

Abstract
The AIDS pandemic continues. Little is understood about how HIV gains access to permissive cells across mucosal surfaces, yet such knowledge is crucial to the development of successful topical anti-HIV-1 agents and mucosal vaccines. HIV-1 rapidly internalizes and integrates into the mucosal keratinocyte genome, and integrated copies of HIV-1 persist upon cell passage. The virus does not appear to replicate, and the infection may become latent. Interactions between HIV-1 and oral keratinocytes have been modeled in the context of key environmental factors, including putative copathogens and saliva. In keratinocytes, HIV-1 internalizes within minutes; in saliva, an infectious fraction escapes inactivation and is harbored and transferable to permissive target cells for up to 48 hours. When incubated with the common oral pathogen Porphyromonas gingivalis, CCR5− oral keratinocytes signal through protease-activated receptors and Toll-like receptors to induce expression of CCR5, which increases selective uptake of infectious R5-tropic HIV-1 into oral keratinocytes and transfer to permissive cells. Hence, oral keratinocytes—like squamous keratinocytes of other tissues—may be targets for low-level HIV-1 internalization and subsequent dissemination by transfer to permissive cells.
Keywords: HIV/AIDS, epithelia, oral epithelium, infectious disease, mucosal immunity, vaccines
In 2007, HIV/AIDS affected more than 33.2 million people worldwide, with approximately 2.5 million newly infected people and 2.1 million fatalities of AIDS-related illnesses (UNAIDS, 2008). Most primary infections with HIV-1 occur at mucosal surfaces. In adults, the mucosal surfaces are exposed during sexual activity (oral, vaginal, and anal). In infants, exposure and infection are a result of breast-feeding; the oral and oropharyngeal mucous membranes are the initial sites exposed to virus. Heterosexual intercourse accounts for more than 80% of current infections, including a dramatic increase in worldwide prevalence among women during the last few years (UNAIDS and World Health Organization, 2006).
Development of Successful Topical Anti-Hiv-1 Agents and Mucosal Vaccines
Among the highest research priorities from the 2008 National Institute of Allergy and Infectious Diseases HIV Vaccine Summit is to “define the first events leading to HIV . . . entering the gut-associated lymphoid tissue” (Fauci et al., 2008). When introduced into the mouth, infectious HIV-1 first encounters the oral and oropharyngeal tissues (Herzberg et al., 2006), but the sites of primary infection are not known. Attempts at vaccine development have proven inadequate in clinical trials in part because the fate of the virus upon exposure to mucosal surfaces is largely unknown. In uninfected men, unprotected oral sex with infected men elicits a salivary IgA1-mediated response that neutralizes HIV-1 (Hasselrot et al., 2009). Although an immune response can result from a direct encounter with virus during primary mucosal infection, whether the squamous epithelial cells serve as a latent HIV-1 reservoir is unclear (Richman et al., 2009). The mobilization of an antiviral immune response can neutralize the expanding pool of infecting virus and thus represent a measure of successful resistance (Li et al., 2009b). If the immune response prevents viral entry into cryptic sites and prevents latent infection, a successful vaccine might augment the induced salivary antiviral response. Recent successes with the development of topical anti-HIV-1 agents in nonhuman primates (Li et al., 2009a) suggest that an anti-HIV-1 response at the mucosal epithelial surface can prevent transmucosal infection by HIV-1 during clinical exposures. There is a growing awareness that HIV-1 encounters with mucosal epithelium are critical to the clinical course of infection.
Fate of HIV-1 in Squamous Epithelia
A multicellular layer of squamous epithelial cells covers the oral and oropharyngeal mucosae and the urogenital mucosae. After primary clinical exposures, R5-tropic HIV-1 systemic infection predominates (Hu et al., 2000), and in HIV-1-seropositive individuals, oral mucosal keratinocytes are infected with HIV-1 on the basis of viral RNA detection using molecular techniques (Rodriguez-Inigo et al., 2005). To study the fate of HIV-1 during encounters with squamous mucosal keratinocytes, we have studied oral keratinocytes that may model the epithelial cells from other squamous mucosae.
We have reported that X4 and R5 HIV-1 internalize and nonproductively infect oral (and oropharyngeal) keratinocytes, as marked by key viral life cycle events, including integration without apparent production of new viral transcripts or virions in vitro (Vacharaksa et al., 2008) (Fig. 1). The receptor for HIV-1 on the surface of oral keratinocytes is unknown. The expression of X4-tropic HIV-1 selective CXCR4 coreceptor notwithstanding, X4- and R5-tropic HIV-1 are both internalized. Nonproductive infection represents one of several possible fates of HIV-1 in oral keratinocytes. Similarly, squamous epithelial cells from the genitourinary tract can be nonpermissively infected by X4- and R5-tropic HIV-1 (Hladik et al., 2007). Like vaginal keratinocytes, HIV-1 internalizes into oral keratinocytes within 1 to 30 minutes (Dietrich et al., manuscript in preparation) and integrates by 3 hours (Vacharaksa et al., 2008). Yet, there is no substantive evidence that HIV-1 can replicate in oral epithelial cells, although earlier reports of the fate of HIV-1 in human oral keratinocytes present conflicting results (Acheampong et al., 2005; Liu et al., 2004). Oral keratinocytes harbor infectious virus, however, and transfer to or trans-infect permissive cells, including peripheral blood mononuclear cells, MOLT-4, and TZM-bl reporter cells (Giacaman et al., 2008; Vacharaksa et al., 2008).
Fig. 1.
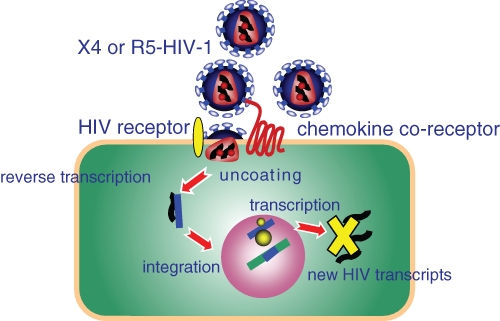
Nonproductive infection of oral keratinocytes by HIV-1. X4- or R5-tropic HIV-1 binds and internalizes into oral keratinocytes in a manner independent of the chemokine coreceptor specificity (CXCR4 or CCR5). A noncanonical HIV-1 receptor likely exists—perhaps, a surrogate for CD4. X4- and R5-tropic HIV-1 internalizes in the keratinocyte and uncoats, and viral RNA is reverse transcribed and integrates into the keratinocyte genome. Integration is stable, but new HIV-1 transcripts are not detected.
Although oral epithelial cells can harbor and trans-infect infectious virions to permissive cells, what is the evidence that HIV exposure of oral epithelial cells results in systemic dissemination? Human in vivo data are not known. In adult and infant nonhuman primates, however, when cell-free SIV is exposed atraumatically to the oral epithelium, infection of the oral epithelium and oropharyngeal tonsils is apparent within a day, with spread to the gastrointestinal mucosa several days later (Ruprecht et al., 1998; Milush et al., 2004). It is unclear whether the disseminating virus is residual from the original primary infecting inoculum or newly replicated SIV. In human tonsils studied ex vivo, HIV readily binds to the epithelium, but trafficking and infection of the organized lymphoid tissues is limited (Maher et al., 2004). HIV-1 replicates in human tonsils but not as effectively as in the rectal mucosa, and it is associated with differential expression of HIV coreceptors and the proximity of CD4 cells (Grivel et al., 2007). Whereas infection of oral keratinocytes is nonproductive, the association with infectious HIV-1 is suggested to be significant nonetheless when trans-infection results in a small population of infected CD4 T cells in the tissue.
Putative Restrictions Against HIV-1 Replication in Oral Keratinocytes
At this time, it is unclear why oral keratinocytes support only nonproductive infection by HIV-1. Potential restrictions against replication could include eukaryotic initiation factor 3 subunit f, which can specifically interfere with the 3′ end processing of HIV-1 mRNAs (Valente et al., 2009); non-Vpu-neutralized tetherin, which impedes release of HIV-1 (Mangeat et al., 2009); TRIM and other capsid-targeting cellular proteins, which contribute to cell cycle–dependent inability to infect (Yamashita and Emerman, 2009); and APOBEC family proteins (Schumacher et al., 2008; Ogawa et al., 2009). How these and other restrictions prevent HIV-1 replication in oral keratinocytes is an important area for future investigation.
Oral Epithelium as a Model for Genitourinary Infections
The genitourinary and oral mucosa are lined by squamous epithelia that are morphologically and phenotypically similar (Hladik and McElrath, 2008). Interspersed between the epithelial cells, Langerhans (immature dendritic) cells are generally localized in the basal and suprabasal layers. Although no quantitative assessment has been reported, Langerhans cells appear to populate the vaginal epithelium at a greater density than in oral epithelium (Cutler and Jotwani, 2006b), although quantitative assessments are not reported. Interestingly, the Langerhans cells of the vaginal mucosa are able to sample antigen at the mucosal surface, but antigen sampling at the surface of the oral mucosa has not been reported.
Although controversial, recent evidence in nonhuman primates (Li et al., 2009a) suggests that HIV-1 translocates intact vaginal and endocervical epithelia HIV to cause a disseminated infection. Like oral keratinocytes (Vacharaksa et al., 2008), ectocervical epithelial cells internalize and transfer intact viral particles to permissive cells (Wu et al., 2003), and vaginal epithelial cells transcytose HIV-1 in a transwell system, albeit at a low rate (Bobardt et al., 2007). Taken together, current evidence suggests that the oral and genitourinary squamous epithelia generally harbor infectious virus without becoming productively infected. Harbored virions in keratinocytes may functionally contribute to dissemination of HIV-1 to permissive cells. In some conditions, perhaps under pressure from coinfecting microorganisms, it is plausible that these mucosal keratinocytes might become productively infected by HIV-1 and amplify virus for systemic spread.
Bathing Fluids
The mucosal secretions contain substances that can modify the infectivity and course of HIV-1 infection. The molecular constituents of these bathing fluids are generally similar, although the proportions might differ from tissue to tissue depending on the glandular and cellular sources. Among the mucosal fluids, whole and fractionated saliva, breast milk, colostrum, seminal plasma, and cervicovaginal secretions all show anti-HIV-1 activity associated with lactoferrin (Kazmi et al., 2006). Seminal plasma and cervicovaginal fluids are less effective against HIV-1 than colostrum, whole milk, and whole saliva.
During primary clinical exposures, viable X4- and R5-tropic HIV-1 is introduced into the oral cavity and oropharynx in breast milk (Van de Perre et al., 1991) or semen (Pasquier et al., 2009). The bolus of HIV-1 is mixed and diluted, however imperfectly, in saliva. Salivary anti-HIV activity is likewise diluted and potentially neutralized given that breast milk and seminal plasma increase the titer of freshly harvested HIV-1 in vitro (Acheampong et al., 2005; Southern and Southern, 2002). When primary HIV-1 infection is diagnosed, virions are no longer detectable in the oral cavity and oropharynx (Herzberg et al., 2006), and shed virions in saliva are not infectious (Goto et al., 1991). Virions are probably inactivated by salivary anti-HIV molecules (Kasmi et al., 2006). In contrast to this conclusion, primary infection in the gastrointestinal mucosa is thought to result when the HIV-1 bolus passes unremarkably through the nonpermissive oral environment and is swallowed (Brenchley et al., 2006).
The actual infectious titer (e.g., TCID50) of HIV-1 in breast milk and semen, containing cell-free and cell-associated virus, is difficult to estimate (Chan, 2005; Yamaguchi et al., 2007). When incubated in fresh crude whole saliva, however, HIV-1 virions remain sufficiently infectious when harbored by oral keratinocytes to trans-infect permissive cells (Dietrich et al., unpublished data). Although saliva is swallowed at an average rate of 0.5 mL per minute (Rudney et al., 1995), the HIV–breast milk or HIV-semen bolus mixes imperfectly with saliva and is able to interface for several minutes or longer with the oral epithelium. In some anatomic sites, the HIV-1 bolus may encounter stasis, remaining in prolonged apposition to mucosal keratinocytes. Indeed, spermatozoa, a surrogate marker for HIV-1 virions in seminal fluid, were retained within tonsillar crypts in ex vivo organ culture experiments (Maher et al., 2004). Hence, infectious inocula may show extended interactions with mucosal surfaces.
Coated with a salivary film, oral mucosal keratinocytes bind high-molecular-weight mucins (MG1; MUC5B) and other salivary molecules (Bradway et al., 1992). Indeed, the salivary mucin glycoprotein MUC5B binds HIV-1 (Habte et al., 2006), which likely docks with cell surface glycoproteins MUC1 and/or gp340 (DMBT1) on the keratinocyte (Stoddard et al., 2007). The fate of HIV-1 in the oral cavity and oropharynx may depend on the rates of inactivation by saliva, clearance by swallowing, and capture by mucosal keratinocytes.
Most of the estimated 300 to 1300 salivary proteins (Guo et al., 2006) are in solution (sol phase) (Glantz et al., 1996), whereas the high-molecular-weight mucin fraction (MG1; MUC5B; > 1000 kDa) self-assembles into a gel phase (Wickstrom et al., 2000). Crude separation of these phases can be accomplished by centrifugation at 10,000 × g for 10 minutes (Herzberg et al., 1979). The partitioned clarified supernatant contains smaller mucin glycoproteins and gp340, sIgA, amylase and other proteins, glycoproteins, and lipids. The sedimented mucous gel phase contains MUC5B and complexed proteins, desquamated oral epithelial cells, bacteria, and viruses. Indeed, mucoid proteins are known to bind influenza virus (Stone, 1949), and saliva is hospitable and transmits many viruses, including members of the herpesvirus family (Miller et al., 2006). As a bathing fluid for HIV-1, saliva maintains a pH of about 6.7 and is hypotonic relative to serum. Admixed breast milk or seminal fluid would increase osmolality to approach serum levels. As the flow rate increases to 1.5 mL per minute, the buffer capacity of saliva approaches that of plasma. HIV-1 should be stable in this fluid environment, except for the presence of antiviral molecules.
Putative salivary anti-HIV-1 molecules (Kazmi et al., 2006) include secretory leukocyte protease inhibitor (McNeely et al., 1995), β-defensin 2 (hBD-2) (Quinones-Mateu et al., 2003), proline-rich proteins (Robinovitch et al., 2001), thrombospondin (Crombie et al., 1998), lysozyme (Lee-Huang et al., 1999), salivary peroxidase and α-defensins (Kazmi et al., 2006), lactoferrin (Saidi et al., 2006), GP340 agglutinin (Wu et al., 2006), and mucins, including MUC5B (Habte et al., 2006). GP340 and MUC5B appear to agglutinate and sequester virus from CD4+ target cells in vitro (Habte et al., 2006; Malamud et al., 1997). In infected individuals, salivary anti-HIV-1 molecules apparently attenuate HIV-1 virions sufficiently enough to virtually eliminate risk of salivary transmission to uninfected individuals. The converse may not be true. The effectiveness of saliva as an anti-HIV-1 agent depends on the mode of action, the kinetics of inactivation, and the ability of the virus to become harbored or shielded from inhibitors. When a bolus of HIV enters the oral cavity of an uninfected individual, saliva may be insufficiently effective (Yeh et al., 1992) to inhibit uptake and infection of mucosal epithelial cells (Dietrich et al., unpublished data).
Coinfecting Mucosal Pathogens
During clinical presentation, a bolus of HIV-1 is invariably exposed to coinfecting commensal bacteria and endogenous pathogens resident in mucosal biofilms. Certainly when existing infection causes mucosal lesions, HIV-1 infection and dissemination are favored. Indeed, ulcerative and nonulcerative sexually transmitted diseases increase the risk for HIV-1 infection. In particular, nonulcerative sexually acquired infection caused by Neisseria gonorrhoeae, Chlamydia trachomatis (Laga et al., 1993), and Trichomonas vaginalis (Buve et al., 2001; Laga et al., 1993) are associated with higher rates of HIV-1 infection. Given that mucosal infections acquired by sexual contact are more prevalent in groups at high risk for HIV-1 infection (Aral et al., 2006), HIV-1 acquisition in association with coinfection is not well explained by mucosal lesions. In general, the risk of acquisition of HIV-1 increases with concomitant sexually transmitted diseases, including gonorrhea (Schwarcz et al., 2002) and HSV-2 (Lingappa and Celum, 2007).
Omnipresent viral infections and HIV-1 appear to exacerbate both infections, suggesting synergistic mechanisms. In HIV-positive individuals, several DNA viruses replicate in the mucosal tissues and shed into the mouth, including HSV-1 and HSV-2; cytomegalovirus; Epstein-Barr virus; human herpes virus 6, 7, and 8; and human papilloma virus (Mbopi-Keou et al., 2002). The DNA viruses induce cytokine release by mucosal cells, which could activate HIV-1 replication and increase risk of HIV-1 infection.
Coinfection with other microorganisms also appears to promote infection by HIV-1. In the oral context, we have shown that Porphyromonas gingivalis, a ubiquitous periodontal pathogen, upregulates expression of CCR5 on oral keratinocytes (Giacaman et al., 2007) (Fig. 2). Healthy oral and oropharyngeal keratinocytes are CD4− CXCR4+ CCR5− in vitro (Vacharaksa et al., 2008) and in vivo (Cutler and Jotwani, 2006a; Kumar et al., 2006). Given that CXCR4 is the coreceptor for infection by X4-tropic HIV-1 and CCR5 for R5-tropic virus, it is unlikely that selective primary clinical infections by R5-tropic HIV-1 could be explained at this level. At the mucosal surface, the receptor tropism of HIV-1 may be modified by endogenous coinfecting microflora, including P. gingivalis. As we report, P. gingivalis signaling through the protease-activated receptors and Toll-like receptors 2 and 4 (Fig. 2A) induces keratinocyte CCR5 appearance as a surface receptor (Giacaman et al., 2007) (Fig. 2B). Expression of CCR5 in response to P. gingivalis increases nonproductive trans-infection of harbored R5-tropic HIV-1 to permissive cells (Giacaman et al., 2008) (Fig. 2C, 2D). This pathway of harboring infectious HIV-1 in oral keratinocytes and trans-infecting permissive cells represents another fate of HIV-1 in oral keratinocytes. Although harboring and trans-infection show no coreceptor tropism, the selective fate of R5-tropic HIV-1 is increased by P. gingivalis, which has been reported to reactivate latent infection of HIV-1 in mononuclear cell lines by chromatin modification through butyrate-specific inhibition of histone deacetylases (Imai et al., 2009). Although numerous reports suggest that the clinical expression of periodontitis and other oral infection is altered in patients with HIV/AIDS, it remains unclear whether preexisting periodontitis increases the risk of acquisition or systemic dissemination of HIV-1. Nonetheless, potential copathogens can exploit alternative mechanisms to promote HIV-1 infection.
Fig. 2.
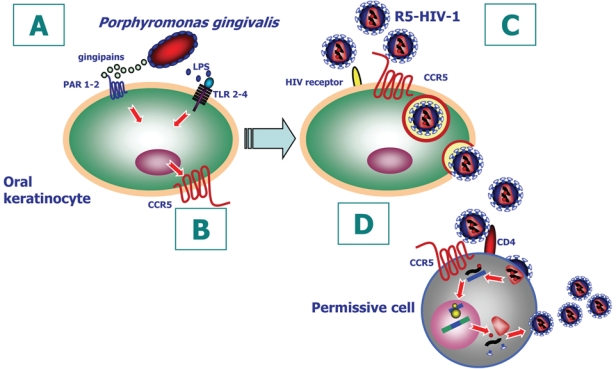
R5-HIV and Porphyromonas gingivalis coinfection of oral epithelial cells: (A) P. gingivalis gingipains signal oral keratinocytes through PAR-1 and PAR-2, and LPS signals through TLR2 and TLR4. (B) Signaling of the oral epithelial cell leads to upregulation and expression of CCR5 on cell surface. (C) P. gingivalis–upregulated CCR5 promotes selective internalization of R5 HIV-1 and intracellular harboring. (D) Harbored infectious virions in oral keratinocytes are trans-infected to permissive cells. R5 HIV-1 is transferred and captured by CD4+ permissive cells, where virus replicates and dissemination can occur.
Mucosal Epithelial Cell Receptors for HIV-1
HIV-1-associated receptors are atypical on squamous keratinocytes when compared to T cells, macrophages, and dendritic cells. Unlike white blood cells, oral squamous epithelial cells are CD4− and CXCR4+ CCR5− in the absence of inflammation (Jotwani et al., 2004; Kumar et al., 2006; Vacharaksa et al., 2008). Whereas oral and vaginal epithelial cells show similar phenotypes, gastrointestinal epithelial cells can express CXCR4 and CCR5. The CD4− gastrointestinal epithelial cells transcytose HIV-1 in a CXCR4- or CCR5-dependent manner (Bomsel, 1997; Bobardt et al., 2007), suggesting that these receptors might mediate HIV-1 entry by a CD4- and membrane fusion–independent mechanism. Although they do not generally express CCR5, oral keratinocytes take up and trans-infect HIV-1 without preference for a particular chemokine receptor-HIV-1 tropism (Vacharaksa et al., 2008). On CD4− cells, the V3 loop domain of gp120 has been suggested to interact with alternative receptors, including galactosylceramide (Cook et al., 1994), whereas a gp120 envelope-independent mechanism of epithelial cell infection also occurs (Pang et al., 2000). On CD4− epithelial cells, alternative receptors may include heparan sulfate proteoglycans (Bobardt et al., 2007), ICAM-1, LFA-1 (Fortin et al., 1998), DC-SIGN (Geijtenbeek et al., 2000), DC-SIGNR (Pohlmann et al., 2001), langerin (de Witte et al., 2007), mannose receptor (Liu et al., 2004), and gp340 (Stoddard et al., 2007). Hence, mucosal keratinocytes may be infected by HIV-1 through several routes, resulting in different intracellular pools of HIV-1.
Primary infection of oral epithelium by HIV-1 through a receptor-mediated mechanism depends on the distribution and density of receptors in proximity to the virus. Although various cells in the gingival tissues collectively express most of the putative HIV-1 receptors and coreceptors—including CXCR4, CCR5, langerin, DC-SIGN, mannose receptor, and galactosylceramide (Jotwani et al., 2004)—the distribution is neither homogeneous nor prominent on the surface keratinocytes. Indeed, the receptor distribution and density changes with inflammation (Jotwani et al., 2004). Inflamed gingival and tonsil epithelia show similar patterns, with variable numbers of CXCR4+ and CCR5+ keratinocytes stretching continuously across the stratified epithelium with greater density in the basal and suprabasal layers (Kumar et al., 2006). Heparan sulfate proteoglycan–positive keratinocytes are prominent in all epithelial layers. Although not reported in the oral epithelium, CXCR4+ CCR5+ cells are identified as CD4+ lymphocytes infiltrating the tonsil epithelium (Kumar et al., 2006). The low density of relevant known HIV-1 receptors in the superficial oral epithelium may represent a restriction to primary HIV-1 infection, whereas noncanonical (CD4− CXCR4+ CCR5− and alterative receptor-independent) uptake pathways may be functional (Vacharaksa et al., 2008).
Transfer to Dendritic Cells
What is the postulated fate of HIV-1 harbored by oral keratinocytes? As we have modeled in vitro and as consistent with primate models, HIV-1 harbored by oral keratinocytes most likely trans-infects Langerhans cells in vivo. Langerhans or immature dendritic cells are resident in the epithelium proximal to basal and suprabasal keratinocytes (Cutler and Jotwani, 2006a), and they form a dendritic network (25 cells/mm3 in patients 14 to 39 years old and 7 cells/mm3 in patients 61 to 74 years old), maximizing the area of antigen capture (Zavala and Cavicchia, 2006). The volume density of keratinocytes is estimated to be at least 1000-fold greater than that of Langerhans cells. Hence, encounters between HIV and keratinocytes may be 1000-fold more likely than with Langerhans cells. Intraepithelial T cells are exceedingly rare in the oral epithelium even during inflammation, whereas dendritic cell–T cell interactions occur in the lamina propria of the oral epithelium (Cutler and Jotwani, 2006b). As we have shown, Langerhans cells and T cells are comparatively more abundant among epithelial cells in tonsils (Kumar et al., 2006). After internalization of HIV-1, immature dendritic cells activate and emigrate from the epithelium to dock with and transfer HIV-1 to CD4+ T cells in the connective tissues (Sugaya et al., 2004). Emigrating DCs and HIV-1-infected T cells disseminate infection to organized mucosal-associated lymphoid tissues.
Oral Epithelium as a “Trojan Horse”
The oral tissues are thought to be protected or nonpermissive to infection and unable to serve as foci of systemic infection. Despite these assumptions, we present cogent evidence from the literature and our own published and unpublished work to show the plausibility of primary R5-tropic HIV-1 infection of the oral epithelium and oropharynx. By extrapolation from atraumatic oral exposure to SIV in the macaque, any amount of infectious HIV-1 that infects and crosses the mucosal epithelial lining to infect a T cell must be considered clinically significant. As suggested for genital epithelia (Hladik and McElrath, 2008), primary exposure may result in HIV-1 reaching immature dendritic cells by a paracellular route, through frank breaches, or upon capture by Langerhans cells (immature dendritic cells) at the mucosal surface. Most likely, HIV-1 internalizes in superficial oral epithelial cells by a noncanonical mechanism, or it moves paracellularly to internalize in suprabasal and basal keratinocytes, which are modeled by cells cultured in vitro. In the presence of saliva and P. gingivalis, R5-tropic infectious virus is therefore likely to be selectively internalized and transferred from oral keratinocytes to immature dendritic cells, setting the stage for systemic dissemination. Hence, the oral environment has the potential to overcome salivary antiviral factors and favor primary clinical infections by R5-tropic HIV-1 resulting in systemic dissemination. Keratinocytes that contain integrated (or perhaps latent) copies of the HIV-1 genome or harbor infectious HIV-1 may also serve to reinfect the host during chronic disease. Furthermore, latent HIV-1 infection can be reactivated by P. gingivalis, suggesting that activation of latent integrated copies of HIV-1 in keratinocytes could be a cryptic source of virus able to reinfect the host. As cryptic sites of HIV-1 infection, oral epithelial keratinocytes may be a threat to current antiviral pharmacologic therapies and so challenge the development of successful vaccines.
The evidence presented in this review supports the plausibility of HIV-1 infection of oral mucosal epithelial cells. The fate of HIV-1 must now be determined more robustly in mucosal epithelial tissues ex vivo and in vivo. Armed with these data, we will better understand the mechanism of primary HIV-1 infection acquired across mucosal surfaces.
Footnotes
Our work on HIV-1 has been supported by National Institutes of Health / National Institute of Dental and Craniofacial Research R01DE015503 (to M.C.H.) and R21 DE015506 (to K.F.R.).
References
- Acheampong EA, Parveen Z, Muthoga LW, Wasmuth-Peroud V, Kalayeh M, Bashir A, et al. (2005). Molecular interactions of human immunodeficiency virus type 1 with primary human oral keratinocytes. J Virol 79:8440-8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aral SO, O’Leary A, Baker C. (2006). Sexually transmitted infections and HIV in the southern United States: an overview. Sex Transm Dis 33 (suppl):1S-5S [DOI] [PubMed] [Google Scholar]
- Bobardt MD, Chatterji U, Selvarajah S, Van der Schueren B, David G, Kahn B, et al. (2007). Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J Virol 81:395-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsel M. (1997). Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat Med 3:42-47 [DOI] [PubMed] [Google Scholar]
- Bradway SD, Bergey EJ, Scannapieco FA, Ramasubbu N, Zawacki S, Levine MJ. (1992). Formation of salivary-mucosal pellicle: the role of transglutaminase. Biochem J 284(pt 2):557-564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Douek DC. (2006). HIV disease: fallout from a mucosal catastrophe? Nat Immunol 7:235-239 [DOI] [PubMed] [Google Scholar]
- Buve A, Weiss HA, Laga M, Van Dyck E, Musonda R, Zekeng L, et al. (2001). The epidemiology of trichomoniasis in women in four African cities. AIDS 15(suppl 4):89-96 [DOI] [PubMed] [Google Scholar]
- Chan DJ. (2005). Pathophysiology of HIV-1 in semen: current evidence for compartmentalisation and penetration by antiretroviral drugs. Curr HIV Res 3:207-222 [DOI] [PubMed] [Google Scholar]
- Cook DG, Fantini J, Spitalnik SL, Gonzalez-Scarano F. (1994). Binding of human immunodeficiency virus type I (HIV-1) gp120 to galactosylceramide (GalCer): relationship to the V3 loop. Virology 201:206-214 [DOI] [PubMed] [Google Scholar]
- Crombie R, Silverstein RL, MacLow C, Pearce SF, Nachman RL, Laurence J. (1998). Identification of a CD36-related thrombospondin 1-binding domain in HIV-1 envelope glycoprotein gp120: relationship to HIV-1-specific inhibitory factors in human saliva. J Exp Med 187:25-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler CW, Jotwani R. (2006a). Dendritic cells at the oral mucosal interface. J Dent Res 85:678-689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler CW, Jotwani R. (2006b). Oral mucosal expression of HIV-1 receptors, co-receptors, and alpha-defensins: tableau of resistance or susceptibility to HIV infection? Adv Dent Res 19:49-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong MA, de Gruijl T, et al. (2007). Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med 13:367-371 [DOI] [PubMed] [Google Scholar]
- Fauci AS, Johnston MI, Dieffenbach CW, Burton DR, Hammer SM, Hoxie JA, et al. (2008). HIV vaccine research: the way forward. Science 321:530-532 [DOI] [PubMed] [Google Scholar]
- Fortin JF, Cantin R, Tremblay MJ. (1998). T cells expressing activated LFA-1 are more susceptible to infection with human immunodeficiency virus type 1 particles bearing host-encoded ICAM-1. J Virol 72:2105-2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, et al. (2000). DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587-597 [DOI] [PubMed] [Google Scholar]
- Giacaman RA, Asrani AC, Gebhard KH, Dietrich EA, Vacharaksa A, Ross KF, et al. (2008). Porphyromonas gingivalis induces CCR5-dependent transfer of infectious HIV-1 from oral keratinocytes to permissive cells. Retrovirology 5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacaman RA, Nobbs AH, Ross KF, Herzberg MC. (2007). Porphyromonas gingivalis selectively up-regulates the HIV-1 coreceptor CCR5 in oral keratinocytes. J Immunol 179:2542-2550 [DOI] [PubMed] [Google Scholar]
- Glantz PO, Baier RE, Christersson CE. (1996). Biochemical and physiological considerations for modeling biofilms in the oral cavity: a review. Dent Mater 12:208-214 [DOI] [PubMed] [Google Scholar]
- Goto Y, Yeh CK, Notkins AL, Prabhakar BS. (1991). Detection of proviral sequences in saliva of patients infected with human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 7:343-347 [DOI] [PubMed] [Google Scholar]
- Grivel JC, Elliott J, Lisco A, Biancotto A, Condack C, Shattock RJ, et al. (2007). HIV-1 pathogenesis differs in rectosigmoid and tonsillar tissues infected ex vivo with CCR5- and CXCR4-tropic HIV-1. AIDS 21:1263-1272 [DOI] [PubMed] [Google Scholar]
- Guo T, Rudnick PA, Wang W, Lee CS, Devoe DL, Balgley BM. (2006). Characterization of the human salivary proteome by capillary isoelectric focusing/nanoreversed-phase liquid chromatography coupled with ESI-tandem MS. J Proteome Res 5:1469-1478 [DOI] [PubMed] [Google Scholar]
- Habte HH, Mall AS, de Beer C, Lotz ZE, Kahn D. (2006). The role of crude human saliva and purified salivary MUC5B and MUC7 mucins in the inhibition of human immunodeficiency virus type 1 in an inhibition assay. Virol J 3:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselrot K, Saberg P, Hirbod T, Soderlund J, Ehnlund M, Bratt G, et al. (2009). Oral HIV-exposure elicits mucosal HIV-neutralizing antibodies in uninfected men who have sex with men. AIDS 23:329-333 [DOI] [PubMed] [Google Scholar]
- Herzberg MC, Levine MJ, Ellison SA, Tabak LA. (1979). Purification and characterization of monkey salivary mucin. J Biol Chem 254:1487-1494 [PubMed] [Google Scholar]
- Herzberg MC, Weinberg A, Wahl SM. (2006). (C3) The oral epithelial cell and first encounters with HIV-1. Adv Dent Res 19:158-166 [DOI] [PubMed] [Google Scholar]
- Hladik F, McElrath MJ. (2008). Setting the stage: host invasion by HIV. Nat Rev Immunol 8:447-457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, Eschenbach D, et al. (2007). Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26:257-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu QX, Barry AP, Wang ZX, Connolly SM, Peiper SC, Greenberg ML. (2000). Evolution of the human immunodeficiency virus type 1 envelope during infection reveals molecular corollaries of specificity for coreceptor utilization and AIDS pathogenesis. J Virol 74:11858-11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Ochiai K, Okamoto T. (2009). Reactivation of latent HIV-1 infection by the periodontopathic bacterium Porphyromonas gingivalis involves histone modification. J Immunol 182:3688-3695 [DOI] [PubMed] [Google Scholar]
- Jotwani R, Muthukuru M, Cutler CW. (2004). Increase in HIV receptors/co-receptors/alpha-defensins in inflamed human gingiva. J Dent Res 83:371-377 [DOI] [PubMed] [Google Scholar]
- Kazmi SH, Naglik JR, Sweet SP, Evans RW, O’Shea S, Banatvala JE, et al. (2006). Comparison of human immunodeficiency virus type 1–specific inhibitory activities in saliva and other human mucosal fluids. Clin Vaccine Immunol 13:1111-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RB, Maher DM, Herzberg MC, Southern PJ. (2006). Expression of HIV receptors, alternate receptors and co-receptors on tonsillar epithelium: implications for HIV binding and primary oral infection. Virol J 3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laga M, Manoka A, Kivuvu M, Malele B, Tuliza M, Nzila N, et al. (1993). Non-ulcerative sexually transmitted diseases as risk factors for HIV-1 transmission in women: results from a cohort study. AIDS 7:95-102 [DOI] [PubMed] [Google Scholar]
- Lee-Huang S, Huang PL, Sun Y, Huang PL, Kung HF, Blithe DL, et al. (1999). Lysozyme and RNases as anti-HIV components in beta-core preparations of human chorionic gonadotropin. Proc Natl Acad Sci U S A 96:2678-2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. (2009a). Glycerol monolaurate prevents mucosal SIV transmission. Nature 458:1034-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Skinner PJ, Ha SJ, Duan L, Mattila TL, Hage A, et al. (2009b). Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science 323:1726-1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingappa JR, Celum C. (2007). Clinical and therapeutic issues for herpes simplex virus-2 and HIV co-infection. Drugs 67:155-174 [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. (2004). CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol 78:4120-4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher D, Wu X, Schacker T, Larson M, Southern P. (2004). A model system of oral HIV exposure, using human palatine tonsil, reveals extensive binding of HIV infectivity, with limited progression to primary infection. J Infect Dis 190:1989-1997 [DOI] [PubMed] [Google Scholar]
- Malamud D, Nagashunmugam T, Davis C, Kennedy S, Abrams WR, Kream R, et al. (1997). Inhibition of HIV infectivity by human saliva. Oral Dis 3(suppl 1):58-63 [DOI] [PubMed] [Google Scholar]
- Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. (2009). HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog 5:e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbopi-Keou FX, Belec L, Teo CG, Scully C, Porter SR. (2002). Synergism between HIV and other viruses in the mouth. Lancet Infect Dis 2:416-424 [DOI] [PubMed] [Google Scholar]
- McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM. (1995). Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J Clin Invest 96:456-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CS, Berger JR, Mootoor Y, Avdiushko SA, Zhu H, Kryscio RJ. (2006). High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus-infected persons in the era of highly active antiretroviral therapy. J Clin Microbiol 44:2409-2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milush JM, Kosub D, Marthas M, Schmidt K, Scott F, Wozniakowski A, et al. (2004). Rapid dissemination of SIV following oral inoculation. AIDS 18:2371-2380 [PubMed] [Google Scholar]
- Ogawa Y, Kawamura T, Kimura T, Ito M, Blauvelt A, Shimada S. (2009). Gram-positive bacteria enhance HIV-1 susceptibility in Langerhans cells, but not in dendritic cells, via Toll-like receptor activation. Blood 113:5157-5166 [DOI] [PubMed] [Google Scholar]
- Pang S, Yu D, An DS, Baldwin GC, Xie Y, Poon B, et al. (2000). Human immunodeficiency virus Env-independent infection of human CD4(-) cells. J Virol 74:10994-11000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier C, Sauné K, Raymond S, Moinard N, Daudin M, Bujan L, et al. (2009). Determining seminal plasma human immunodeficiency virus type 1 load in the context of efficient highly active antiretroviral therapy. J Clin Microbiol 47:2883-2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlmann S, Soilleux EJ, Baribaud F, Leslie GJ, Morris LS, Trowsdale J, et al. (2001). DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc Natl Acad Sci U S A 98:2670-2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, et al. (2003). Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17:F39-F48 [DOI] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. (2009). The challenge of finding a cure for HIV infection. Science 323:1304-1307 [DOI] [PubMed] [Google Scholar]
- Robinovitch MR, Ashley RL, Iversen JM, Vigoren EM, Oppenheim FG, Lamkin M. (2001). Parotid salivary basic proline-rich proteins inhibit HIV-I infectivity. Oral Dis 7:86-93 [PubMed] [Google Scholar]
- Rodriguez-Inigo E, Jimenez E, Bartolome J, Ortiz-Movilla N, Bartolome Villar B, Jose Arrieta J, et al. (2005). Detection of human immunodeficiency virus type 1 RNA by in situ hybridization in oral mucosa epithelial cells from anti-HIV-1 positive patients. J Med Virol 77:17-22 [DOI] [PubMed] [Google Scholar]
- Rudney JD, Ji Z, Larson CJ. (1995). The prediction of saliva swallowing frequency in humans from estimates of salivary flow rate and the volume of saliva swallowed. Arch Oral Biol 40:507-512 [DOI] [PubMed] [Google Scholar]
- Ruprecht RM, Baba TW, Liska V, Ayehunie S, Andersen J, Montefiori DC, et al. (1998). Oral SIV, SHIV, and HIV type 1 infection. AIDS Res Hum Retroviruses 14(suppl 1):97-103 [PubMed] [Google Scholar]
- Saidi H, Eslaphazir J, Carbonneil C, Carthagena L, Requena M, Nassreddine N, et al. (2006). Differential modulation of human lactoferrin activity against both R5 and X4-HIV-1 adsorption on epithelial cells and dendritic cells by natural antibodies. J Immunol 177:5540-5549 [DOI] [PubMed] [Google Scholar]
- Schumacher AJ, Haché G, Macduff DA, Brown WL, Harris RS. (2008). The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J Virol 82:2652-2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz SK, Kellogg TA, McFarland W, Louie B, Klausner J, Withum DG, et al. (2002). Characterization of sexually transmitted disease clinic patients with recent human immunodeficiency virus infection. J Infect Dis 186:1019-1022 [DOI] [PubMed] [Google Scholar]
- Southern S, Southern P. (2002). Cellular mechanism for milk-borne transmission of HIV and HTLV. Adv Exp Med Biol 503:183-190 [DOI] [PubMed] [Google Scholar]
- Stoddard E, Cannon G, Ni H, Kariko K, Capodici J, Malamud D, et al. (2007). gp340 expressed on human genital epithelia binds HIV-1 envelope protein and facilitates viral transmission. J Immunol 179:3126-3132 [DOI] [PubMed] [Google Scholar]
- Stone JD. (1949). Inhibition of influenza virus haemagglutination by mucoids: conversion of virus to indicator for inhibitor. Aust J Exp Biol Med Sci 27:337-352 [DOI] [PubMed] [Google Scholar]
- Sugaya M, Lore K, Koup RA, Douek DC, Blauvelt A. (2004). HIV-infected Langerhans cells preferentially transmit virus to proliferating autologous CD4+ memory T cells located within Langerhans cell-T cell clusters. J Immunol 172:2219-2224 [DOI] [PubMed] [Google Scholar]
- UNAIDS (2008). Report on the global AIDS epidemic. http://www.unaids.org/en/KnowledgeCentre/HIVData/GlobalReport/2008
- UNAIDS, World Health Organization (2006). AIDS epidemic update: December 2006. http://data.unaids.org/pub/EpiReport/2006/2006_EpiUpdate_en.pdf Accessed November 9, 2010
- Vacharaksa A, Asrani AC, Gebhard KH, Fasching CE, Giacaman RA, Janoff EN, et al. (2008). Oral keratinocytes support non-replicative infection and transfer of harbored HIV-1 to permissive cells. Retrovirology 5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente ST, Gilmartin GM, Mott C, Falkard B, Goff SP. (2009). Inhibition of HIV-1 replication by eIF3f. Proc Natl Acad Sci U S A 106:4071-4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Perre P, Simonon A, Msellati P, Hitimana DG, Vaira D, Bazubagira A, et al. (1991). Postnatal transmission of human immunodeficiency virus type 1 from mother to infant: a prospective cohort study in Kigali, Rwanda. N Engl J Med 325:593-598 [DOI] [PubMed] [Google Scholar]
- Wickstrom C, Christersson C, Davies JR, Carlstedt I. (2000). Macromolecular organization of saliva: identification of “insoluble” MUC5B assemblies and non-mucin proteins in the gel phase. Biochem J 351(pt 2):421-428 [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Chen Z, Phillips DM. (2003). Human genital epithelial cells capture cell-free human immunodeficiency virus type 1 and transmit the virus to CD4+ cells: implications for mechanisms of sexual transmission. J Infect Dis 188:1473-1482 [DOI] [PubMed] [Google Scholar]
- Wu Z, Lee S, Abrams W, Weissman D, Malamud D. (2006). The N-terminal SRCR-SID domain of gp-340 interacts with HIV type 1 gp120 sequences and inhibits viral infection. AIDS Res Hum Retroviruses 22:508-515 [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Sugiyama T, Takizawa M, Yamamoto N, Honda M, Natori M. (2007). Viability of infectious viral particles of HIV and BMCs in breast milk. J Clin Virol 39:222-225 [DOI] [PubMed] [Google Scholar]
- Yamashita M, Emerman M. (2009). Cellular restriction targeting viral capsids perturbs human immunodeficiency virus type 1 infection of nondividing cells. J Virol 83:9835-9843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CK, Handelman B, Fox PC, Baum BJ. (1992). Further studies of salivary inhibition of HIV-1 infectivity. J Acquir Immune Defic Syndr 5:898-903 [PubMed] [Google Scholar]
- Zavala WD, Cavicchia JC. (2006). Deterioration of the Langerhans cell network of the human gingival epithelium with aging. Arch Oral Biol 51:1150-1155 [DOI] [PubMed] [Google Scholar]