancer were observed in the FDRs of BRCA2 mutation carriers or in individuals without a BRCA1 or BRCA2 germline mutation. This observation regarding ovarian cancer in BRCA2 mutation carriers fits well with a previous observation that we made in an earlier study on a mainly separate set of BRCA1-associated and BRCA2-associated families. In that study, we did not observe any increased risk of ovarian cancer in members of BRCA2-mutation families if the cases that led to the identification of the families were excluded from the analysis [32]. Interestingly, the increased risk of prostate cancer and colorectal cancer that has been observed in some studies of BRCA mutation carriers [9,32,33] was not observed in the present material. The fact that prostate cancer and colorectal cancer were not excessive in the material could be due to the relatively small size of the study and to insufficient statistical power and too few years of follow-up in FDRs at relevant ages. However, it may also be an important observation with implications for male mutation carriers in BRCA1 and BRCA2 mutation-positive families. This situation might change with prolonged follow-up.
Considering the fact that the excess risk of ovarian cancer in FDRs of mutation carriers was picked up in the present study, it does seem justified to draw the conclusion that the risks of prostate cancer and of colorectal cancer up to the age of 79 years in BRCA1 and BRCA2 families are significantly lower than the risk of ovarian cancer in FDRs of BRCA mutation carriers. This may support the opinion that the need for controls aiming at early detection of these diagnoses in mutation carriers may not be clinically warranted. Finally, a cumulative breast cancer incidence at ages 50 and 70 years was generated for mutation carriers of BRCA1 and BRCA2, and was found to be 32.9% and 50.2%, respectively. This moderate risk estimate fits well into the previously observed pattern of extremely high penetrance estimates based on selected high-risk families [9,10], and more moderate estimations stemming from materials that are population based [11-14]. However, since the number of included relatives of mutation carriers is relatively limited, the estimate must be regarded with some caution. This is especially true for the incidence older than age 50 years, where a limited number of person-years were observed (n = 651) and only two breast cancers were recorded. Most of these person-years refer to mothers of the index individuals.
The cumulative breast cancer risk at ages 50 and 70 years was analysed in the three groups of FDRs. The practice of using incident breast cancer cases when estimating cumulative breast cancer risk has been criticized due to the risk of overestimation since nongenetic risk factors are likely to be over-represented in such series [34]. We have tried to minimize this bias by excluding index individuals from all estimations. In the present study of cumulative breast cancer incidence, BRCA1 and BRCA2 mutation carriers were analysed jointly because of the limited number of available cases. This may generate a less informative penetrance estimate related to the fact that the penetrance of breast cancer in BRCA1 and BRCA2 mutation carriers may differ due to differences in gene-specific properties regarding breast cancer risk. In addition, the number of cases analysed may be considered low, but the set of BRCA mutation carriers upon which the estimate is based is larger than or similar to those of several previous studies in the field [13,14,35]. The cumulative incidence curves of the different study groups suggest that the annual risk of breast cancer in BRCA1 and BRCA2 is largest premenopausally, and that this may differ from what might be expected in genes that are detected in the future, which may display a risk that is approximately the same before and after menopause.
Several genes and chromosomal loci have been suggested to be the molecular genetic basis for the increased risk of breast cancer in non-BRCA1/non-BRCA2 breast cancer families. Genes such as p53 [36-38], CHK2 [39,40], PTEN [41] and CDKN2A/p16 [42] may be associated with some of the remaining high-risk families. In addition, there is some evidence that families with Peutz–Jeghers syndrome and a mutation in LKB1 [43] have an increased risk of breast cancer and ovarian tumours [44,45], even if the major clinical consequences of this syndrome are related to the gastrointestinal tract, making it more likely that such families are identified through these manifestations than as breast cancer or ovarian cancer families. The genes BARD1 [46], RAD51 [47], BACH1 [48] and ATM [49] are additional genes that have been suggested to be associated with hereditary breast cancer. However, their contribution is likely to be small. Germline mutations in the E-cadherin gene are associated with not only lobular breast cancer risk, but also with risk of diffuse stomach cancer, and they may also contribute to inherited breast cancer risk [50].
Hitherto unidentified genes have, based on linkage analysis of limited chromosomal regions or genome-wide scanning, been suggested to be located on chromosomal loci on 2q [17], 6q [18], 8p [19,20], and 13q21 [21]. In a UK study of 112 ovarian and breast cancer–ovarian cancer families, most breast cancer–ovarian cancer and ovarian cancer families were likely to be associated with BRCA1 and BRCA2 [51]. A chromosomal locus on 3p25-p22 has been linked to families with an increased prevalence of ovarian cancer in a study on Japanese ovarian cancer families [22]. These chromosomal loci may harbour genes that, after future identification and characterization, may prove useful in the investigation and counselling of individuals seeking medical advice for a suspicion of an increased hereditary risk of breast cancer.
The current study indicates that prostate cancer risk may be an associated risk on one or more future breast cancer susceptibility genes. Until these genes are molecularly and genetically characterized, we shall have to rely on epidemiological studies such as the present one for the estimation of breast cancer risk in healthy relatives of breast cancer patients without mutations in known susceptibility genes.
Conclusions
Breast cancer risk in FDRs of early-onset breast cancer cases is elevated irrespective of the BRCA mutation status. The risk appears to increase with decreasing age and increasing number of affected relatives. In relatives of breast cancer cases younger than age 36 years, an association with prostate cancer and nonmelanoma skin cancer is suggested. There is no indication of an increased risk of ovarian cancer in families without a BRCA mutation. The cumulative breast cancer risk at age 50 years is likely to be higher in FDRs of BRCA mutation carriers compared with FDRs of carriers of hitherto unidentified breast cancer susceptibility genes.
Competing interests
None declared.
Abbreviations
BRCA1 = breast cancer susceptibility gene 1; BRCA2 = breast cancer susceptibility gene 2; BRCA = both breast cancer susceptibility gene 1 and breast cancer susceptibility gene 2; CI = confidence interval; FDR = first-degree relative; NHL = non-Hodgkin's lymphoma; SIR = standardized incidence ratio.
Acknowledgments
Acknowledgements
Paul Pharoah and Antonis Antoniou are thanked for valuable discussions on penetrance analysis. Financial support for this study was provided by grants from the Swedish Cancer Society, from the Mrs Bertha Kamprad Foundation, from the Gunnar, Arvid, and Elisabeth Nilsson Foundation, from the John & Augusta Persson Foundation, from the Hospital of Lund Foundations, from the FM Bergqvist foundation, from King Gustav V's Jubilee Foundation and from the Nordic Cancer Union.
References
- Pharoah PDP, Day NE, Duffy S, Easton D, Ponder BAJ. Family history and risk of breast cancer: a systematic review and meta-analysis. Int J Cancer. 1997;71:800–809. doi: 10.1002/(sici)1097-0215(19970529)71:5<800::aid-ijc18>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Andersson H, Bladström A, Olsson H, Möller TR. Familial breast and ovarian cancer: a Swedish population-based register study. Am J Epidemiol. 2000;152:1154–1163. doi: 10.1093/aje/152.12.1154. [DOI] [PubMed] [Google Scholar]
- Claus E, Risch N, Thompson W. Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer. 1994;73:643–651. doi: 10.1002/1097-0142(19940201)73:3<643::aid-cncr2820730323>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, Mulvihill JJ. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–1886. doi: 10.1093/jnci/81.24.1879. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G, Barfoot R, Hamoudi R, Patel S, Rice C, Biggs P, Hashim Y, Smith A, Connor F, Arason A, Gudmundsson J, Ficenec D, Kelsell D, Ford D, Tonin P, Bishop DT, Spurr NK, Ponder BAJ, Eeles R, Peto J, Devilee P, Cornelisse C, Lynch H, Narod S, Lenoir G, Egilsson V, Barkadottir RB, Easton DF, Bentley DR, Futreal PA, Ashworth A, Stratton MR. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Simmard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, Merajver S, Thorlacius S, Offit K, Stoppa-Lyonnet D, Belanger C, Bell R, Berry S, Bogden R, Chen Q, Davies T, Dumont M, Frye C, Hattier T, Jammulapati S, Janecki T, Jiang P, Kehrer R, Leblanc J-F, Mitchell JT, McArthur-Morrison J, Nguyen K, Peng Y, Samson C, Schroeder M, Snyder SC, Steele L, Stringfellow M, Stroup C, Swedlund B, Swensen J, Teng D, Thomas A, Tran T, Tranchant T, Weaver-Feldhaus J, Wong AKC, Shizuya H, Eyfjord JE, Cannon-Albright L, Labrie F, Skolnick MH, Weber B, Kamb A, Goldgar DE. The complete BRCA2 gene and mutations in chomosome13q linked kindreds. Nat Genet. 1996;12:333–337. doi: 10.1038/ng0396-333. [DOI] [PubMed] [Google Scholar]
- Dörum A, Heimdal K, Hovig E, Inganäs M, Möller P. Penetrance of BRCA1 1675delA and 1135 insA with respect to breast cancer and ovarian cancer. Am J Hum Genet. 1999;65:671–679. doi: 10.1086/302530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE, Breast Cancer Linkage Consortium Risks of cancer in BRCA1-mutation carriers. Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Neuhausen SL, Steele L, Fields P, Ward JH, Tran T, Ngyuen K, Stratton MR, Easton DF. A 45-year follow-up of kindred 107 and the search for BRCA2. J Natl Cancer Inst Monogr. 1995;17:15–19. [PubMed] [Google Scholar]
- Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA. The risk of cancer associated with specific mutations of BRCA1 and BRCA2among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- Thorlacius S, Struewing JP, Hartge P, Olafsdottir GH, Sigvaldason H, Tryggvadottir L, Wacholder S, Tulinius H, Eyrfjörd JE. Population-basd study of risk of breast cancer in carriers of BRCA2 mutation. Lancet. 1998;352:1337–1339. doi: 10.1016/s0140-6736(98)03300-5. [DOI] [PubMed] [Google Scholar]
- Hopper JL, Southey MC, Dite GS, Jolley DJ, Giles GG, McCredie MR, Easton DF, Venter DJ. Population-based estimate of the average age-specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Australian Breast Cancer Family Study. Cancer Epidemiol Biomarkers Prev. 1999;8:741–747. [PubMed] [Google Scholar]
- Anglian Breast Cancer Study Group Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000;83:1301–1308. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper J, Loman N, Olsson H, Johansson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles D, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Tulinius H, Thorlacius S, Nevanlinna H, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with mutations in BRCA1 or BRCA2 detected in case series unselected for family history: a combined analysis of 22 studies. Am Hum Genet. 2003;75:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94:1365–1372. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- Huusko P, Gillanders E, Vahteristo P-S, Sarantaus L, Jou S, Kainu T, Rapakko K, Jones M, Markey C, Eerola H, Allinen M, Vehmanen P, Leisti J, Blanco G, Blomqvist C, Trent J, Bailey-Wilson J, Winqvist R, Nevanlinna H, Kallioniemi O. Genome-wide scanning for linkage in Finnish breast cancer families [abstract] Annual Meeting of the American Society of Human Genetics: San Diego. 2001. http://www.faseb.org/genetics/ashg01/f149.htm [DOI] [PubMed]
- Zuppan P, Hall JM, Lee MK, Ponglikitmongkol M, King M-C. Possible linkage of the estrogen receptor gene to breast cancer in a family with late-onset disease. Am J Hum Genet. 1991;48:1065–1068. [PMC free article] [PubMed] [Google Scholar]
- Imbert A, Chaffanet M, Essioux L, Noguchi T, Adelaide J, Kerangueven F, Pasilier DL, Bonaiti-Pellie C, Sobol H, Birnboum D, Pebusque M. Integrated map of the chromosome 8p12-p21 region, a region involved in human cancers and Werner syndrome. Genomics. 1996;32:29–32. doi: 10.1006/geno.1996.0073. [DOI] [PubMed] [Google Scholar]
- Seitz S, Rohde K, Bender E, Nothnagel A, Kolble K, Schlag P, Scherneck S. Strong indication for a breast cancer susceptibility gene on chromosome 8p12-p22: linkage analysis in German breast cancer families. Oncogene. 1997;14:741–746. doi: 10.1038/sj.onc.1200881. [DOI] [PubMed] [Google Scholar]
- Kainu T, Juo S, Desper R, Schaffer A, Gillanders E, Rozenblum E, Freas-Lutz D, Weaver D, Stephan D, Bailey-Wilson J, Kallioniemi O, Tirkkonen M, Syrjakoski K, Kuukasjarvi T, Koivisto P, Karhu R, Holli K, Arason A, Johannesdottir G, Bergthorsson J, Johannsdottir H, Egilsson V, Barkardottir R, Johannsson O, Haraldsson K, Sandberg T, Holmberg E, Gronberg H, Olsson H, Borg A, Vehmanen P, Eerola H, Heikkila P, Pyrhonen S, Nevanlinna H. Somatic deletions in hereditary breast cancers implicate 13q21 as a putative novel breast cancer susceptibility locus. Proc Natl Acad Sci USA. 2000;97:9603–9608. doi: 10.1073/pnas.97.17.9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine M, Nagata H, Tsuji S, Hirai Y, Fujimoto S, Hatae M, Kobayashi I, Fujii T, Ngata I, Ushijima K, Obata K, Suzuki M, Yoshinaga M, Umesaki H, Satoh S, Enomoto T, Motoyama S, Tanaka K. Localization of a novel susceptibility gene for familial ovarian cancer to chromosome 3p22-p25. Hum Mol Genet. 2001;10:1421–1429. doi: 10.1093/hmg/10.13.1421. [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Pharoah PDP, McMullan G, Day NE, Ponder BAJ, Easton D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet Epidemiol. 2001;21:1–18. doi: 10.1002/gepi.1014. [DOI] [PubMed] [Google Scholar]
- Loman N, Johannsson O, Kristoffersson U, Olsson H, Borg Å. Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population-based series of early-onset breast cancer. J Natl Cancer Inst. 2001;93:1215–1223. doi: 10.1093/jnci/93.16.1215. [DOI] [PubMed] [Google Scholar]
- StataCorp Stata Statistical Software, release 60 College Station, TX: Stata Corporation. 1999.
- Bratt O, Kristoffersson U, Lundgren R, Olsson H. The risk of malignant tumours in first-degree relatives of men with early onset prostate cancer: a population-based cohort study. Eur J. 1997;33:2237–2240. doi: 10.1016/s0959-8049(97)00320-1. [DOI] [PubMed] [Google Scholar]
- Kalish LA, McDougal WS, McKinlay JB. Family history and the risk of prostate cancer. Urology. 2000;56:803–806. doi: 10.1016/s0090-4295(00)00780-9. [DOI] [PubMed] [Google Scholar]
- Hayes RB, Liff JM, Pottern LM, Greenberg RS, Schoenberg JB, Schwartz AG, Swanson GM, Silverman DT, Brown LM, Hoover RN, Fraumeni JF. Prostate cancer risk in US blacks and whites with a family history of cancer. Int J Cancer. 1995;60:361–364. doi: 10.1002/ijc.2910600315. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86:1600–1608. doi: 10.1093/jnci/86.21.1600. [DOI] [PubMed] [Google Scholar]
- Tulinius H, Egilsson V, Olafsdottir GH, Sigvaldason H. Risk of prostate, ovarian and endometrial cancer among relatives of women with breast cancer. BMJ. 1992;305:855–857. doi: 10.1136/bmj.305.6858.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium Cancer risk in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- Johannsson O, Loman N, Möller T, Kristoffersson U, Borg Å, Olsson H. Incidence of malignant tumours in relatives of BRCA1 and BRCA2 germline mutation carriers. Eur J Cancer. 1999;35:1248–1257. doi: 10.1016/s0959-8049(99)00135-5. [DOI] [PubMed] [Google Scholar]
- Thompson D, Easton D, Consortium BCL. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet. 2001;68:410–419. doi: 10.1086/318181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg CB. On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst. 2002;94:1221–1226. doi: 10.1093/jnci/94.16.1221. [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Gayther SA, Stratton JF, Ponder BA, Easton DF. Risk models for familial ovarian and breast cancer. Genet Epidemiol. 2000;18:173–190. doi: 10.1002/(SICI)1098-2272(200002)18:2<173::AID-GEPI6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Börresen A-L, Andersen TI, Garber J, Barbeier-Piraux N, Thorlacius S, Eyfjörd J, Ottestad L, Smith-Sörensen B, Holvig E, Malkin D, Friend SH. Screening for germline TP53 mutations in breast cancer patients. Cancer Res. 1992;52:3234–3236. [PubMed] [Google Scholar]
- Prosser J, Porter D, Coles C, Thompson AM, Chetty U, Steel CM, Evans HJ. Constitutional p53 mutations in a non-Li–Fraumeni cancer family. Br J Cancer. 1992;65:527–528. doi: 10.1038/bjc.1992.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, Davidson N. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992;52:2984–2986. [PubMed] [Google Scholar]
- Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- Vahteristo P, Bartkova J, Eerola H, Syrjakoski K, Ojala S, Kilpivaara O, Tamminen A, Kononen J, Aittomaki K, Heikkila P, Holli K, Blomqvist C, Bartek J, Kallioniemi OP, Nevanlinna H. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet. 2002;71:432–438. doi: 10.1086/341943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald MG, Marsh DJ, Wahrer D, Bell D, Caron S, Shannon KE, Ishioka C, Isselbacher KJ, Garber JE, Eng C, Haber DA. Germline mutations in PTEN are an infrequent cause of genetic predisposition to breast cancer. Oncogene. 1998;17:727–731. doi: 10.1038/sj.onc.1201984. [DOI] [PubMed] [Google Scholar]
- Borg Å, Sandberg T, Nilsson K, Johannsson O, Klinker M, Måsbäck A, Westerdahl J, Olsson H, Ingvar C. High frequency of multiple melanomas and breast and pancreas carcinomnas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst. 2000;92:1260–1266. doi: 10.1093/jnci/92.15.1260. [DOI] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Thomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen A, Stratton M, Chapelle Adl, Aaltonen L. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Spigelman A, Murday V, Phillips R. Cancer and the Peutz–Jeghers syndrome. Gut. 1989;30:1588–1590. doi: 10.1136/gut.30.11.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick D, Prior J. Peutz–Jeghers syndrom. A clinicopathologic study of a large family with a 27-year follow-up. Cancer. 1982;50:2139–2146. doi: 10.1002/1097-0142(19821115)50:10<2139::aid-cncr2820501028>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Thai TH, Du F, Tsan JT, Jin Y, Phung A, Spillman MA, Massa HF, Müller CY, Ashfaq R, Hathis JM, Miller DS, Trask BJ, Baer R, Bowcock AM. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum Mol Genet. 1998;7:195–202. doi: 10.1093/hmg/7.2.195. [DOI] [PubMed] [Google Scholar]
- Kato M, Yano K, Matsuo F, Saito H, Katagiri T, Kurumizaka H, Yoshimoto M, Kasumi F, Akiyama F, Sakamoto G, Nagawa H, Nakamura Y, Miki Y. Identification of Rad51 alteration in patients with bilateral breast cancer. J Hum Genet. 2000;45:133–137. doi: 10.1007/s100380050199. [DOI] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- Athma P, Rappaport R, Swift M. Molecular genotypingshows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet Cytogenet. 1996;92:130–134. doi: 10.1016/s0165-4608(96)00328-7. [DOI] [PubMed] [Google Scholar]
- Keller G, Vogelsang H, Becker I, Hutter J, Ott K, Candidus S, Grundei T, Becker KF, Mueller J, Siewert JR, Hofler H. Diffuse type gastric and lobular breast carcinoma in a familial gastric cancer patient with an E-cadherin germline mutation. Am J. 1999;155:337–342. doi: 10.1016/S0002-9440(10)65129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther S, Russel P, Harrington P, Antoniu A, Easton D, Ponder B. The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: no evidence for other ovarian cancer-susceptibility genes. Am J Hum Genet. 1999;65:1021–1029. doi: 10.1086/302583. [DOI] [PMC free article] [PubMed] [Google Scholar]