

Abstract
Background
Currently, there are no specific therapies available to treat cardiac dysfunction due to sepsis and other chronic inflammatory conditions. Activation of toll-like receptor 4 (TLR4) by lipopolysaccharide (LPS) is an early event in gram-negative bacterial sepsis, triggering a robust inflammatory response and changes in metabolism. PPARγ coactivator-1 (PGC-1) α and β serve as critical physiological regulators of energy metabolic gene expression in heart.
Methods and Results
Injection of mice with LPS triggered a myocardial fuel switch similar to that of the failing heart; reduced mitochondrial substrate flux and myocyte lipid accumulation. The LPS-induced metabolic changes were associated with diminished ventricular function and suppression of the genes encoding PGC-1α and β, known transcriptional regulators of mitochondrial function. This cascade of events required TLR4 and NF-κB activation. Restoration of PGC-1β expression in cardiac myocytes in culture and in vivo in mice reversed the gene regulatory, metabolic, and functional derangements triggered by LPS. Interestingly, the effects of PGC-1β overexpression were independent of the upstream inflammatory response, highlighting the potential utility of modulating downstream metabolic derangements in cardiac myocytes as a novel strategy to prevent or treat sepsis-induced heart failure.
Conclusions
LPS triggers cardiac energy metabolic reprogramming via suppression of PGC-1 coactivators in the cardiac myocyte. Reactivation of PGC-1β expression can reverse the metabolic and functional derangements caused by LPS-TLR4 activation, identifying the PGC-1 axis as a candidate therapeutic target for sepsis-induced heart failure.
Keywords: sepsis, toll-like receptor 4, heart failure, inflammation, metabolism
Bacterial sepsis is a common clinical disorder that accounts for greater than 200,000 deaths in the United States annually.1 The septic syndrome is characterized by robust systemic inflammation and end-organ dysfunction. The heart is affected in at least 50% of patients with sepsis and its presence portends a worse prognosis.2 Alterations in myocardial metabolism are known to occur during sepsis;2,3 however, the links between inflammatory signaling and myocardial fuel and energy metabolism remain unclear. To date, attempts to modulate myocardial metabolism in the setting of sepsis have not been reported.
The heart’s high capacity ATP-generating mitochondrial system is under the control of a gene regulatory circuit.4 Members of the nuclear receptor transcription factor superfamily, including peroxisome proliferator-activated receptors (PPARs) and the estrogen-related receptors (ERRs), control the expression of many genes involved in mitochondrial energy transduction pathways including fatty acid β-oxidation (FAO), the electron transport complex, and oxidative phosphorylation.5,6 The nuclear respiratory factors [NRF-1 (Nrf1) and NRF-2 (Gabpa)] coordinately regulate transcription of nuclear and mitochondrial genes involved in electron transport and mitochondrial DNA replication.7 The expression of several of these genes has been shown to be suppressed by inflammation at the transcriptional level; however, the upstream signals and functional relevance of this response are unclear.8,9
The activity of the PPARs, ERRs, and NRFs is orchestrated by PGC-1α and β (Ppargc1a and b), inducible, cardiac-enriched transcriptional coactivators.10,11 Targeted disruption of the PGC-1α and PGC-1β genes in mice causes mitochondrial dysfunction and heart failure, evidence that deactivation of this pathway can lead to myocyte contractile dysfunction.12,13 The PGC-1 gene regulatory circuit is also deactivated in heart failure through mechanisms that have not been defined.14 Inflammatory signaling has been reported to both augment or suppress the expression or activity of the PGC-1 coactivators.8,15-17 The interpretation of these studies is further confounded by the lack of corroborative functional and metabolic data. Thus, questions remain regarding the regulation and physiologic impact of the PGC-1 coactivators during times of inflammatory stress.
The inflammatory response and left ventricular dysfunction induced by LPS is mediated, in large part, by the host molecule toll-like receptor 4 (TLR4).18 Interestingly, TLR4 signaling is also important in other cardiovascular diseases including ischemia-reperfusion injury and cardiac hypertrophy.19-21 Activation of TLR4 leads to the nuclear translocation of NF-κB, which drives the expression of inflammatory cytokines.22 We hypothesized that TLR4-mediated inflammation could serve as the upstream signal that modulates myocardial energy metabolism in sepsis. Our data demonstrated that the acute administration of LPS recapitulated aspects of the metabolic reprogramming known to occur in the failing heart, including downregulation of PGC-1α and β gene expression leading to reduced mitochondrial substrate flux and lipid accumulation. The suppression of PGC-1 gene expression was shown to occur through a TLR4- and NF-κB-dependent mechanism. Restoration of PGC-1 coactivator activity in cardiac myocytes during acute inflammation reversed LPS-induced alterations in cardiac fuel metabolism in vitro and in vivo.
Methods
Animal Experiments and Generation
Adult (8-12 weeks) male C57BL/6 mice were obtained from Jackson Laboratory. TLR4 and TLR2 knockout mice (C57BL/6 background) were purchased from Oriental BioService, Inc (Japan). Adult mice (8-16 weeks of age) were used for experiments. To generate the TRE-PGC-1β line, Ppargc1b cDNA was cloned into the pTRE-2 vector (Clontech), linearized with AatII and AseI, and injected into fertilized mouse eggs. Positive founders were identified by transgene-specific PCR using the following primers: Forward 5’-TCCGTGAGTTACCATTCTGAT-3’, Reverse 5’-GAGGAGACAATGGTTGTCAAC-3’. The TRE-PGC-1β mice were crossed to the MHC-rtTA23 line to produce TRE-PGC-1βMHC-rtTA mice. TRE-PGC-1βMHC-rtTA mice (FVB background) were fed chow containing doxycycline (Research Diets C11300-200i) adlib for 2 days. Mice (10-16 weeks of age) were given intraperitoneal injections of 150 μg (FVB) or 200 μg (C57BL/6) of E.coli 0111:B4 LPS (Sigma) resuspended in PBS or an equal volume of PBS alone. Injections were performed in the morning and food was removed from all animals immediately after the injection to control for the anorexia induced by LPS.
All animal experiments were conducted in strict accordance with NIH guidelines for humane treatment of animals and were reviewed by the Animal Studies Committee of Washington University School of Medicine.
Statistical analysis was performed using GraphPad Prism 5. Two-way ANOVA with Bonferroni post-test analysis or repeated measures ANOVA with Bonferroni post-testing was used. Student’s unpaired t-test using Microsoft Excel was used for simple comparisons. See Figure Legends for more detail.
An expanded materials and methods, including all procedures used in this manuscript, is available online.
Results
LPS stimulation suppresses cardiac mitochondrial fatty acid oxidation leading to myocyte lipid accumulation
To assess the impact of inflammation on myocardial fuel metabolism, a sub-lethal dose of LPS was administered to adult male C57BL/6 mice. LPS stimulation led to rapid induction of myocardial cytokine gene expression, including TNFα, followed by an increase in the levels of mRNAs encoding macrophage chemoattractant protein 1[MCP1 (Ccl2)], interleukin 6 (IL-6), and nitric oxide synthase 2 (NOS2 or iNOS; Figure 1a). Left ventricular fractional shortening was significantly decreased 6h after LPS injection with recovery by 24h, consistent with observed transient myocardial inflammatory response (Figure 1b).
Figure 1. LPS induces myocardial inflammation, transient LV dysfunction, and reduces myocardial FAO.
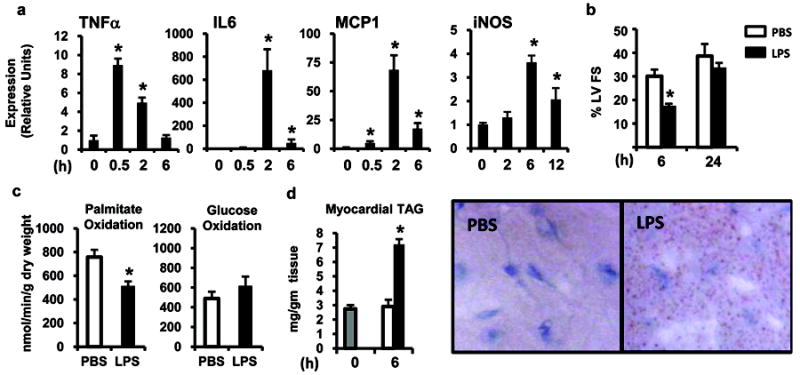
(a) C57BL/6 mice were injected with LPS (black bars) and gene expression of inflammatory cytokines in the myocardium was quantified by qRT-PCR at the indicated time points. All values were normalized to 36B4. For each timepoint, graph shows mean ± SE, n=4/group; * p≤ 0.05 compared to time 0. (b) C57BL/6 mice were injected with LPS (black bars) or PBS (white bars) and 2-D echocardiography was performed to determine left ventricular fractional shortening (%LVFS) at the indicated time points. For each timepoint, graph shows mean ± SE, n=4/group. (c) Myocardial palmitate and glucose oxidization rates were determined using an isolated working heart system 12h after the injection of LPS or PBS (n=6-8 mice per group). (d) Myocardial triglyceride (TAG) accumulation was assessed by direct quantification (left) and oil Red O staining (right) at baseline (gray bar) and 6h after LPS or PBS injection. For each timepoint, graph shows mean ± SE, n=4/group; * p≤ 0.05 for LPS vs. PBS injected (one-way ANOVA (a), repeated measures ANOVA (b), and Student’s t-test (c,d)).
Fuel oxidation rates were assessed in working hearts isolated from mice 12h after injection of LPS or vehicle. LPS treatment resulted in significantly lower myocardial palmitate oxidation rates compared to vehicle-injected mice (Figure 1c). Glucose oxidation rates were low in both groups, likely related to the fasting state of the animals which increases expression of pyruvate dehydrogenase kinase 4, an enzyme that suppresses glucose oxidation (Figure 1c). Serum free fatty acid and triglyceride levels were increased 6h following LPS injection compared to controls (1.17 ± 0.06mg/dl vs. 1.58 ± 0.07mg/dl, p ≤ 0.05 and 115 ± 6.8mg/dl vs. 148 ± 8.7mg/dl, p ≤ 0.05, respectively). Myocardial triglyceride levels were elevated after LPS injection as evidenced by oil red O staining of neutral lipids in cardiac myocytes and direct quantification (Figure 1d).
LPS-triggered activation of inflammatory pathways in the cardiac myocyte results in suppression of the PGC-1 gene regulatory cascade
Given the regulatory role of PGC-1 coactivators in maintaining a high capacity mitochondrial system, we next assessed the effect of LPS on the myocardial expression of the PGC-1α and β genes. LPS injection led to a rapid downregulation of PGC-1α and β mRNA levels, with PGC-1β being the most dramatically affected (reduced >90%; Figure 2a). PGC-1α protein levels were also significantly diminished (Figure 2a).
Figure 2. LPS downregulates PGC-1α and β and their FAO and mitochondrial gene targets.
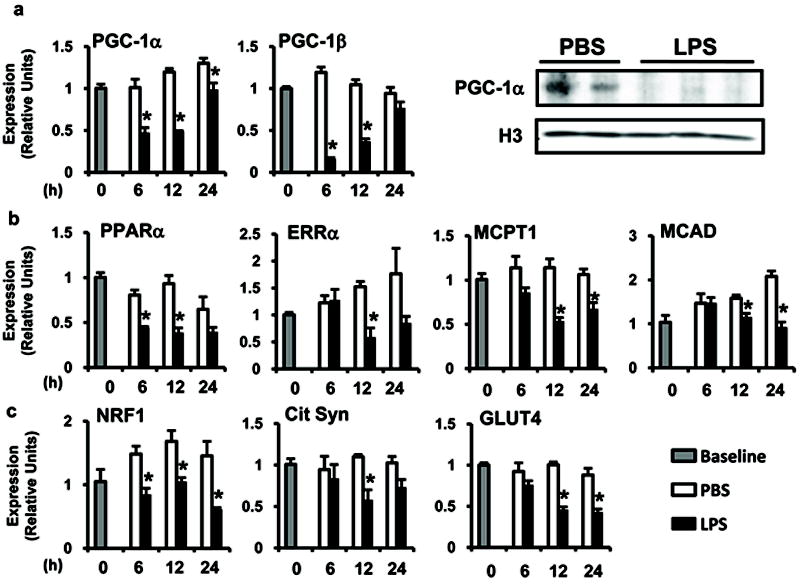
C57BL/6 mice were injected with LPS (black bars) or PBS (white bars) and mRNA or protein expression of (a) PGC-1α and β nuclear receptor coactivators, (b) FAO gene targets, and (c) mitochondrial and glucose import gene targets was quantified using qRT-PCR or Western blotting (12h) at the indicated time points. Gray bars represent baseline animals (PBS injection, time 0). All results are normalized to 36B4. For all groups, n=4 and values represent mean ± SE; * p≤ 0.05 for LPS vs. PBS (two-way ANOVA).
PGC-1 coactivators modulate the expression of metabolic transcription factors [PPARα, ERRα, NRF1 (Nrf1), NRF2 (Gabpa)] and their downstream gene targets involved in FAO [MCPT-1 (Cpt1b), MCAD (Acadm), LCAD (Acadl), VLCAD (Acadvl)] and mitochondrial function [COX4 (Cox4i1), ATP Synthase β (Atp5b)] (Figure 2b,c and Supplemental Figure 1a). Similar to the effects on PGC-1 coactivators, the expression of all of these metabolic genes was reduced at the mRNA level following LPS injection. In addition, expression of the PGC-1 target gene encoding the glucose transporter, GLUT4 (Slc2a4), was also decreased by LPS instillation. Other genes involved in the regulation of FAO such as PPARβ/δ and the myocardial FA transporter, CD36 (Cd36), were not differentially expressed in the inflamed myocardium (Supplemental Figure 1a). These gene expression results are similar to those observed in the hearts of PGC-1α and β double knockout mice,13 supporting the concept that LPS-mediated inflammation leads to rapid deactivation of the PGC-1 metabolic gene regulatory circuit.
To assess whether the observed effects of LPS occur in cardiac myocytes independent of systemic alterations such as ventricular dysfunction, LPS stimulation experiments were conducted with primary neonatal rat cardiac myocytes in culture. The effects of LPS on metabolic gene expression paralleled that seen in the heart in vivo, including reduced expression of genes encoding PGC-1α and β, PPARα, ERRα, FAO enzymes, and GLUT4 (Figure 3a,b,c). To determine whether the LPS-mediated suppression of PGC-1α gene expression involved transcriptional mechanisms, a mouse PGC-1α promoter luciferase reporter (PGC.Luc.2112) was used for transient transfection studies in cardiac myocytes. LPS reduced the activity of PGC.Luc.2112 by approximately 50%, but activated an NF-κB responsive control reporter (Figure 3d).
Figure 3. LPS induces inflammatory cytokines and leads to the downregulation of FAO gene expression in cardiac myocytes.
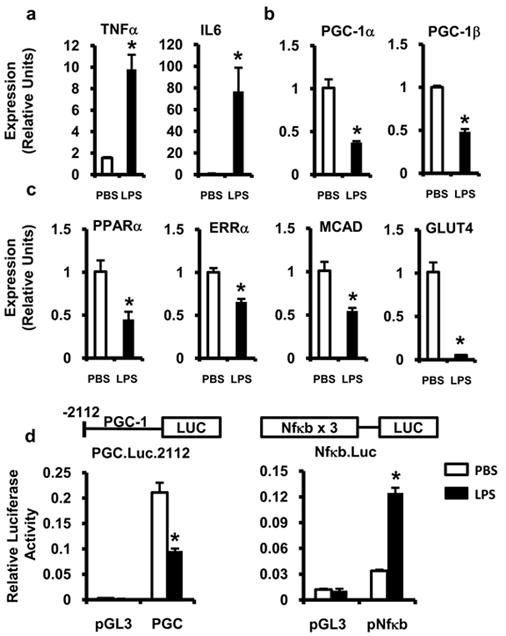
Neonatal rat cardiac myocytes were stimulated with LPS (black bars) or PBS (white bars) for 18h after which gene expression of inflammatory cytokines (a), PGC-1α and β (b), and PGC-1-target metabolic genes (c) were determined by qRT-PCR. All samples were normalized to 36B4. (d) Cardiac myocytes were transfected with a PGC-1α promoter luciferase construct (PGC), NF-κB reporter construct (pNF-κB), or empty vector (pGL3) followed by treatment with LPS (black bars) or PBS (white bars). Relative luciferase activity was determined 18h after stimulation and normalized to SV40 renilla luciferase expression. Experiments were performed in triplicate and reported values represent mean ± SE; * p≤ 0.05 for LPS vs. PBS (Student’s t-test (a-c), two-way ANOVA (d)).
AMP-activated protein kinase (AMPK) is an energy-sensing kinase known to stimulate expression of the PGC-1α gene.24,25 To determine the effects of LPS on AMPK, the level of AMPK phosphorylation was determined following LPS stimulation. Phosphorylated AMPK was increased by LPS treatment arguing that AMPK is not upstream of the observed metabolic changes (Supplemental Figure 1b).
TLR4 and NF-κB are required for LPS-mediated suppression of PGC-1α and β gene expression
To address whether the changes in PGC-1 coactivator expression were dependent on functional TLR4, LPS challenge experiments were conducted with wild-type, TLR4-/-, and TLR2-/- mice followed by assessment of gene expression and myocardial triglyceride levels at 6h. The absence of TLR4 abolished LPS-induced downregulation of PGC-1α and significantly blunted the downregulation of PGC-1β (Figure 4a). The increase in serum and myocardial lipids was also attenuated in mice lacking TLR4 (Figure 4a and data not shown). In contrast, TLR2-/- mice behaved identically to wild-type animals. Similar results were obtained in cell culture using a specific inhibitor of TLR4 signaling, CLI9526 (Figure 4b). Thus, TLR4 is an obligate component of the inflammatory and metabolic events induced by LPS in the myocardium.
Figure 4. The cardiometabolic effects of LPS are mediated through TLR4 and NF-κB.
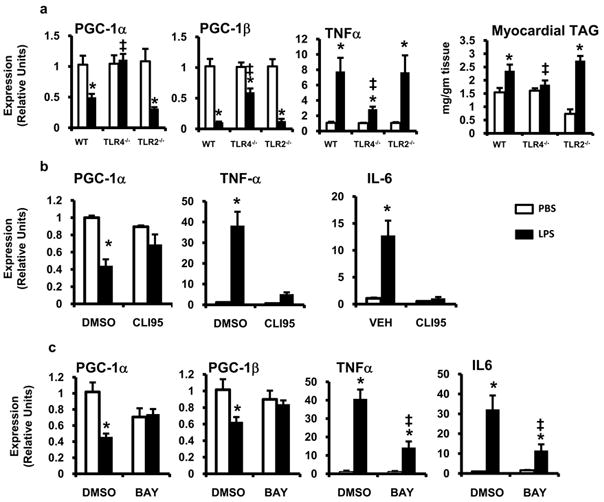
(a) Wild-type (WT) , TLR4-/- , and TLR2-/- mice were treated with LPS (black bars) or PBS (white bars) for 6h and myocardial gene expression of PGC-1α, PGC-1β, and TNF-α was assessed by qRT-PCR. All results were normalized to 36B4. Myocardial triglyceride (TAG) was quantified 6h after LPS/PBS treatment. Myocytes were pretreated with 10 μMol/L of CLI95 (b), 6.25 μMol/L BAY 11-7085 (c), or DMSO after which the cells were stimulated with LPS (black bars) or PBS (white bars) for 18h and mRNA expression was quantified via qRT-PCR for the genes denoted. All samples were run in quadruplicate; * p≤ 0.05 for LPS vs. PBS, ‡ p≤ 0.05 for TLR4-/- or inhibitor vs. wild-type or DMSO (two-way ANOVA).
NF-κB is a key downstream target of TLR4 signaling. To determine the importance of NF-κB activation in this system, cardiac myocytes were pre-treated with a chemical inhibitor of I-κ Kinase, Bay 11-7085. Compared to DMSO treated controls, Bay 11-7085 completely blocked the suppression of PGC-1α and β expression following LPS-stimulation (Figure 4c). As expected, Bay 11-7085 also decreased the expression of the NF-κB regulated cytokines, TNFα, and IL-6 (Figure 4c). TNFα, IL-6, and iNOS are induced by LPS in a TLR4 and NF-κB dependent manner and have all been implicated in sepsis-induced cardio-depression.27,28 To determine the role of TNFα and IL-6 in the suppression of PGC-1α, we used inhibitory antibodies to block their biologic activity. Neither inhibition of TNFα or IL-6 prevented the downregulation of PGC-1α by LPS (Supplemental Figure 2a,b). Cardiac myocytes were also pre-treated with the iNOS inhibitor L-NIL which, despite preventing LPS-induced nitric oxide production, had no affect on PGC-1α gene expression (Supplemental Figure 2c). Together this data argues that TLR4 and NF-κB are required for the suppression of PGC-1α expression by LPS; however, this effect is largely independent of TNF, IL-6, and nitric oxide.
Cardiac-specific induction of PGC-1β reverses LPS-triggered suppression of myocardial fatty acid oxidation and ventricular dysfunction
To further explore the role of PGC-1 coactivators in the cardiometabolic effects of LPS, we assessed the impact of re-activating PGC-1β expression in cardiac myocytes during TLR4-mediated inflammatory stress. To force PGC-1β expression in LPS-treated cardiac myocytes, the cells were infected with an adenovirus expressing PGC-1β and green fluorescent protein (GFP) (Ad-PGC-1β) or GFP alone (Ad-GFP) for 24h prior to LPS treatment. Ad-PGC-1β-infected cardiac myocytes were resistant to LPS-mediated suppression of PPARα, ERRα, and FAO gene expression (Figure 5). Importantly, LPS-mediated suppression of PGC-1α expression was preserved in Ad-PGC-1β-infected cells, indicating that the upstream events involved in suppressing PGC-1 gene expression were unaffected by PGC-1β overexpression. Similarly, inflammatory cytokine expression was not attenuated; rather it was augmented in Ad-PGC-1β-infected cells.
Figure 5. LPS-induced downregulation of FAO and glucose metabolic genes is rescued by PGC-1β expression in vitro.
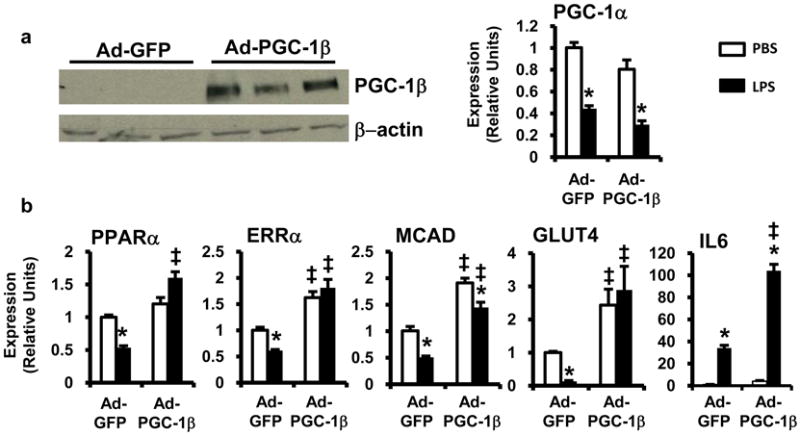
Cardiac myocytes were infected with adenovirus expressing GFP (Ad-GFP) or PGC-1β + GFP (Ad-PGC-1β) and stimulated with LPS (black bars) or PBS (white bars). (a) (Left) Western blot demonstrating overexpression of PGC-1β by Ad-PGC-1β. (Right) mRNA expression of PGC-1α determined by qRT-PCR. (b) mRNA expression of downstream metabolic gene targets was determined by qRT-PCR after treatment with LPS or PBS and infected with adenovirus as defined. Results were normalized to 36B4; n=4 per group. Bars represent mean ± SE; * p <0.05 PBS vs. LPS, ‡ p <0.05 Ad-GFP vs. Ad-PGC-1β two-way ANOVA .
To extend the rescue studies to an in vivo system, we employed a doxycycline inducible, dual transgenic, mouse system to force expression of PGC-1β specifically in cardiac myocytes (TRE-PGC-1βMHCrtTA mice23). For these experiments, non-transgenic and TRE-PGC-1βMHCrtTA mice were given doxycycline-containing chow 48h prior to PBS or LPS injection. In hearts of non-transgenic mice, LPS led to profound downregulation of PGC-1β mRNA levels. As expected, myocardial expression of PGC-1β in TRE-PGC-1βMHCrtTA mice was induced ~6-7 fold and the expression level was unaffected by LPS. Myocardial PGC-1α gene expression was equally suppressed by LPS treatment in both non-transgenic and TRE-PGC-1βMHCrtTA mice (Figure 6). Interestingly, the suppression of PGC-1 gene targets by LPS was largely reversed by induction of PGC-1β (Figure 6). Notably, LPS-mediated induction of inflammatory gene expression (TNFα, iNOS, IL-6) occurred to comparable or greater levels in TRE-PGC-1βMHCrtTA mice relative to non-transgenic animals (Supplemental Figure 3). These results provide further evidence that downregulation of the PGC-1 circuit is a critical metabolic event downstream of TLR4-mediated inflammation.
Figure 6. LPS-induced downregulation of FAO and glucose metabolic genes is rescued by PGC-1β expression in vivo.
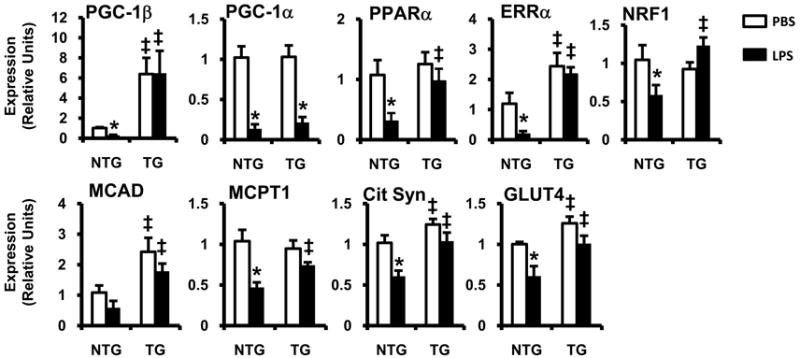
Male and female TRE-PGC-1βMHCrtTA transgenic (TG) or non-transgenic (NTG) littermate controls were fed doxycycline for 48h after which they were stimulated with LPS (black bars) or PBS (white bars). Myocardial mRNA was isolated 6h (PGC-1α/β) or 12h (all others) after LPS injection and gene expression was assessed by qRT-PCR. All results are normalized to 36B4; n=5-8 per group, bars represent mean ± SE; * p <0.05 PBS vs. LPS, ‡ p <0.05 NTG vs. TG (two-way ANOVA).
To assess the metabolic consequences of PGC-1β induction in the setting of LPS stimulation, myocardial palmitate oxidation rates were determined in hearts isolated from non-transgenic and TRE-PGC-1βMHCrtTA mice treated with LPS or PBS. In non-transgenic mice, we observed the expected decline in FAO rates with LPS treatment. In contrast, TRE-PGC-1βMHCrtTA animals were protected from this effect (Figure 7a). The LPS-mediated increase in myocardial triglyceride accumulation was also reduced by ~50% in TRE-PGC-1βMHCrtTA mice. Importantly, serum lipid levels were similar in non-transgenic and TRE-PGC-1βMHCrtTA mice, demonstrating that the reduction in myocardial triglyceride is not a consequence of altered lipid delivery to the heart (Figure 7b).
Figure 7. PGC-1β expression in cardiac myocytes reverses the suppression of FAO induced by LPS.
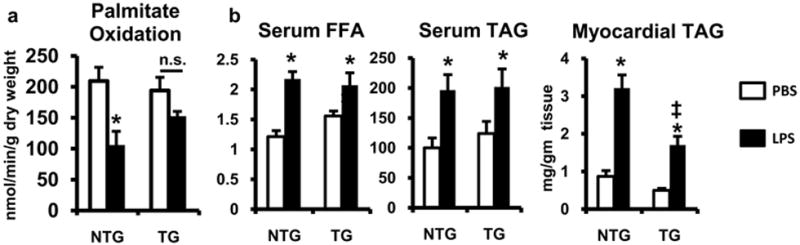
(a) Palmitate oxidation rates were determined by isolated working heart analysis performed on doxycycline-treated TRE-PGC-1βMHCrtTA (TG) or non-transgenic (NTG) animals 12h after treatment with LPS (white bars) or PBS (white bars); n=4-5 mice per group. (b) Plasma and myocardial free fatty acid (FFA) and triglyceride (TAG) levels were quantified 6h after the injection of LPS or PBS in mice; n=4-6 mice per group; * p <0.05 PBS vs. LPS, ‡ p <0.05 NTG vs. TG (two-way ANOVA).
To investigate whether cardiac myocyte expression of PGC-1β modulates cardiac contractile function during LPS-induced inflammatory stress, echocardiography was performed on non-transgenic and TRE-PGC-1βMHCrtTA mice after LPS injection. PGC-1β overexpression by itself did not produce structural or functional cardiac abnormalities as illustrated in the PBS controls (Table, Figure 8a,b). Non-transgenic mice (FVB) injected with LPS had a significant decline in cardiac function as evidenced by a reduction in left ventricular fractional shortening (52.6 ± 0.6% to 24.0±1.3%). These results were similar to those seen with C57BL/6 mice (Figure 1b), except the baseline anesthesia effects on cardiac function are more pronounced in the C57BL/6 strain. In striking contrast, TRE-PGC-1βMHCrtTA mice exhibited only a modest decrease in cardiac function compared to PBS injected littermates (50.8±1.2% vs. 37.9±0.9%; Table, Figure 8a,b).
Table.
Echocardiographic measurements in NTG and TRE-PGC-1βMHCrtTA mice at baseline and with LPS
| NTG (PBS) | NTG (LPS) | PGC-1β (PBS) | PGC-1β (LPS) | |
|---|---|---|---|---|
| HR, beats/min | 393±17 | 463±11* | 385±23 | 468±9* |
| LVEDD (mm) | 3.57±0.14 | 3.65±0.09 | 3.65±0.09 | 3.52±0.06 |
| LVESD (mm) | 1.69±0.06 | 2.78±0.10* | 1.80±0.07 | 2.19±0.06*‡ |
| FS (%) | 52.5±0.6 | 24.0±1.4* | 50.8±1.2 | 37.9±0.9*‡ |
| LPWT (mm) | 0.91±0.03 | 0.96±0.02 | 0.94±0.03 | 0.94±0.03 |
NTG (non-transgenic); HR (heart rate); LVEDD (left ventricular end-diastolic diameter); LVESD (left ventricular end-systolic diameter); FS (fractional shortening); LPWT (left ventricular posterior wall thickness); PGC-1β (TRE-PGC-1βMHCrtTA mice);
p <0.05 PBS vs. LPS,
p <0.05 NTG vs. PGC-1β via two-way ANOVA analysis.
Figure 8. PGC-1β expression in cardiac myocytes attenuates the myocardial dysfunction and ROS induced by LPS.
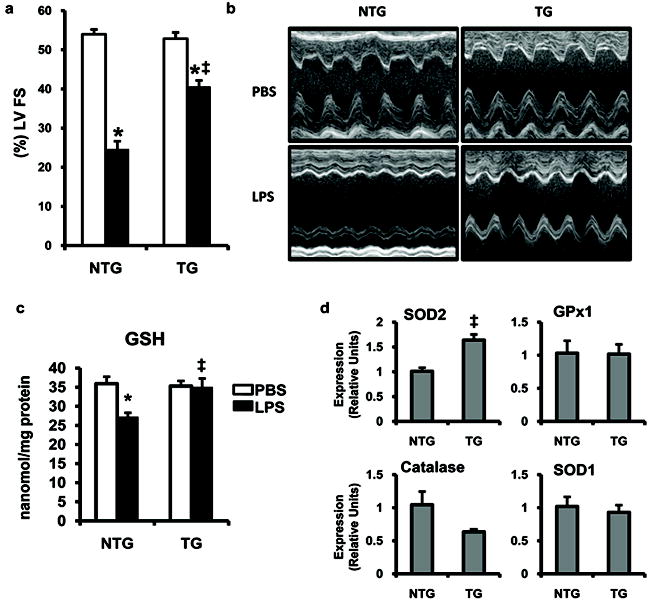
2D echocardiograms were performed 6h after LPS (black bars) or PBS (white bars) injection in TRE-PGC-1βMHCrtTA (TG) or non-transgenic (NTG) mice. Percent left ventricular fractional shortening (%LV FS) (a) and M-mode echocardiographic images (b) are shown; n=7-15 per group. (c) Myocardial tissue was harvested 6h after the injection of LPS or PBS in mice. Bars represent GSH levels quantified as a measure of oxidative stress (n=5-6 mice per group). (d) qRT-PCR assessment of ROS scavenging enzymes from NTG and TG mice after 48h of PGC-1β induction with doxycycline (n=3-6 mice per group). * p <0.05 PBS vs. LPS, ‡ p <0.05 NTG vs. TG (two-way ANOVA).
In addition to enhancing mitochondrial function and metabolism, PGC-1 coactivators can induce the expression of reactive oxygen species (ROS) scavengers.29 The generation of ROS in the myocardium occurs rapidly after LPS administration and is linked to myocardial dysfunction.15,30,31 To assess the effects of PGC-1β overexpression on myocardial oxidative stress after LPS injection, we determined tissue levels of reduced glutathione (GSH) in non-transgenic and TRE-PGC-1βMHCrtTA mice after PBS or LPS injection. As shown in Figure 8c, LPS caused a drop in myocardial GSH levels 6h after injection, consistent with increased levels of oxidative stress. In contrast, LPS did not induce a decrease in myocardial GSH levels in transgenic animals. The mRNA expression of ROS scavengers known to be influenced by PGC-1 revealed that the mitochondrial enzyme superoxide dismutase (SOD)2 was upregulated in TRE-PGC-1βMHCrtTA mice (Figure 8d). This data suggests that, in addition to their metabolic effects, the PGC-1 coactivators likely influence oxidative stress levels in the myocardium.
Discussion
Sepsis is a common clinical disease characterized by widespread inflammation and end-organ dysfunction. The heart is frequently involved in this syndrome and contributes to the likelihood of mortality. At present, there are no effective therapies to improve myocardial function during sepsis. TLR4 plays a key role in initiating the inflammatory response during gram-negative bacteremia by stimulating the production of NF-κB-dependent cytokines. Attempts to treat sepsis by preventing upstream activation of TLR4 and/or cytokine signaling have been largely unsuccessful in patients.32 Therefore, identification of potential targets downstream of the inflammatory response is of significant interest. Sepsis is known to diminish oxidative metabolism in the heart and other tissues,2 suggesting that targeting inflammatory-metabolic “crosstalk” could improve outcome in this syndrome. Herein, we describe a cascade of events that links TLR4 activation to myocardial metabolism through downregulation of PGC-1 coactivators.
Previous studies have reported variable effects of LPS on cardiac fuel and energy metabolism.33,34 Using an isolated working heart system, we demonstrated that LPS led to a consistent decrease in palmitate oxidation rates compared to controls. Together with an increase in circulating lipids, the reduction in myocardial capacity for fat oxidation promotes the development of myocardial steatosis, a pathologic finding also observed in the hearts of humans with sepsis.4-6,35 Consistent with these findings, TLR4 was recently shown to mediate a similar fuel utilization shift in human primary skeletal muscle cells in culture.36
The presence of increased myocardial lipids normally triggers a compensatory increase in the transcriptional activity of PPARα and ERRα, and the expression of their coactivators PGC-1α and β leading to an increase in capacity for FAO.7 However, in our system LPS diminished myocardial FAO rates despite an elevation in plasma lipids, suggesting a direct effect of inflammation on the machinery that regulates FAO enzyme expression. To determine the mechanism by which LPS alters myocardial metabolism, we initially focused our investigation on PGC-1α and β given their ability to “boost” the activity of several transcription factors that regulate mitochondrial FAO. Previous studies have demonstrated both up- and down-regulation of PGC-1 coactivator expression during inflammation, which may be related to the kinetics of the PGC-1 response and/or the model of systemic inflammation.8,16,37 The data presented herein provides strong evidence that the PGC-1 axis is deactivated in the cardiac myocyte early after LPS stimulation. Specifically, the expression of PGC-1α and β was reduced by LPS in cardiac myocytes in vivo and in culture. Furthermore, LPS decreased the expression of known PGC-1 gene targets with a pattern similar to that of PGC-1-deficient hearts.13 Interestingly, this gene expression profile is reminiscent of that seen in other forms of heart failure. Importantly, activation of TLR4 in cardiac myocytes, where the systemic effects of LPS are removed, still triggered the downregulation of PGC-1α/β and their target genes, thereby confirming the direct relationship between these inflammatory signaling-metabolic events.
The suppression of PGC-1α gene expression by LPS was found to occur, at least in part, at the transcriptional level. Moreover, blocking the TLR4:NFκB axis prevented the downregulation of PGC-1 coactivator expression by LPS. We excluded AMPK, a known activator of PGC-1α,10 as well as TNFα, IL-6, and nitric oxide as mediators of LPS-induced suppression of PGC-1 gene expression. Whether NF-κB directly mediates the suppression of PGC-1 gene transcription, as has been recently proposed,38 or acts indirectly through the production of another molecule is an area of active investigation.
The conflicting data surrounding the effects of sepsis and LPS on the expression and activity of PGC-1 coactivators has been confounded, in part, by the lack of corroborative functional and physiological studies. To determine whether the suppression of myocardial FAO triggered by LPS:TLR4 signaling requires deactivation of the PGC-1 axis, we utilized PGC-1β gain-of-function strategies. Forced PGC-1β expression in cardiac myocytes in culture and in vivo reversed the suppression of FAO/mitochondrial gene expression induced by LPS. Importantly, reactivation of PGC-1β did not alter the induction of upstream inflammatory mediators or the downregulation of the related coactivator, PGC-1α, following LPS challenge. To validate the physiologic relevance of these gene expression changes, we demonstrated that forced PGC-1β expression protected mice from LPS-induced suppression of mitochondrial FAO. Consistent with this finding, myocyte triglyceride accumulation was significantly decreased after LPS stimulation in TRE-PGC-1βMHCrtTA compared to non-transgenic mice despite similar increases in the levels of circulating lipids. That the LPS-mediated downregulation of PGC-1α is still observed in TRE-PGC-1βMHCrtTA mice argues that the rescue of metabolism by PGC-1β is not a secondary consequence of disrupting upstream cytokine signaling or improving cardiac function. Although the use of a non-physiological overexpression system to deliver PGC-1β is a limitation of this model, it serves as an important proof-of-concept experiment for future efforts towards targeting this gene regulatory circuit as a therapeutic strategy.
Myocardial dysfunction is a common and important finding in patients with sepsis, yet there are currently no therapies directed towards improving cardiac function in this syndrome.39 In this study, we demonstrate that cardiac myocyte-specific PGC-1β overexpression leads to significantly improved cardiac function following LPS treatment. The mechanistic basis for this finding is likely multifactorial, but may relate to the normalization of metabolic derangements and/or a reduction in oxidative stress levels. Previous studies have demonstrated that LPS injection rapidly triggers ROS generation in the myocardium15,30 and PGC-1 coactivators regulate the expression of several ROS scavenging enzymes.29,40 Consistent with this, we observed an increase in ROS with LPS that was reversed by PGC-1β overexpression. Gene expression analysis revealed that of the PGC-1 ROS scavenger targets, only SOD2 was significantly modulated with PGC-1β overexpression. Thus, PGC-1 appears to influence oxidative stress generation in addition to modulating metabolism in response to inflammatory stress. These findings, combined with evidence that “recovery” of PGC-1 expression in human patients with sepsis is predictive of survival, suggest that strategies to target PGC-1 expression/activity in cardiac myocytes may prove to be a novel therapeutic approach for the prevention and treatment of sepsis-induced cardiac dysfunction.41 Moreover, the suppression of PGC-1 expression by inflammatory signals may be a common theme in more diverse forms of heart failure.
Supplementary Material
Clinical Summary.
Sepsis syndrome is a significant cause of morbidity and mortality worldwide. Myocardial dysfunction is a common feature of this condition and is a predictor of worse clinical outcome. At present there are no specific therapeutic targets for the cardiodepressant effects of systemic inflammation. Attempts to disrupt the early inflammatory phase of sepsis have been unsuccessful at improving outcomes. Therefore, targeting pathologic pathways downstream of the inflammatory cascade, such as cellular metabolic derangements, has the potential to be a more fruitful approach. Previous studies have suggested that organ failure in sepsis may be related to dysfunctional mitochondrial metabolism, leading to a reduction in energy production and an increase in reactive oxygen species generation. Herein we demonstrate that systemic inflammation activated by bacterial LPS reduces oxidative metabolism in the heart. This metabolic shift is associated with deactivation of the PPAR gamma coactivator-1 (PGC-1) transcriptional circuit, a key regulator of mitochondrial function. Using both cell culture and in vivo approaches we demonstrate that reactivation of PGC-1 during acute inflammation restores the expression of its downstream gene targets, reverses the decline in fatty acid oxidation, reduces oxidative stress, and improves cardiac function. These data suggest that the PGC-1 axis represents a novel therapeutic target for sepsis-induced cardiac dysfunction.
Acknowledgments
We thank Genevieve DeMaria for help with manuscript preparation.
Sources of Funding This work was supported by Washington University’s Clinical Nutrition Research Unit (P30 DK56341) and Digestive Disease Research Core Center (DK052574), and by NIH grants HL077113, HL058493, and K08 HL098373 (JDS).
Footnotes
Disclosures Dr. Kelly has received consulting fees for his advisory board relationships with Eli Lilly and Company, and Johnson & Johnson.
References
- 1.Maeder M, Fehr T, Rickli H, Ammann P. Sepsis-associated myocardial dysfunction: Diagnostic and prognostic impact of cardiac troponins and natriuretic peptides. Chest. 2006;129:1349–1366. doi: 10.1378/chest.129.5.1349. [DOI] [PubMed] [Google Scholar]
- 2.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Evans RD. Effect of endotoxin and platelet-activating factor on lipid oxidation in the rat heart. J Mol Cell Cardiol. 1997;29:1915–1926. doi: 10.1006/jmcc.1997.0430. [DOI] [PubMed] [Google Scholar]
- 4.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 5.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 6.Madrazo JA, Kelly DP. The PPAR trio: Regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968–975. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 8.Feingold K, Kim MS, Shigenaga J, Moser A, Grunfeld C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;286:E201–207. doi: 10.1152/ajpendo.00205.2003. [DOI] [PubMed] [Google Scholar]
- 9.Feingold KR, Wang Y, Moser A, Shigenaga JK, Grunfeld C. LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J Lipid Res. 2008;49:2179–2187. doi: 10.1194/jlr.M800233-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 11.Finck BN, Kelly DP. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehman JJ, Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail Rev. 2002;7:175–185. doi: 10.1023/a:1015332726303. [DOI] [PubMed] [Google Scholar]
- 15.Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 2004;64:279–288. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 map kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 17.Planavila A, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Atorvastatin prevents peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) downregulation in lipopolysaccharide-stimulated H9C2 cells. Biochim Biophys Acta. 2005;1736:120–127. doi: 10.1016/j.bbalip.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Nemoto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA, Mann DL. Escherichia coli LPS-induced LV dysfunction: Role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–2323. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 19.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 20.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G, de Kleijn DP. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 21.Ha T, Li Y, Hua F, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, William Browder I, Kao RL, Li C. Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovasc Res. 2005;68:224–234. doi: 10.1016/j.cardiores.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Akira S. Tlr signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 24.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, Lee KU, Park JY. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 27.Tavener SA, Kubes P. Cellular and molecular mechanisms underlying LPS-associated myocyte impairment. Am J Physiol Heart Circ Physiol. 2006;290:H800–806. doi: 10.1152/ajpheart.00701.2005. [DOI] [PubMed] [Google Scholar]
- 28.Ullrich R, Scherrer-Crosbie M, Bloch KD, Ichinose F, Nakajima H, Picard MH, Zapol WM, Quezado ZM. Congenital deficiency of nitric oxide synthase 2 protects against endotoxin-induced myocardial dysfunction in mice. Circulation. 2000;102:1440–1446. doi: 10.1161/01.cir.102.12.1440. [DOI] [PubMed] [Google Scholar]
- 29.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 30.Ceylan-Isik AF, Zhao P, Zhang B, Xiao X, Su G, Ren J. Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. J Mol Cell Cardiol. 2010;48:367–378. doi: 10.1016/j.yjmcc.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamion F, Bauer F, Richard V, Laude K, Renet S, Slama M, Thuillez C. Myocardial dysfunction in early state of endotoxemia role of heme-oxygenase-1. J Surg Res. 2010;158:94–103. doi: 10.1016/j.jss.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Marshall JC. Such stuff as dreams are made on: Mediator-directed therapy in sepsis. Nat Rev Drug Discov. 2003;2:391–405. doi: 10.1038/nrd1084. [DOI] [PubMed] [Google Scholar]
- 33.Liu MS, Spitzer JJ. Myocardial fatty acid and lactate metabolism after E. Coli endotoxin administration. Circ Shock. 1977;4:191–200. [PubMed] [Google Scholar]
- 34.Tappy L, Chiolero R. Substrate utilization in sepsis and multiple organ failure. Crit Care Med. 2007;35:S531–534. doi: 10.1097/01.CCM.0000278062.28122.A4. [DOI] [PubMed] [Google Scholar]
- 35.Rossi MA, Celes MR, Prado CM, Saggioro FP. Myocardial structural changes in long-term human severe sepsis/septic shock may be responsible for cardiac dysfunction. Shock. 2007;27:10–18. doi: 10.1097/01.shk.0000235141.05528.47. [DOI] [PubMed] [Google Scholar]
- 36.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, Heilbronn L, Haynie K, Muoio B, Li L, Hulver MW. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab. 2010;298:E988–998. doi: 10.1152/ajpendo.00307.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol. 2008;44:411–418. doi: 10.1016/j.yjmcc.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, Laguna JC, Vazquez-Carrera M. The p65 subunit of NF-{kappa}B binds to PGC-1{alpha}, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc Res. 2010;87(3):445–58. doi: 10.1093/cvr/cvq080. [DOI] [PubMed] [Google Scholar]
- 39.Merx MW, Weber C. Sepsis and the heart. Circulation. 2007;116:793–802. doi: 10.1161/CIRCULATIONAHA.106.678359. [DOI] [PubMed] [Google Scholar]
- 40.Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182:745–51. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.