

Abstract
Aggregatibacter actinomycetemcomitans is a Gram-negative bacterium that colonizes the human oral cavity and is the causative agent for localized aggressive periodontitis (LAP), an aggressive form of periodontal disease that occurs in adolescents. A. actinomycetemcomitans secretes a protein toxin, leukotoxin (LtxA), which helps the bacterium evade the host immune response during infection. LtxA is a membrane-active toxin that specifically targets white blood cells (WBCs). In this review, we discuss recent developments in this field, including the identification and characterization of genes and proteins involved in secretion, regulation of LtxA, biosynthesis, newly described activities of LtxA, and how LtxA may be used as a therapy for the treatment of diseases.
Keywords: leukemia, periodontitis, RTX toxin, LAP
Introduction
Aggregatibacter actinomycetemcomitans is a Gram-negative bacterium that inhabits the mouths of one-third or more of the population (Leys et al., 1994; Lamell et al., 2000). In a small number of adolescents, the bacterium can cause a rapidly progressing form of periodontal disease, localized aggressive periodontitis (LAP) (Newman et al., 1976; Zambon, 1985; Slots and Ting, 1999). In contrast to adult or generalized periodontitis, LAP affects the central incisors and first molars. Individuals with LAP have breakdown of the supporting structures of the teeth, and tooth loss occurs when the condition is left untreated. Furthermore, individuals of African-American descent have a 10- to 15-fold greater risk of developing the disease than do Caucasian Americans. Approximately 70,000 adolescents in the U.S. develop the disease per year (Löe and Brown, 1991). In addition to causing oral disease, A. actinomycetemcomitans is a member of the HACEK group of bacteria, which includes the Gram-negative bacteria that cause infective endocarditis (Das et al., 1997). A. actinomycetemcomitans has also been isolated from numerous other diseased sites of the body and is therefore considered a systemic pathogen. Indeed, the link between oral and systemic health is becoming well-established as the molecular and immunological basis for cardiovascular diseases is further studied (van Winkelhoff and Slots, 1999; Slots, 2003; Beck et al., 2005).
To colonize, cause disease, and persist in a host, a bacterial pathogen must maintain an arsenal of virulence factors. A. actinomycetemcomitans produces numerous factors that have been well-characterized, including adherence proteins, biofilm polysaccharides, LPS, and toxins (reviewed in Fine et al., 2006). Specifically, 2 protein toxins have been described for A. actinomycetemcomitans, cytolethal distending toxin (CDT) (Shenker et al., 1999, 2005) and leukotoxin (LtxA) (Kolodrubetz et al., 1989; Lally et al., 1989b). Both proteins are thought to play a role in immune evasion, but may differ in their target-cell specificity and pattern of expression during disease. This review will focus on the biology of LtxA, with an emphasis on more recent studies.
The ability of A. actinomycetemcomitans extracts to cause the death of leukocytes was first described 30 years ago (Baehni et al., 1979, 1981). Soon after this observation, the toxin was isolated from the bacterium (Tsai et al., 1979, 1984). It was shown that leukotoxic activity was due to a protein from A. actinomycetemcomitans, leukotoxin. The DNA sequence of the leukotoxin gene (ltxA) was revealed by two different groups at about the same time (Kolodrubetz et al., 1989; Lally et al., 1989a; Kraig et al., 1990), and it was found to be similar to other RTX (repeat in toxin) toxin genes. The LtxA protein is ~113 kDa (based on amino acid sequence), shares ~51% amino acid identity with E. coli alpha-hemolysin, and is 43% identical to Mannheimia haemolytica leukotoxin. The RTX toxin family of proteins includes membrane-active toxins that are found in many Gram-negative pathogens. While there are similarities among the RTX toxins of Gram-negative bacteria, important differences exist, such as host cell specificity and mechanisms by which they interact with and affect host cells.
RTX Toxins
While none of the RTX toxin three-dimensional structures has yet been determined, different domains of the molecule have been mapped, based on experimental evidence and modeling of proteins with similar sequences. At the N-terminus of the toxin are amphipathic helices, and analysis of the data suggests that these helices interact with the host cell membrane and receptor (Lally et al., 1999). In the middle of the protein are 2 lysine residues that are covalently modified with fatty acid residues (discussed below and reviewed in Stanley et al., 1998). RTX toxins received their designation based on the nonapeptide glycine-rich repeats that occur in the proteins and are responsible for binding calcium ions. Calcium is required for activity of RTX toxins. One calcium ion binds each repeat, and binding causes the formation of a “spring-like” beta-superhelix structure (Baumann et al., 1993). These repeats have recently been shown also to play a role in interaction with the host cell surface (Sanchez-Magraner et al., 2007), and LtxA has been reported to have 12 such repeats (Kraig et al., 1990). At the extreme C-terminus is a ~100-amino-acid domain that is involved in secretion of the toxin by a type I secretion system (discussed below).
The ltx Promoter
It was recognized that A. actinomycetemcomitans exhibits 2 different leukotoxic phenotypes: minimally leukotoxic (652 strains) and highly leukotoxic (JP2 strains) (Spitznagel et al., 1991). Highly leukotoxic strains produced more LtxA protein and ltx mRNA than minimally leukotoxic strains, and an important discovery was the revelation of 2 different types of promoters that control the expression of ltx genes (Brogan et al., 1994). DNA sequence analysis of the leukotoxin promoter regions from these 2 types of strains revealed that the highly leukotoxic strain was missing ~500 bp at the 3′ end. Thus, the minimally leukotoxic strain harbors a full-length (~1 kb) promoter region, while the highly leukotoxic strain has a truncation (Fig. 1). Leukotoxin promoters are now referred to as either JP2-type or 652-type.
Figure 1.

Leukotoxin operon from the 652 strain harboring the full-length promoter region and JP2 strain with a 530-bp deletion at the 3′ end of the promoter region.
The reason for greater production of LtxA in JP2-like strains is still not completely understood. A logical hypothesis is that the region missing from the JP2-type promoter normally binds a regulatory protein that regulates transcription of the downstream ltx genes. In 2003, Mitchell et al. identified a factor that binds to the ltx promoter region, but the nature of that protein has not yet been identified. Hence, a ltx repressor protein has thus far remained elusive. Alternatively, the deleted region may result in a shorter distance between the RNA polymerase binding site and ltx structural genes and, consequently, increased levels of transcription.
In 1999, He et al. identified, in Japanese individuals, highly leukotoxic strains of A. actinomycetemcomitans that did not possess the JP2 promoter. The promoter region in these strains was 1926 bp in length and contained a DNA transposable element (IS1301) that was required for high leukotoxicity. More recently, Schaeffer et al. (2008) characterized the mechanism that leads to overproduction of LtxA in these strains. They reported that both the transposase promoter and a protein encoded by an open reading frame just upstream of ltxC (OrfA) are required for maximal transcriptional activity of the operon. They proposed that OrfA may act as a regulator protein by binding near the -35 sequence to cause increased transcription. Ongoing studies to characterize the ltx promoter region should reveal mechanisms by which A. actinomycetemcomitans controls the expression of LtxA in all strains.
Secretion of LtxA
For a long time, LtxA was considered to be the only RTX toxin not secreted from bacterial cells, and instead remained associated with the bacterial cell surface. Hence, it was also assumed that a protein required for secretion of other RTX toxins, TolC, was missing in A. actinomycetemcomitans. In 2000, it was reported that LtxA was secreted from A. actinomycetemcomitans cells (Kachlany et al., 2000). A. actinomycetemcomitans actively transports LtxA from the cell, and this secretion is sensitive to numerous factors, including the way the growth medium is prepared, the age of the medium, and the age of the growing culture. All strains that were examined to date secrete LtxA, regardless of their serotype or ltx promoter type (Diaz et al., 2006).
In addition to being released into the cell-free supernatant, LtxA has been detected in lipid vesicles that appear to be derived from the outer bacterial membrane (Kato et al., 2002) as well as attached to the cell surfaces of bacteria (Berthold et al., 1992; Johansson et al., 2003; Diaz et al., 2006). Ohta et al. (1991) found that LtxA could be released from whole cells by treatment with nucleases, suggesting that nucleic acids may also play a role in binding of LtxA to the bacterial cell surface. The significance of the membrane-bound form of the toxin is unknown, but other bacteria ‘package’ their toxins in vesicles as a mechanism of toxin delivery to host cells (Kesty et al., 2004; Balsalobre et al., 2006).
The genes required for the secretion of E. coli HlyA have been well-characterized and serve as a model for related systems (Schlor et al., 1997; Holland et al., 2005). HlyA is secreted via a type I secretion system that uses an uncleaved C-terminal secretion signal. The type I secretion apparatus in E. coli includes 3 different proteins that traverse the inner membrane (HlyB), periplasm (HlyD), and outer membrane (TolC) to form a continuous channel from the cytosol to the extracellular environment. Therefore, proteins secreted via type I secretion do not assume a periplasmic intermediate.
The sequences of the genes, ltxB and ltxD, were published in 1990, and their function was presumed based on sequence similarity and mutational experiments (Guthmiller et al., 1990a,b, 1995). In addition, Lally et al. (1991) showed that LtxB and LtxD are involved in the translocation and insertion of LtxA into the membrane of E. coli. However, because LtxA secretion was only recently revealed in A. actinomycetemcomitans, little is known about the native secretion process. In an attempt at full characterization of the secretion system for A. actinomycetemcomitans LtxA without bias, a random mutagenesis screen was carried out on blood agar plates (Isaza et al., 2008). Non-hemolytic mutants were chosen for study, since these bacteria have a defect in some aspect of LtxA biosynthesis (see below). Through this screen, we identified the secretion proteins LtxB, LtxD, and a third protein, TdeA (for Toxin and Drug Export), which we discovered at about the same time using an independent method (Crosby and Kachlany, 2007).
All 3 proteins—LtxB, LtxD, and TdeA—are required for the secretion of LtxA (Crosby and Kachlany, 2007; Isaza et al., 2008). LtxB is predicted to localize to the inner membrane (IM) and interact with LtxD, which is attached to the periplasmic side of the IM. LtxB has a predicted ATP-binding and hydrolysis site, which would provide energy for the secretion process. Based on the model (Fig. 2), LtxA interacts with LtxB in the cytosol, causing a conformational change in LtxD to allow contact between LtxD and TdeA in the outer membrane. TdeA is a TolC-like protein that likely forms a trimeric pore in the outer membrane. In addition, Gallant et al. (2008) recently reported that an inner membrane protein, MorC, is involved in LtxA secretion, but not transcription. MorC affects the structure of the bacterial outer membrane, and loss of MorC resulted in a defect in LtxA secretion. The mechanism by which MorC influences LtxA production is not known; however, MorC may interact directly with other secretion components, or its general effect on the cell membrane could affect assembly of the type I secretion complex.
Figure 2.
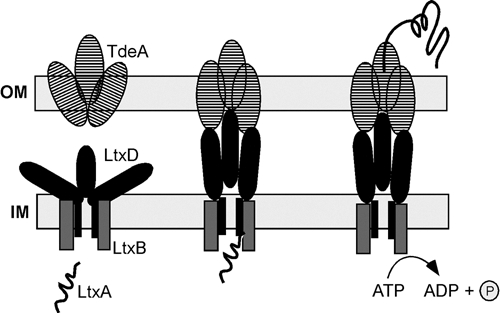
Type I secretion system for export of LtxA from A. actinomycetemcomitans. The model is based on experimental evidence and data from other systems. Reproduced in modified form from Kuhnert and Christensen (2008) with permission.
Like E. coli mutants of tolC (Wandersman and Delepelaire, 1990; Nishino et al., 2003, 2006; Tamura et al., 2005), A. actinomycetemcomitans tdeA mutants are significantly more sensitive to antimicrobial agents than are wild-type strains (Crosby and Kachlany, 2007). The reason for this is that TdeA is also part of one or more drug efflux pumps, similar to other TolC proteins. A mechanism used by A. actinomycetemcomitans to resist antibiotics and other toxic agents is the active export of these agents after they enter the cell, and drug efflux pumps are responsible for this process. Thus, targeting TdeA would render bacteria not only defective for LtxA secretion, but also highly sensitive to conventional antimicrobial agents, and could be an effective strategy for targeting A. actinomycetemcomitans infections in the future.
Regulation of Leukotoxicity
While an ltx regulatory protein has not yet been characterized, numerous external factors are known to affect the production or activity of LtxA. These are discussed below.
Carbohydrates
In 2001, Inoue et al. reported that the level of fermentable sugars regulates the production of LtxA. They found that growth in a fructose chemostat resulted in low levels of LtxA production and decreased levels of intracellular cAMP. More recently, the genetic system that is responsible for the regulation of ltxA production by certain sugars has been identified (Isaza et al., 2008). The connection between sugars and cAMP is through the phosphotransferase system (PTS). The PTS is responsible for the transfer of phosphate to sugars during their active transport into the cell. PTS mutants (ptsH and ptsI) were attenuated for LtxA production, and cAMP levels in these mutants were lower compared with those in wild-type strains. Interestingly, wild-type production of LtxA could be restored in the PTS mutants by providing exogenous cAMP in the culture medium. In other bacteria, cAMP is able to regulate gene expression by binding to cAMP receptor protein (CRP), which binds to a CRP binding site on the DNA. A CRP binding site has not been identified in the ltx operon (Inoue et al., 2001), and so whether A. actinomycetemcomitans CRP binds to a different consensus sequence or acts through a novel mechanism is not yet clear.
Fatty Acid Modification
A unique property of RTX toxins is the covalent attachment of fatty acid residues to the protein. Two other RTX toxins, E. coli HlyA and B. pertussis CyaA, have been shown to be modified with fatty acid residues at internal lysines. Modification is required for full activity of these toxins, and the fatty acid residues are proposed to play a role in the insertion of toxin into the lipid membrane of the host cell (Stanley et al., 1994, 1998). Recently, it was reported that A. actinomycetemcomitans LtxA is also modified at two internal lysine residues; however, the nature of the fatty acids is currently unknown (Balashova et al., 2009). As in E. coli and B. pertussis, the ‘c’ gene of the operon (ltxC) is responsible for carrying out this modification (Lally et al., 1994; Balashova et al., 2009). An A. actinomycetemcomitans ltxC mutant was able to secrete LtxA at levels similar to the wild-type strain, suggesting that modification is not required for secretion. As expected, LtxA from the ltxC mutant did not kill cells, nor did it cause an elevation in intracellular calcium, an event that occurs early during LtxA-mediated intoxication of a cell.
Iron
A unique feature of LtxA is the high specificity for human and Old World primate WBCs (Taichman et al., 1980, 1987; Simpson et al., 1988). It was noted early on that LtxA can target WBCs, but not RBCs (Tsai et al., 1979, 1984), and in the literature, A. actinomycetemcomitans is listed as being non-beta-hemolytic (Bergey and Holt, 1994; Avila-Campos, 1995). However, recently, it was reported that A. actinomycetemcomitans exhibits beta-hemolysis on certain types of growth media (Balashova et al., 2006a). Mutants lacking LtxA are non-hemolytic, and purified LtxA has hemolytic properties. Interestingly, the ability of A. actinomycetemcomitans colonies to appear beta-hemolytic is dependent on the type of medium used (Fig. 3) and may explain why hemolysis was previously not observed (Balashova et al., 2006a). In contrast to cytolysis of WBCs, hemolysis appears to be non-specific, since human, sheep, and horse RBCs underwent lysis by LtxA, and hemolysis requires a higher dose of LtxA than the killing of WBCs. The nature of the RBC receptor for LtxA is unknown, but is the focus of ongoing studies in our laboratory.
Figure 3.

Hemolytic properties of A. actinomycetemcomitans on different growth media. The names on the left indicate different strains of A. actinomycetemcomitans, and the letters refer to the media. All media contained 5% sheep blood. Lane A, Columbia agar from Accumedia; lane B, Columbia agar from BBL-Becton Dickinson; lane C, Trypticase soy agar from BBL-Becton Dickinson; lane D, AAGM. Reproduced with permission from Balashova et al. (2006a).
It is not yet known why LtxA-mediated hemolysis is dependent on medium type, but one explanation is that the growth medium affects the expression or activity of LtxA. Another possibility is that the medium somehow “protects” the RBCs, rendering them resistant to the hemolytic effects of LtxA. The fact that purified LtxA is able to mediate lysis of washed RBCs suggests that the growth medium does not render RBCs sensitive to LtxA.
The discovery that LtxA could cause lysis of RBCs raised new questions about other potential functions of LtxA. A. actinomycetemcomitans has been detected in the blood, and the bacterium is not only an oral pathogen, but can also cause numerous systemic diseases, most notably, infective endocarditis (Das et al., 1997). While in the blood and heart tissue, A. actinomycetemcomitans could acquire iron by lysis of RBCs to release heme and hemoglobin (Balashova et al., 2006a). Because there is essentially no free iron available in the body, bacteria have evolved several ways of “stealing” iron from host iron-binding proteins. These include the production of siderophores and proteins that can metabolize heme and hemoglobin to release iron. Nagata et al. (2002) reported that at least one strain of A. actinomycetemcomitans can bind hemoglobin; but, interestingly, JP2 strains, in contrast to 652 strains, do not produce a functional hemoglobin-binding protein (HgpA) and can thus not utilize hemoglobin as an iron source (Hayashida et al., 2002). In addition, A. actinomycetemcomitans is unable to obtain iron from human transferrin or lactoferrin (Winston et al., 1993; Hayashida et al., 2002) and does not produce siderophores (Winston et al., 1993; Rhodes et al., 2005). Thus, JP2 strains have an impaired ability to utilize complexed iron from host proteins. We have proposed that perhaps high leukotoxicity (and hence deletion of the promoter region) was strongly selected for in these strains that lacked HgpA, resulting in the JP2 phenotype (Balashova et al., 2006b). Increased lysis of RBCs would result in greater release of heme and hemoglobin and a more favorable chance that some could be used by the bacterium.
If LtxA plays a role in iron acquisition, one possible scenario is that iron could play a regulatory role in the production of LtxA. Indeed, iron regulates secretion, not transcription, of LtxA (Balashova et al., 2006b). In the presence of excess iron, but not other metal ions, LtxA remains intracellular; but under iron-limiting conditions, LtxA is essentially fully secreted. Controlling the secretion step rather than transcription allows the bacterium to respond rapidly to changes in the environment, since transcription and translation of a protein require more time to complete than just secretion. Thus, the data are consistent with the hypothesis that LtxA also plays a role in iron acquisition during infection. The mechanism by which iron regulates secretion is not known, but iron may interact with TdeA to cause a decrease in the pore size to prevent the passage of LtxA out of the cell (Crosby and Kachlany, 2007).
Superoxide Dismutase
After being secreted, LtxA appears to be stabilized by interaction with another A. actinomycetemcomitans protein, Cu,Zn superoxide dismutase (SOD). SOD is a bacterial protein that is required for the detoxification of the superoxide radicals that are generated during a respiratory burst by host cells. SOD interacts with LtxA, and SOD mutants are highly sensitive to superoxide radicals (Balashova et al., 2007). In addition, degradation of LtxA protein occurs more rapidly after exposure to reactive oxygen species (ROS) and reactive nitrogen species (RNS). It has been reported that LtxA and other RTX toxins induce the production of ROS from host cells (Bhakdi and Martin, 1991; Yamaguchi et al., 2001). Hence, during intoxication of host cells, LtxA would likely be exposed to the damaging effects of its own doing. Interaction between LtxA and SOD may result in the stabilization of the toxin during the LtxA-mediated respiratory burst. It is not yet clear where the interaction between LtxA and SOD takes place. It is possible that SOD is secreted at later times, after LtxA has had a chance to act on host cells. Alternatively, SOD may be released from cells that have undergone lysis. SOD is also found associated with the membrane in A. actinomycetemcomitans (Fletcher et al., 2001; Balashova et al., 2007), and so it is possible that LtxA interacts with SOD at the cell surface.
LtxA SOD may also have a second function. Interestingly, Cu,Zn SOD from Haemophilus ducreyi has been shown to bind heme, and this interaction may not only provide a source of iron for the bacterium, but it may also serve as a mechanism for the detoxification of toxic oxyradicals formed from heme iron and oxygen (Pacello et al., 2001). To date, A. actinomycetemcomitans SOD has not been shown to bind heme, although this remains an interesting possibility, given that LtxA can also cause lysis of RBCs. A summary of how different factors regulate leukotoxicity and the proteins required for LtxA production is shown in Fig. 4.
Figure 4.
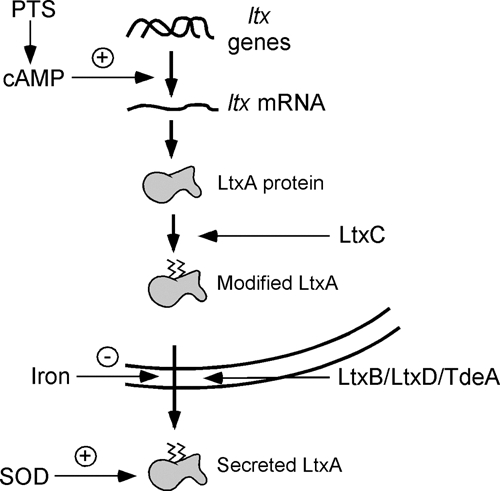
Regulation of LtxA biosynthesis. Activation of the PTS results in the production of cAMP, which has a positive effect on the expression of ltx genes. LtxA protein is modified with fatty acid residues by LtxC and then secreted through a channel consisting of LtxB, LtxD, and TdeA. The presence of iron blocks the secretion of LtxA. After being secreted into the environment, LtxA may be stabilized by SOD and protected from the ROS and RNS released by host cells.
In addition, it has been shown that LtxA is regulated by other environmental factors, such as pH and oxygen. Lower pH values (6.0-7.0) and anaerobic conditions have positive effects on LtxA production and/or stability (Hritz et al., 1996; Ohta et al., 1996; Kolodrubetz et al., 2003), and these have been discussed in greater detail in a previous review (Inoue et al., 2006). Like many other bacterial virulence factors, LtxA is also regulated by quorum-sensing. Quorum-sensing is the mechanism by which bacteria can detect the numbers of other bacteria in their environment by chemical signals. Fong et al. (2001) reported that the autoinducer, LuxS, increased production of LtxA. Interestingly, LuxS also increased production of AfuA, a protein involved in iron acquisition (Fong et al., 2001).
Interaction with Host Cells
The primary role of LtxA is in immune evasion. The immune response against A. actinomycetemcomitans after it migrates into the periodontal pocket is the infiltration of PMNs to engulf and destroy the bacterium. LtxA is the counter-defense for A. actinomycetemcomitans. The goal of a pathogen is not to eliminate its host, but rather to live in symbiosis with it. Hence, A. actinomycetemcomitans is an outstanding example of a successful pathogen. By targeting only certain WBCs, the bacterium ensures that the effects of the toxin are limited to only the cells that are most immunologically relevant. In 1997, Lally et al. identified leukocyte function antigen-1 (LFA-1) as the cellular receptor for LtxA. LFA-1 is expressed only on cells of hematopoietic origin; hence, LFA-1 serves as an ideal marker for WBCs.
LFA-1 is a heterodimer composed of 2 proteins, CD11a and CD18, which assemble on the surfaces of essentially all human WBCs at some level. While both molecules are required for LtxA to interact with cells, it was found that CD18 confers species specificity on the toxin (Dileepan et al., 2007). Cells expressing bovine CD11a/human CD18 were sensitive to LtxA, but human CD11a/bovine CD18 cells were resistant. Further, the cysteine-rich tandem repeats encompassing integrin-epidermal growth-factor-like domains 2, 3, and 4 of the extracellular region of human CD18 are critical for conferring susceptibility to LtxA (Dileepan et al., 2007). In addition, Kieba et al. (2007) showed that a 128-amino-acid domain encompassing the beta-propeller domain of CD11a is important for recognition by LtxA. Thus, LtxA comes into contact with both the CD11a and CD18 domains of LFA-1.
The normal function of LFA-1 is to mediate adhesion to vascular endothelial cells and then migration and extravasation of immune cells during infection (Hogg et al., 2004; Kinashi, 2005). LFA-1 assumes at least two conformations, a resting state and an activated state (also called low-affinity and high-affinity states, respectively). In the resting state, LFA-1 is tucked in toward the cell surface and unavailable for binding to other molecules. However, upon activation of the cell by cytokines (such as during an infection), LFA-1 is extended and becomes available for binding. The natural ligand for LFA-1 is intercellular adhesion molecule-1 (ICAM-1), located on the surfaces of vascular endothelial cells. Activated LFA-1 binds ICAM-1 and then mediates the migration of the cell out of the blood vessel and into infected tissue. Thus, cells that are most immunocompetent are “tagged” with an activated form of LFA-1.
In a recent publication, we examined the relationship between levels of surface LFA-1 and sensitivity to LtxA, and we observed a direct correlation (Kachlany et al., 2009). For example, THP-1 cells had highest levels of CD11a and CD18 on their surfaces, as determined by flow cytometry, and were most sensitive to LtxA. Additionally, LtxA preferentially targets cells with activated LFA-1. Given the natural role of the toxin in immune evasion, this observation makes logical sense, since the WBCs that LtxA encounters will have been activated. A model showing the interplay between A. actinomycetemcomitans LtxA and the host immune system in the gingival crevice is shown in Fig. 5. It is not known if the actual levels of LtxA produced by 652 strains in vivo are as high as JP2 strains. Examination of gingival crevicular fluid or saliva in persons with LAP may address this issue. Also, bacterial cells can interact directly with neutrophils, and thus, cell-bound LtxA would be important for mediating cell death via direct contact (Permpanich et al., 2006).
Figure 5.

Host response to A. actinomycetemcomitans infection in the mouth. Interaction between bacterial cells and gingival tissue causes the release of cytokine signals by lymphocytes and other WBCs. Cytokine signaling results in the activation of circulating WBCs in the underlying vasculature. Activation results in the immunological priming of WBCs and a conformational change of LFA-1 from a low-affinity state to a high-affinity, exposed state. High-affinity, activated LFA-1 can bind ICAM-1 expressed by vascular endothelial cells. Interaction between ICAM-1 and LFA-1 results in the migration of WBCs into the infected tissue via the process of diapedesis. In the gingival tissue, WBCs are incapacitated by secreted LtxA binding to activated LFA-1, allowing A. actinomycetemcomitans to colonize, persist, and invade deeper into the periodontal pocket.
While infiltration of PMNs represents the initial response in the periodontal pocket to A. actinomycetemcomitans infection, and thus would be considered the intended biologic target for LtxA, other WBCs are equally susceptible to LtxA in vitro (Taichman et al., 1980, 1987; Kachlany et al., 2009). However, because LtxA would be delivered locally by the bacteria, other WBC types that do not access the diseased site in the body may not be affected by the toxin.
At high doses, LtxA destroys cells by forming pores in the membrane and causing necrosis; at lower (and probably more physiologically representative) doses, LtxA induces apoptosis (Korostoff et al., 1998, 2000). The mechanism by which LtxA interacts with and ultimately kills host cells is poorly understood. Based on the well-accepted model, the initial step involves binding of LtxA to host cells in a non-specific, reversible manner. Fong et al. (2006) has shown that the first step to cellular intoxication by LtxA is an increase in intracellular calcium levels, even before interaction with the LFA-1 receptor. The mechanism by which this occurs is unknown. The rise in calcium then leads to activation of calpain, which cleaves talin. LFA-1 is then released from talin protein and clusters into lipid rafts. LtxA interacts with clustered LFA-1, and this interaction may then stimulate an integrin signaling pathway. Morova et al. (2008) recently reported that RTX toxins, including LtxA, recognize oligosaccharide subunits on LFA-1; however, LtxA was still partly active, even after the oligosaccharides were removed from WBCs with glycosidases, suggesting that either deglycosylation was not complete or that LtxA may recognize other entities as well. Bound LtxA destroys host cells by apoptosis (Mangan et al., 1991; Korostoff et al., 1998; Lally et al., 1999; Yamaguchi et al., 2001) or activation of caspase 1 through a process that differs from classic apoptosis (Kelk et al., 2003). Others have shown that LtxA affects the mitochondrial membrane potential and causes the release of cytochrome c from the mitochondrial intermembrane space and into the cytosol, where it likely activates an apoptosis cascade (Korostoff et al., 2000; Yamaguchi et al., 2001). A proposed model of LtxA action on host cells is shown in Fig. 6. The question marks indicate proteins or pathways that are currently undefined. Structural changes that LtxA necessarily undergoes have been removed for clarity.
Figure 6.

A model for the mechanism of how LtxA interacts with host cells and ultimately causes cellular intoxication by apoptosis. See text for details.
Relevance to Disease
Because of the high specificity of LtxA for primate WBCs, it is difficult to test the contribution of this putative virulence factor in well-established mouse and rat models for periodontal disease (Evans et al., 1992; Baker et al., 2002; Niederman et al., 2002; Lalla et al., 2003; Schreiner et al., 2003; Graves et al., 2008; Lee et al., 2009; Polak et al., 2009). However, analysis of clinical data indicates that LtxA is an important virulence factor for A. actinomycetemcomitans. For example, highly leukotoxic strains (JP2 strains) are more often associated with severe disease than minimally leukotoxic strains (652 strains), especially in individuals of African-American descent (Haubek et al., 1997, 2001, 2008; Macheleidt et al., 1999; Haraszthy et al., 2000). In a recent study (Haubek et al., 2008) in Morocco, individuals who harbored the JP2 strains had significantly increased risk of periodontal attachment loss than those with the 652 strains. However, minimally leukotoxic strains are also found in persons with LAP (Kaplan et al., 2002; Fine et al., 2007). Fine et al. (2007) studied adolescents in Newark, NJ, and found that the majority of individuals who developed bone loss did not harbor the JP2 strain in their mouths during the study period. Thus, these authors concluded that it is currently not possible to determine if the JP2 strains are more virulent than other minimally leukotoxic strains. It is also important to note that the locations of these studies and ethnicity of the populations differ and could account for clonal variations. However, a rational biological explanation for the observations by Fine et al. (2007) is that LtxA is up-regulated to higher levels in the mouth. Thus, strains with the 652 promoter are minimally leukotoxic under laboratory conditions, but produce levels similar to those of JP2 strains in the physiological environment. Studies to investigate the actual levels of LtxA in the periodontal pockets of individuals with disease could test this hypothesis.
Potential Therapeutic Use of LtxA
While LtxA has been studied solely because of the threat it poses to the host immune system, we asked if the unique and natural properties of the toxin could be exploited for therapeutic use. Several bacterial toxins have been used clinically, including Clostridium botulinum neurotoxin (BOTOX) for the treatment of neuromuscular disorders (Jankovic, 2004) and Corynebacterium diphtheriae toxin (ONTAK) for the treatment of T-cell lymphoma (Olsen et al., 2001; Duvic et al., 2002).
Hematologic malignancies include leukemia, lymphoma, and myeloma and are defined as cancer of WBCs. It has been shown that LFA-1 can be up-regulated in many leukemias and lymphomas (Inghirami et al., 1988; Horst et al., 1991; Bechter et al., 1999). Thus, malignant cells may be more sensitive to LtxA-mediated cytotoxicity than normal counterparts. Indeed, healthy PBMCs are relatively resistant to LtxA, while primary PBMCs from leukemia patients are highly sensitive to the toxin (Kachlany et al., 2009). In addition, LtxA is highly effective in a SCID mouse xenograft model for human leukemia (Fig. 7), and the toxin shows targeted activity and safety when administered intravenously to a non-human primate (Kachlany et al., 2009). In the rhesus macaque, a relatively low dose of LtxA (22 μg/kg) caused a decrease in WBCs, but no change in hemoglobin values, RBCs, or platelets, or other diagnostic markers of toxicity. Thus, while conventional chemotherapeutic agents show little specificity and high general toxicity, LtxA may represent novel targeted biotherapy for WBC disorders, and LtxA is currently in pre-clinical pharmaceutical development.
Figure 7.
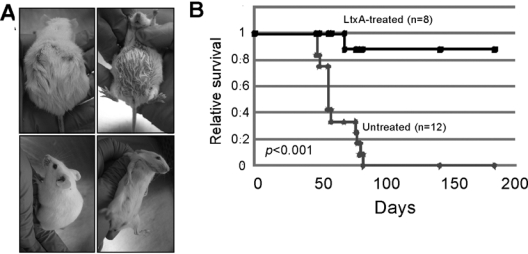
Efficacy of LtxA therapy in a humanized mouse model for leukemia. (A) SCID mice were injected with the human leukemia cell line, HL-60, in the peritoneal cavity and then remained untreated (top) or were treated with 3 doses of LtxA (bottom). (B) Kaplan-Meier survival plot shows that 7/8 leukemic mice that received LtxA were long-term survivors and remained disease-free, while the untreated leukemic mice all succumbed to disease. Reproduced with permission from Kachlany et al. (2009).
While leukemia is unrelated to LAP and oral disease, we believe that understanding the latter could help in the development of LtxA as a therapeutic agent for the former. Just as dispersin B from A. actinomycetemcomitans is being investigated for the treatment of wound infections (Izano et al., 2007; Kaplan, 2009), we hope to employ the bacterium for yet another of its natural products. In essence, because of exquisite co-evolution between pathogen and human host, the bacterium has carried out the “research and discovery” phase of the pharmaceutical development process, and we plan to take advantage of the work A. actinomycetemcomitans has already done for us.
Summary
Since the discovery of A. actinomycetemcomitans leukotoxic activity 30 years ago, much has been learned about the protein, LtxA, which is responsible for this important pathogenic property. Knowledge from other RTX toxins has also shed light on the biology of LtxA; however, important differences exist between LtxA and other RTX toxins, such as regulation of expression and secretion, target cell specificity, host receptor, and structural properties. Thus, simple extrapolation of knowledge from other systems may not be possible in many cases. Important questions that still remain include the mechanism of LtxA action on host cells, the physiological contribution of LtxA to disease, and how LtxA recognizes and targets RBCs. Furthermore, because of the natural toxicity and specificity of the toxin for certain WBCs, LtxA may show significant therapeutic potential for the treatment of WBC diseases.
Acknowledgments
The author thanks the numerous members of his laboratory and collaborators who have contributed to our studies described here.
Footnotes
Work in the author’s laboratory has been generously supported by grants from the National Institute of Dental and Craniofacial Research, National Institute of Allergy and Infectious Diseases, New Jersey Commission on Science and Technology, and the Foundation of UMDNJ.
S.C.K. owns stock in a company that has licensed patents from UMDNJ for the clinical use of leukotoxin.
References
- Avila-Campos MJ. (1995). Haemolytic activity of Actinobacillus actinomycetemcomitans strains on different blood types. Rev Inst Med Trop São Paulo 37:215-217 [DOI] [PubMed] [Google Scholar]
- Baehni P, Tsai CC, McArthur WP, Hammond BF, Taichman NS. (1979). Interaction of inflammatory cells and oral microorganisms. VIII. Detection of leukotoxic activity of a plaque-derived Gram-negative microorganism. Infect Immun 24:233-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehni PC, Tsai CC, McArthur WP, Hammond BF, Shenker BJ, Taichman NS. (1981). Leukotoxic activity in different strains of the bacterium Actinobacillus actinomycetemcomitans isolated from juvenile periodontitis in man. Arch Oral Biol 26:671-676 [DOI] [PubMed] [Google Scholar]
- Baker PJ, Howe L, Garneau J, Roopenian DC. (2002). T cell knockout mice have diminished alveolar bone loss after oral infection with Porphyromonas gingivalis. FEMS Immunol Med Microbiol 34:45-50 [DOI] [PubMed] [Google Scholar]
- Balashova NV, Crosby JA, Al Ghofaily L, Kachlany SC. (2006a). Leukotoxin confers beta-hemolytic activity to Actinobacillus actinomycetemcomitans. Infect Immun 74:2015-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashova NV, Diaz R, Balashov SV, Crosby JA, Kachlany SC. (2006b). Regulation of Aggregatibacter (Actinobacillus) actinomycetemcomitans leukotoxin secretion by iron. J Bacteriol 188:8658-8661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashova NV, Park DH, Patel JK, Figurski DH, Kachlany SC. (2007). Interaction between leukotoxin and Cu, Zn superoxide dismutase in Aggregatibacter actinomycetemcomitans. Infect Immun 75:4490-4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashova NV, Shah C, Patel JK, Megalla S, Kachlany SC. (2009). Aggregatibacter actinomycetemcomitans LtxC is required for leukotoxin activity and initial interaction between toxin and host cells. Gene 443:42-47 [DOI] [PubMed] [Google Scholar]
- Balsalobre C, Silvan JM, Berglund S, Mizunoe Y, Uhlin BE, Wai SN. (2006). Release of the type I secreted alpha-haemolysin via outer membrane vesicles from Escherichia coli. Mol Microbiol 59:99-112 [DOI] [PubMed] [Google Scholar]
- Baumann U, Wu S, Flaherty KM, McKay DB. (1993). Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium binding parallel beta roll motif. EMBO J 12:3357-3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechter OE, Eisterer W, Dirnhofer S, Pall G, Kuhr T, Stauder R, et al. (1999). Expression of LFA-1 identifies different prognostic subgroups in patients with advanced follicle center lymphoma (FCL). Leuk Res 23:483-488 [DOI] [PubMed] [Google Scholar]
- Beck JD, Eke P, Heiss G, Madianos P, Couper D, Lin D, et al. (2005). Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation 112:19-24 [DOI] [PubMed] [Google Scholar]
- Bergey DH, Holt JG. (1994). Bergey’s manual of determinative bacteriology. 9th ed. Baltimore, MD, USA: Lippincot, Williams & Wilkins [Google Scholar]
- Berthold P, Forti D, Kieba IR, Rosenbloom J, Taichman NS, Lally ET. (1992). Electron immunocytochemical localization of Actinobacillus actinomycetemcomitans leukotoxin. Oral Microbiol Immunol 7:24-27 [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Martin E. (1991). Superoxide generation by human neutrophils induced by low doses of Escherichia coli hemolysin. Infect Immun 59:2955-2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. (1994). Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect Immun 62:501-508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby JA, Kachlany SC. (2007). TdeA, a TolC-like protein required for toxin and drug export in Aggregatibacter (Actinobacillus) actinomycetemcomitans. Gene 388:83-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Badley AD, Cockerill FR, Steckelberg JM, Wilson WR. (1997). Infective endocarditis caused by HACEK microorganisms. Annu Rev Med 48:25-33 [DOI] [PubMed] [Google Scholar]
- Diaz R, Ghofaily LA, Patel J, Balashova NV, Freitas AC, Labib I, et al. (2006). Characterization of leukotoxin from a clinical strain of Actinobacillus actinomycetemcomitans. Microb Pathog 40:48-55 [DOI] [PubMed] [Google Scholar]
- Dileepan T, Kachlany SC, Balashova NV, Patel J, Maheswaran SK. (2007). Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infect Immun 75:4851-4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M, Kuzel TM, Olsen EA, Martin AG, Foss FM, Kim YH, et al. (2002). Quality-of-life improvements in cutaneous T-cell lymphoma patients treated with denileukin diftitox (ONTAK). Clin Lymphoma 2:222-228 [DOI] [PubMed] [Google Scholar]
- Evans RT, Klausen B, Ramamurthy NS, Golub LM, Sfintescu C, Genco RJ. (1992). Periodontopathic potential of two strains of Porphyromonas gingivalis in gnotobiotic rats. Arch Oral Biol 37:813-819 [DOI] [PubMed] [Google Scholar]
- Fine DH, Kaplan JB, Kachlany SC, Schreiner HC. (2006). How we got attached to Actinobacillus actinomycetemcomitans: a model for infectious diseases. Periodontol 2000 42:114-157 [DOI] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. (2007). Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol 45:3859-3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Nair SP, Ward JM, Henderson B, Wilson M. (2001). Analysis of the effect of changing environmental conditions on the expression patterns of exported surface-associated proteins of the oral pathogen Actinobacillus actinomycetemcomitans. Microb Pathog 30:359-368 [DOI] [PubMed] [Google Scholar]
- Fong KP, Chung WO, Lamont RJ, Demuth DR. (2001). Intra- and interspecies regulation of gene expression by Actinobacillus actinomycetemcomitans LuxS. Infect Immun 69:7625-7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong KP, Pacheco CM, Otis LL, Baranwal S, Kieba IR, Harrison G, et al. (2006). Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cell Microbiol 8:1753-1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant CV, Sedic M, Chicoine EA, Ruiz T, Mintz KP. (2008). Membrane morphology and leukotoxin secretion are associated with a novel membrane protein of Aggregatibacter actinomycetemcomitans. J Bacteriol 190:5972-5980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. (2008). The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol 35:89-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthmiller JM, Kolodrubetz D, Cagle MP, Kraig E. (1990a). Sequence of the lktB gene from Actinobacillus actinomycetemcomitans. Nucleic Acids Res 18:5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthmiller JM, Kraig E, Cagle MP, Kolodrubetz D. (1990b). Sequence of the lktD gene from Actinobacillus actinomycetemcomitans. Nucleic Acids Res 18:5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthmiller JM, Kolodrubetz D, Kraig E. (1995). Mutational analysis of the putative leukotoxin transport genes in Actinobacillus actinomycetemcomitans. Microb Pathog 18:307-321 [DOI] [PubMed] [Google Scholar]
- Haraszthy VI, Hariharan G, Tinoco EM, Cortelli JR, Lally ET, Davis E, et al. (2000). Evidence for the role of highly leukotoxic Actinobacillus actinomycetemcomitans in the pathogenesis of localized juvenile and other forms of early-onset periodontitis. J Periodontol 71:912-922 [DOI] [PubMed] [Google Scholar]
- Haubek D, Dirienzo JM, Tinoco EM, Westergaard J, Lopez NJ, Chung CP, et al. (1997). Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J Clin Microbiol 35:3037-3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Poulsen K, Poulsen S, Benzarti N, Kilian M. (2001). Early-onset periodontitis in Morocco is associated with the highly leukotoxic clone of Actinobacillus actinomycetemcomitans. J Dent Res 80:1580-1583 [DOI] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. (2008). Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet 371:237-242 [DOI] [PubMed] [Google Scholar]
- Hayashida H, Poulsen K, Kilian M. (2002). Differences in iron acquisition from human haemoglobin among strains of Actinobacillus actinomycetemcomitans. Microbiology 148(Pt 12):3993-4001 [DOI] [PubMed] [Google Scholar]
- He T, Nishihara T, Demuth DR, Ishikawa I. (1999). A novel insertion sequence increases the expression of leukotoxicity in Actinobacillus actinomycetemcomitans clinical isolates. J Periodontol 70:1261-1268 [DOI] [PubMed] [Google Scholar]
- Hogg N, Smith A, McDowall A, Giles K, Stanley P, Laschinger M, et al. (2004). How T cells use LFA-1 to attach and migrate. Immunol Lett 92:51-54 [DOI] [PubMed] [Google Scholar]
- Holland IB, Schmitt L, Young J. (2005). Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol Membr Biol 22:29-39 [DOI] [PubMed] [Google Scholar]
- Horst E, Radaszkiewicz T, Hooftman-den Otter A, Pieters R, van Dongen JJ, Meijer CJ, et al. (1991). Expression of the leucocyte integrin LFA-1 (CD11a/CD18) and its ligand ICAM-1 (CD54) in lymphoid malignancies is related to lineage derivation and stage of differentiation but not to tumor grade. Leukemia 5:848-853 [PubMed] [Google Scholar]
- Hritz M, Fisher E, Demuth DR. (1996). Differential regulation of the leukotoxin operon in highly leukotoxic and minimally leukotoxic strains of Actinobacillus actinomycetemcomitans. Infect Immun 64:2724-2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inghirami G, Wieczorek R, Zhu BY, Silber R, Dalla-Favera R, Knowles DM. (1988). Differential expression of LFA-1 molecules in non-Hodgkin’s lymphoma and lymphoid leukemia. Blood 72:1431-1434 [PubMed] [Google Scholar]
- Inoue T, Tanimoto I, Tada T, Ohashi T, Fukui K, Ohta H. (2001). Fermentable-sugar-level-dependent regulation of leukotoxin synthesis in a variably toxic strain of Actinobacillus actinomycetemcomitans. Microbiology 147(Pt 10):2749-2756 [DOI] [PubMed] [Google Scholar]
- Inoue T, Fukui K, Ohta H. (2006). Leukotoxin production by Actinobacillus actinomycetemcomitans. Toxin Reviews 25:1-18 [Google Scholar]
- Isaza MP, Duncan MS, Kaplan JB, Kachlany SC. (2008). Screen for leukotoxin mutants in Aggregatibacter actinomycetemcomitans: genes of the phosphotransferase system are required for leukotoxin biosynthesis. Infect Immun 76:3561-3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izano EA, Wang H, Ragunath C, Ramasubbu N, Kaplan JB. (2007). Detachment and killing of Aggregatibacter actinomycetemcomitans biofilms by dispersin B and SDS. J Dent Res 86:618-622 [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2004). Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry 75:951-957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A, Claesson R, Hanstrom L, Kalfas S. (2003). Serum-mediated release of leukotoxin from the cell surface of the periodontal pathogen Actinobacillus actinomycetemcomitans. Eur J Oral Sci 111:209-215 [DOI] [PubMed] [Google Scholar]
- Kachlany SC, Fine DH, Figurski DH. (2000). Secretion of RTX leukotoxin by Actinobacillus actinomycetemcomitans. Infect Immun 68:6094-6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachlany SC, Schwartz AB, Balashova NV, Hioe CE, Tuen M, Le A, et al. (2009). Anti-leukemia activity of a bacterial toxin with natural specificity for LFA-1 on white blood cells. Leukemia Res [Epub ahead of print] September 9, 2009. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB. (2009). Therapeutic potential of biofilm-dispersing enzymes. Int J Artif Org 32:545-554 [DOI] [PubMed] [Google Scholar]
- Kaplan JB, Schreiner HC, Furgang D, Fine DH. (2002). Population structure and genetic diversity of Actinobacillus actinomycetemcomitans strains isolated from localized juvenile periodontitis patients. J Clin Microbiol 40:1181-1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Kowashi Y, Demuth DR. (2002). Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb Pathog 32:1-13 [DOI] [PubMed] [Google Scholar]
- Kelk P, Johansson A, Claesson R, Hanstrom L, Kalfas S. (2003). Caspase 1 involvement in human monocyte lysis induced by Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun 71:4448-4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. (2004). Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J 23:4538-4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieba IR, Fong KP, Tang HY, Hoffman KE, Speicher DW, Klickstein LB, et al. (2007). Aggregatibacter actinomycetemcomitans leukotoxin requires beta-sheets 1 and 2 of the human CD11a beta-propeller for cytotoxicity. Cell Microbiol 9:2689-2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi T. (2005). Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol 5:546-559 [DOI] [PubMed] [Google Scholar]
- Kolodrubetz D, Dailey T, Ebersole J, Kraig E. (1989). Cloning and expression of the leukotoxin gene from Actinobacillus actinomycetemcomitans. Infect Immun 57:1465-1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodrubetz D, Phillips L, Jacobs C, Burgum A, Kraig E. (2003). Anaerobic regulation of Actinobacillus actinomycetemcomitans leukotoxin transcription is ArcA/FnrA-independent and requires a novel promoter element. Res Microbiol 154:645-653 [DOI] [PubMed] [Google Scholar]
- Korostoff J, Wang JF, Kieba I, Miller M, Shenker BJ, Lally ET. (1998). Actinobacillus actinomycetemcomitans leukotoxin induces apoptosis in HL-60 cells. Infect Immun 66:4474-4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostoff J, Yamaguchi N, Miller M, Kieba I, Lally ET. (2000). Perturbation of mitochondrial structure and function plays a central role in Actinobacillus actinomycetemcomitans leukotoxin-induced apoptosis. Microb Pathog 29:267-278 [DOI] [PubMed] [Google Scholar]
- Kraig E, Dailey T, Kolodrubetz D. (1990). Nucleotide sequence of the leukotoxin gene from Actinobacillus actinomycetemcomitans: homology to the alpha-hemolysin/leukotoxin gene family. Infect Immun 58:920-929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert P, Christensen H, editors (2008). Pasteurellaceae: biology, genomics and molecular aspects. Norfolk, UK: Caister Academic Press [Google Scholar]
- Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, et al. (2003). Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 23:1405-1411 [DOI] [PubMed] [Google Scholar]
- Lally ET, Golub EE, Kieba IR, Taichman NS, Rosenbloom J, Rosenbloom JC, et al. (1989a). Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J Biol Chem 264:15451-15456 [PubMed] [Google Scholar]
- Lally ET, Kieba IR, Demuth DR, Rosenbloom J, Golub EE, Taichman NS, et al. (1989b). Identification and expression of the Actinobacillus actinomycetemcomitans leukotoxin gene. Biochem Biophys Res Commun 159:256-262 [DOI] [PubMed] [Google Scholar]
- Lally ET, Golub EE, Kieba IR, Taichman NS, Decker S, Berthold P, et al. (1991). Structure and function of the B and D genes of the Actinobacillus actinomycetemcomitans leukotoxin complex. Microb Pathog 11:111-121 [DOI] [PubMed] [Google Scholar]
- Lally ET, Golub EE, Kieba IR. (1994). Identification and immunological characterization of the domain of Actinobacillus actinomycetemcomitans leukotoxin that determines its specificity for human target cells. J Biol Chem 269:31289-31295 [PubMed] [Google Scholar]
- Lally ET, Kieba IR, Sato A, Green CL, Rosenbloom J, Korostoff J, et al. (1997). RTX toxins recognize a beta2 integrin on the surface of human target cells. J Biol Chem 272:30463-30469 [DOI] [PubMed] [Google Scholar]
- Lally ET, Hill RB, Kieba IR, Korostoff J. (1999). The interaction between RTX toxins and target cells. Trends Microbiol 7:356-361 [DOI] [PubMed] [Google Scholar]
- Lamell CW, Griffen AL, McClellan DL, Leys EJ. (2000). Acquisition and colonization stability of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in children. J Clin Microbiol 38:1196-1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SF, Andrian E, Rowland E, Marquez IC. (2009). Immune response and alveolar bone resorption in a mouse model of Treponema denticola infection. Infect Immun 77:694-698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys EJ, Griffen AL, Strong SJ, Fuerst PA. (1994). Detection and strain identification of Actinobacillus actinomycetemcomitans by nested PCR. J Clin Microbiol 32:1288-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löe H, Brown LJ. (1991). Early onset periodontitis in the United States of America. J Periodontol 62:608-616 [DOI] [PubMed] [Google Scholar]
- Macheleidt A, Muller HP, Eger T, Putzker M, Fuhrmann A, Zoller L. (1999). Absence of an especially toxic clone among isolates of Actinobacillus actinomycetemcomitans recovered from army recruits. Clin Oral Investig 3:161-167 [DOI] [PubMed] [Google Scholar]
- Mangan DF, Taichman NS, Lally ET, Wahl SM. (1991). Lethal effects of Actinobacillus actinomycetemcomitans leukotoxin on human T lymphocytes. Infect Immun 59:3267-3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, Gao L, Demuth DR. (2003). Positive and negative cis-acting regulatory sequences control expression of leukotoxin in Actinobacillus actinomycetemcomitans 652. Infect Immun 71:5640-5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morova J, Osicka R, Masin J, Sebo P. (2008). RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc Natl Acad Sci USA 105:5355-5360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata H, Ikawa Y, Kuboniwa M, Maeda K, Shizukuishi S. (2002). Characterization of hemoglobin binding to Actinobacillus actinomycetemcomitans. Anaerobe 8:109-114 [DOI] [PubMed] [Google Scholar]
- Newman MG, Socransky SS, Savitt ED, Propas DA, Crawford A. (1976). Studies of the microbiology of periodontosis. J Periodontol 47:373-379 [DOI] [PubMed] [Google Scholar]
- Niederman R, Kelderman H, Socransky S, Ostroff G, Genco C, Kent R, Jr, et al. (2002). Enhanced neutrophil emigration and Porphyromonas gingivalis reduction following PGG-glucan treatment of mice. Arch Oral Biol 47:613-618 [DOI] [PubMed] [Google Scholar]
- Nishino K, Yamada J, Hirakawa H, Hirata T, Yamaguchi A. (2003). Roles of TolC-dependent multidrug transporters of Escherichia coli in resistance to beta-lactams. Antimicrob Agents Chemother 47:3030-3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Latifi T, Groisman EA. (2006). Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol Microbiol 59:126-141 [DOI] [PubMed] [Google Scholar]
- Ohta H, Kato K, Kokeguchi S, Hara H, Fukui K, Murayama Y. (1991). Nuclease-sensitive binding of an Actinobacillus actinomycetemcomitans leukotoxin to the bacterial cell surface. Infect Immun 59:4599-4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Miyagi A, Kato K, Fukui K. (1996). The relationships between leukotoxin production, growth rate and the bicarbonate concentration in a toxin-production-variable strain of Actinobacillus actinomycetemcomitans. Microbiology 142(Pt 4):963-970 [DOI] [PubMed] [Google Scholar]
- Olsen E, Duvic M, Frankel A, Kim Y, Martin A, Vonderheid E, et al. (2001). Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol 19:376-388 [DOI] [PubMed] [Google Scholar]
- Pacello F, Langford PR, Kroll JS, Indiani C, Smulevich G, Desideri A, et al. (2001). A novel heme protein, the Cu, Zn-superoxide dismutase from Haemophilus ducreyi. J Biol Chem 276:30326-30334 [DOI] [PubMed] [Google Scholar]
- Permpanich P, Kowolik MJ, Galli DM. (2006). Resistance of fluorescent-labelled Actinobacillus actinomycetemcomitans strains to phagocytosis and killing by human neutrophils. Cell Microbiol 8:72-84 [DOI] [PubMed] [Google Scholar]
- Polak D, Wilensky A, Shapira L, Halabi A, Goldstein D, Weiss EI, et al. (2009). Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J Clin Periodontol 36:406-410 [DOI] [PubMed] [Google Scholar]
- Rhodes ER, Tomaras AP, McGillivary G, Connerly PL, Actis LA. (2005). Genetic and functional analyses of the Actinobacillus actinomycetemcomitans AfeABCD siderophore-independent iron acquisition system. Infect Immun 73:3758-3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Magraner L, Viguera AR, Garcia-Pacios M, Garcillan MP, Arrondo JL, de la Cruz F, et al. (2007). The calcium-binding C-terminal domain of Escherichia coli alpha-hemolysin is a major determinant in the surface-active properties of the protein. J Biol Chem 282:11827-11835 [DOI] [PubMed] [Google Scholar]
- Schaeffer LM, Schmidt ML, Demuth DR. (2008). Induction of Aggregatibacter actinomycetemcomitans leukotoxin expression by IS1301 and orfA. Microbiology 154(Pt 2):528-538 [DOI] [PubMed] [Google Scholar]
- Schlor S, Schmidt A, Maier E, Benz R, Goebel W, Gentschev I. (1997). In vivo and in vitro studies on interactions between the components of the hemolysin (HlyA) secretion machinery of Escherichia coli. Mol Gen Genet 256:306-319 [DOI] [PubMed] [Google Scholar]
- Schreiner HC, Sinatra K, Kaplan JB, Furgang D, Kachlany SC, Planet PJ, et al. (2003). Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc Natl Acad Sci USA 100:7295-7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker BJ, McKay T, Datar S, Miller M, Chowhan R, Demuth D. (1999). Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol 162:4773-4780 [PubMed] [Google Scholar]
- Shenker BJ, Besack D, McKay T, Pankoski L, Zekavat A, Demuth DR. (2005). Induction of cell cycle arrest in lymphocytes by Actinobacillus actinomycetemcomitans cytolethal distending toxin requires three sub-units for maximum activity. J Immunol 174:2228-2234 [DOI] [PubMed] [Google Scholar]
- Simpson DL, Berthold P, Taichman NS. (1988). Killing of human myelomonocytic leukemia and lymphocytic cell lines by Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun 56:1162-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slots J. (2003). Update on general health risk of periodontal disease. Int Dent J 53(Suppl 3):200-207 [DOI] [PubMed] [Google Scholar]
- Slots J, Ting M. (1999). Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: occurrence and treatment. Periodontol 2000 20:82-121 [DOI] [PubMed] [Google Scholar]
- Spitznagel J, Jr, Kraig E, Kolodrubetz D. (1991). Regulation of leukotoxin in leukotoxic and nonleukotoxic strains of Actinobacillus actinomycetemcomitans. Infect Immun 59:1394-1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Packman LC, Koronakis V, Hughes C. (1994). Fatty acylation of two internal lysine residues required for the toxic activity of Escherichia coli hemolysin. Science 266:1992-1996 [DOI] [PubMed] [Google Scholar]
- Stanley P, Koronakis V, Hughes C. (1998). Acylation of Escherichia coli hemolysin: a unique protein lipidation mechanism underlying toxin function. Microbiol Mol Biol Rev 62:309-333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman NS, Dean RT, Sanderson CJ. (1980). Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect Immun 28:258-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman NS, Simpson DL, Sakurada S, Cranfield M, DiRienzo J, Slots J. (1987). Comparative studies on the biology of Actinobacillus actinomycetemcomitans leukotoxin in primates. Oral Microbiol Immunol 2:97-104 [DOI] [PubMed] [Google Scholar]
- Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. (2005). Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry 44:11115-11121 [DOI] [PubMed] [Google Scholar]
- Tsai CC, McArthur WP, Baehni PC, Hammond BF, Taichman NS. (1979). Extraction and partial characterization of a leukotoxin from a plaque-derived Gram-negative microorganism. Infect Immun 25:427-439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CC, Shenker BJ, DiRienzo JM, Malamud D, Taichman NS. (1984). Extraction and isolation of a leukotoxin from Actinobacillus actinomycetemcomitans with polymyxin B. Infect Immun 43:700-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Winkelhoff AJ, Slots J. (1999). Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in nonoral infections. Periodontol 2000 20:122-135 [DOI] [PubMed] [Google Scholar]
- Wandersman C, Delepelaire P. (1990). TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc Natl Acad Sci USA 87:4776-4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston JL, Chen CK, Neiders ME, Dyer DW. (1993). Membrane protein expression by Actinobacillus actinomycetemcomitans in response to iron availability. J Dent Res 72:1366-1373 [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Kieba IR, Korostoff J, Howard PS, Shenker BJ, Lally ET. (2001). Maintenance of oxidative phosphorylation protects cells from Actinobacillus actinomycetemcomitans leukotoxin-induced apoptosis. Cell Microbiol 3:811-823 [DOI] [PubMed] [Google Scholar]
- Zambon JJ. (1985). Actinobacillus actinomycetemcomitans in human periodontal disease. J Clin Periodontol 12:1-20 [DOI] [PubMed] [Google Scholar]