

Abstract
Resistance to treatment and the appearance of secondary tumors in head and neck squamous cell carcinomas (HNSCC) have been attributed to the presence of cells with stem-cell-like properties in the basal layer of the epithelium at the site of the lesion. In this study, we tested the hypothesis that these putative cancer stem cells (CSC) in HNSCC could be specifically targeted and inhibited. We found that 9 of 10 head and neck tumor biopsies contained a subpopulation of cells that expressed CD133, an unusual surface-exposed membrane-spanning glycoprotein associated with CSC. A genetically modified cytolethal distending toxin (Cdt), from the periodontal pathogen Aggregatibacter actinomycetemcomitans, was conjugated to an anti-human CD133 monoclonal antibody (MAb). The Cdt-MAb complex preferentially inhibited the proliferation of CD133+ cells in cultures of established cell lines derived from HNSCC. Inhibition of the CD133+ cells was rate- and dose-dependent. Saturation kinetics indicated that the response to the Cdt-MAb complex was specific. Healthy primary gingival epithelial cells that are native targets of the wild-type Cdt were not affected. Analysis of these data provides a foundation for the future development of new therapies to target CSC in the early treatment of HNSCC. Abbreviations: Cdt, cytolethal distending toxin; CSC, cancer stem cells; HNSCC, head and neck squamous cell carcinoma; MAb, monoclonal antibody.
Keywords: cancer stem cells, CD133, cytolethal distending toxin, squamous cell carcinoma
Introduction
Head and neck squamous cell cancer afflicts over half a million people worldwide. Efforts to reduce the morbidity and mortality (40% survival at 5 yrs) of the disease are compromised by the development of resistance to chemotherapeutic agents and the occurrence of secondary tumors.
Resistance to treatment and the appearance of secondary tumors have been attributed to the presence of cancer cells that have stem-cell-like properties (Fiala, 1968). Cell lines derived from primary head and neck squamous cell carcinomas (HNSCC) contain a subpopulation of cells with clonogenic characteristics (Mackenzie, 2004) and can retain this subpopulation years after isolation and in vitro passage (Locke et al., 2005; Harper et al., 2007). Stem cells present in the basal layer of the epithelium mutate to create a clonal unit that becomes an expanding field. Clonal selection, in combination with other factors, contributes to the development of a carcinoma. Secondary tumors that arise can be clonally related and derived from expansion of the original clone (Braakhuis et al., 2004). Therapies that target cells with acquired stem-cell-like properties early in tumor development may have utility in the treatment of early-stage head and neck cancer (Jones et al., 2004; Choi and Myers, 2008).
The objective of this study was to examine, in vitro, a novel strategy designed specifically to target and inhibit cells with acquired stem-cell-like properties in HNSCC using a genotoxin produced by the periodontal pathogen Aggregatibacter actinomycetemcomitans (Mayer et al., 1999). This cytolethal distending toxin (Cdt) is composed of CdtA (18-25 kDa) and CdtC (21 kDa) subunits, which bind to a receptor on the cell surface, and CdtB (31 kDa), which is related to a superfamily of Mg2+-dependent phosphohydrolases (Hofmann et al., 2000). CdtB appears to act as a neutral, cation-dependent nuclease that damages host DNA (Elwell and Dreyfus, 2000; Lara-Tejero and Galán, 2000). DNA damage leads to growth arrest at the G2/M interphase of the cell cycle, due to inhibition of dephosphorylation of the checkpoint protein kinase Cdc2 (Alby et al., 2001). Cells of epithelial origin appear to be natural targets of the A. actinomycetemcomitans Cdt (Kanno et al., 2005).
In this report, we provide evidence that cell lines and tissue derived from HNSCC contain a subpopulation of cells that express a unique antigen, known as CD133. We show that an anti-human CD133 MAb conjugated to a genetically modified Cdt specifically targets and inhibits the proliferation of this subpopulation of cells. Analysis of these data provides an important foundation for the future development of approaches designed to attack cells with acquired stem-cell-like properties for the early treatment of HNSCC.
Materials & Methods
HNSCC Cell Lines and Tumor Biopsies
CAL-27 and the mouse B-cell hybridoma cell line HB-12346™ (AC133.1) were cultured in DMEM and FaDu, RPMI 2650, and Caco-2 in EMEM media. These cell lines were obtained from the ATCC (Rockville, MD, USA). JHU006 and JHU012 were cultured in RPMI 1640 medium (Khan et al., 2010). Media were supplemented with 10-20% fetal calf serum (FCS). Primary HGEC were isolated (Oda and Watson, 1990) from non-inflamed gingival tissue from periodontal surgeries conducted in the University of Pennsylvania School of Dental Medicine Graduate Periodontics Clinic. Institutional Review Board approval was obtained, and donors provided informed consent. The cells were Ep-CAM+ (human epithelial cell adhesion molecule) and Ab-1- (CD90/Thy-1). Primary HGEC were immortalized with pSG5T DNA, containing the SV40 large T-antigen gene, with use of the Effectene Transfection Reagent (QIAGEN, Valencia, CA, USA).
Ten HNSCC samples, stored as paraffin-embedded blocks, were obtained from the University of Pennsylvania Head and Neck Tissue and Serum Bank. Sections were prepared by the Tissue Processing Service at the School of Dental Medicine.
Immunofluorescence Staining
Immunofluorescence microscopy was performed as described previously (Kanno et al., 2005). Cells (1 × 104 per well) and tissue sections were stained with mouse anti-human CD133/1 monoclonal antibody (MAb) and Alexa Fluor 488 goat anti-mouse IgG conjugate (Invitrogen/Molecular Probes®, Carlsbad, CA, USA). Slides were mounted with Prolong Gold containing DAPI (Invitrogen/Molecular Probes®) and viewed with a Nikon Eclipse 80i fluorescence microscope. Digital images were recorded with Spot Advanced 4.6 software (Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
For flow cytometry, 1 × 106 cells were incubated with either: (i) no antibody (unstained), (ii) Alexa Fluor 488 goat anti-mouse IgG conjugate (Invitrogen) (Isotype FITC), (iii) AC133.1 MAb IgG and Alexa Fluor 488 (CD133MAb), or (iv) mouse anti-human CD133/1 MAb conjugated to R-phycoerythrin (PE) (Miltenyi Biotec, Auburn, CA, USA). The data from 50,000 events were analyzed with FlowJo 8.8.6 (Tree Star, Inc., Ashland, OR, USA).
Real-time PCR
Total RNA was isolated from cells with the Trizol reagent, and cDNA was prepared from 2 µg of RNA with oligo(dT) and the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). Reactions were preformed in an ABI 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with primers 5′-GAGAAAGTGGCATCGTGCAA-3′ (forward) and 5′-TGCCAAACCAAAACAAATTCAA-3′ (reverse). TATA-binding protein (TBP) mRNA, amplified with 5′-GGAGCTGTGATGTGAAGTTTCCTA-3′ (forward) and 5′-CCAGGAAATAACTCTGGCTCATAAC-3′ (reverse) primers, served as a normalizing control. A negative PCR control without template cDNA was included.
Western Blotting
CD133 was detected in cell lysates with an Enhanced Chemiluminescence Western Blotting Detection Kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA) after binding of mouse anti-human CD133/1 (AC133) MAb (Miltenyi Biotec) and horseradish peroxidase (HPR)-conjugated anti-mouse IgG (Novagen, San Diego, CA, USA) (Mao and DiRienzo, 2002). A lysate of Caco-2 cells was used as a positive control (Corbeil et al., 2000). Actin was labeled with rabbit anti-human actin polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and HRP-conjugated anti-rabbit IgG (GE Healthcare, Piscataway NJ, USA).
Site-specific Mutagenesis of CdtA
C178, in CdtA, was replaced with alanine by site-directed mutagenesis with the primers 5′-AAAAGTGTGTCACAAGGACGTGCAGTCACTTATAATCCTGTAAGTCC-3′ (forward) and 5′-GGACTTACAGGATTATAAGTGACTGCACGTCCTTGTGACACACTTTT-3′ (reverse) (alanine codons underlined). Mutated DNA strands were made with PfuUltra DNA polymerase in PCR (Stratagene, La Jolla, CA, USA). Template DNA was obtained from pMUTc149cdtA that contained the amino acid substitution C149A in CdtA (Cao et al., 2006). Methylated parental DNA strands were digested with DpnI (New England Biolabs, Beverly, MA, USA) prior to transformation of Escherichia coli TOP10 competent cells (Invitrogen). The mutation was confirmed by DNA sequencing, and E. coli BL-21 competent cells (Novagen) were transformed for isolation of the mutated gene product, designated CdtAC149A, C178A.
Protein Isolation and Toxin Reconstitution
Recombinant Cdt proteins were isolated by affinity chromatography as described previously (Cao et al., 2005). Heterotrimers were reconstituted, in a refolding buffer, from wild-type subunits, CdtAC149A, C178A-His6 and MAb-conjugated CdtAC149A, C178A-His6 (Mao and DiRienzo, 2002). Heterotrimer formation was confirmed by differential dialysis followed by Western blotting (Cao et al., 2005).
CdtA Binding Assay
We used a cell enzyme-linked immunosorbent assay (CELISA) to examine binding kinetics of CdtAC149A, C178A-His6 (Cao et al., 2005). We seeded 96-well microtiter plates with 1.5 × 104 RPMI 2650 or FaDu cells/well in growth medium and incubated them for 48 hrs. The cells were fixed, and a 0-2 µg quantity of purified CdtAC149A, C178A-His6 or CdtA-His6 was added to each well. Bound protein was detected, after 1 hr, with anti-His●Tag MAb (Novagen).
Monoclonal Antibody Conjugation to CdtA
IgG1 was purified from AC133.1 mouse hybridoma culture medium with the Montage™ Antibody Purification PROSEP-A Kit (Millipore, Billerica, MA, USA). Antibody concentration was determined according to the Beer-Lambert Law [IgG (mg/mL) = A280 × 0.72], and a titer was obtained with a whole-cell lysate of Caco-2 cells. The IgG1 was conjugated to CdtAC149A, C178A-His6, with 4-succinimidyloxycarbonyl-α-methyl-α-(2-pyridyldithio)toluene (SMPT; Pierce, Rockford, IL, USA; 1 mg SMPT/10 mg of IgG) as described previously (Ghetie et al., 1991). CdtAC149A, C178A-His6 (0.5 mg) was mixed with SMPT-MAb, and the preparation was incubated for 48 hrs at 4°C. Un conjugated excess SMTP-MAb was removed on a nickel-iminodiacetic acid column. Unconjugated CdtAC149A, C178A-His6 was removed by dialysis, and the final protein concentration was determined with a Micro BCA Protein Assay Kit (Pierce).
Cell Cycle Analysis
Cell cycle arrest was determined by flow cytometry (Cao et al., 2005). Cells were grown for 24 hrs (80-90% confluent) and incubated for 24 or 36 hrs with 2.5 or 10 µg/mL of one of the following preparations: (i) reconstituted wild-type heterotoxin (CdtABC), (ii) heterotoxin made with MAb-conjugated mutated CdtA (CdtAC149A, C178A BC-CD133MAb), (iii) heterotoxin made with unconjugated mutated CdtA (CdtAC149A, C178A BC), or (iv) purified AC133.1 MAb (CD133MAb). Input protein concentration was based on dose-response data from Cao et al. (2005). Propidium-iodide-stained nuclei were examined on a FACSCalibur four-color dual-laser flow cytometer (BD Biosciences) at the Abramson Cancer Center Flow Cytometry and Cell Sorting Resource Laboratory of the University of Pennsylvania. Data from 30,000 events were analyzed with ModFit LT 3.2 (Verity Software House, Topsham, ME, USA). A dose-response curve was obtained with the RPMI 2650 cell line with 0-32 µg/mL of CdtAC149A, C178ABC-CD133MAb for 36 hrs.
Cell Viability Assay
Cultures of RPMI 2650 (5 × 105 cells) were treated with 10 µg/mL of CdtABC, CdtAC149A, C178ABC-CD133MAb, or CdtAC149A, C178ABC for 0 to 72 hrs and then stained with a 0.4% solution of trypan blue (Invitrogen). Live cells were counted in a hemocytometer.
Results
Gene Expression and Cell-surface Display of CD133 in Oral Cancer Cells
Nine of 10 biopsies, from different patients, contained CD133+ cells (Fig. 1A) in the following distributions: > 75% (n = 2), 30-75% (n = 1), 10-30% (n = 5), and < 10% (n = 1). Among the 5 cell lines examined, CD133+ cells were detected by immunofluorescence in JHU006, JHU012, and RPMI 2650 cultures (Fig. 1B).
Figure 1.
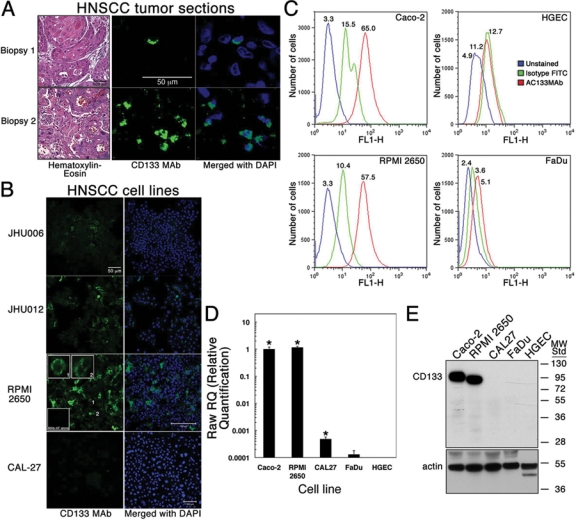
Detection of CD133+ cells in HNSCC biopsied tissue and cell lines. (A) Tissue sections from tumor biopsies were labeled with mouse anti-human CD133/1 MAb (1:200; CD133 MAb) and goat anti-mouse IgG conjugated to Alexa Fluor (MAb-AF; 1:400). Nuclei were stained with DAPI (Merged). Two representative biopsies are shown. Magnification 60x. Separate sections were stained with hematoxylin-eosin. Magnification 40x. (B) HNSCC cell lines JHU006, JHU012, RPMI 2650, and CAL-27 were treated as in panel A. Magnification was 20x except for RPMI 2650 (40x). Insets show enlarged images of labeled cells (1 and 2) and cells treated only with MAb-AF. Experiments were performed 3 times. (C) Change in fluorescence intensity was measured in flow cytometry for unlabeled RPMI 2650 cells (blue peaks) and cells labeled with AC133.1 MAb IgG and Alexa Fluor 488 goat anti-mouse IgG (red peaks) or Alexa Fluor 488 conjugate alone (green peaks). Gated runs were analyzed with FlowJo modeling software. The number above each peak is the fluorescence value below which 50% of the events were found (median). (D) Real-time (RT)-PCR showing amplification of CD133 mRNA from representative HNSCC cell lines and HGEC. PCR primers and conditions are described in Materials & Methods. Values for the amount of PCR amplicon were normalized to that for the ubiquitous, constitutively expressed TATA-binding protein (TBP) gene mRNA. Values were plotted relative to that for Caco-2 CD133 mRNA (positive control). Mean values ± SD are shown (n = 3 in each group). Asterisks mark statistically significant differences between the relative amount of the CD133 amplicon from the HNSCC line mRNA and that from HGEC mRNA (no expression). Experiments were performed twice. (E) CD133 detected in cell lysates on a Western blot with mouse anti-human CD133/1 MAb (1:100) and HPR-conjugated anti-mouse IgG (1:2000). The Caco-2 cell lysate was a positive control. Molecular-weight standards are in kDa. Actin expression, detected with a polyclonal antibody (1:1000) and HRP-conjugated anti-rabbit IgG (1:1000), was used to evaluate sample loading. Three actin polypeptides were observed in HGEC.
CD133 was detected on the surface of RPMI 2650 cells by flow cytometry (Fig. 1C). RPMI 2650 cells exhibited a median fluorescence (MF) of 57.5 compared with 65.0 for the Caco-2 positive control cells and 5.1 for the CD133- FaDu cells. Primary HGEC had a MF value of 12.7. Similar results were obtained when RPMI 2650 cells were labeled with a CD133 MAb directly conjugated to PE (MF = 64.8; data not shown). Real-time (RT)-PCR showed that the RPMI 2650 cells expressed the CD133 gene at a level comparable with that of Caco-2 cells (Fig. 1D). CD133 expression by CAL-27 was orders of magnitude lower. The small amount of amplicon obtained from FaDu cDNA was not statistically significant. A CD133 mRNA was not detected in primary HGEC. These results matched exactly with the heterogeneous distribution of the CD133 protein as determined by Western blotting (Fig. 1E).
Targeting the Cytolethal Distending Toxin (Cdt) to CD133+ HNSCC Cells
The recombinant A. actinomycetemcomitans Cdt inhibited the proliferation of all epithelioid cells examined to date. To target the Cdt specifically to CD133-expressing cells, we constructed a mutated CdtA subunit protein that no longer bound to the native toxin receptor and contained a single molecular surface exposed cysteine (C197) for conjugating the anti-human CD133 MAb (Fig. 2A). This mutant, CdtAC149A, C178A, lost the ability to bind to Cdt-sensitive RPMI 2650 and FaDu cells (Fig. 2B). The reduction in cell binding was statistically significant and observed over input protein concentrations of ≥ 250 ng per well. CdtAC149A, C178A retained the ability to form a heterotrimer with wild-type CdtB and CdtC (Fig. 2B inset).
Figure 2.
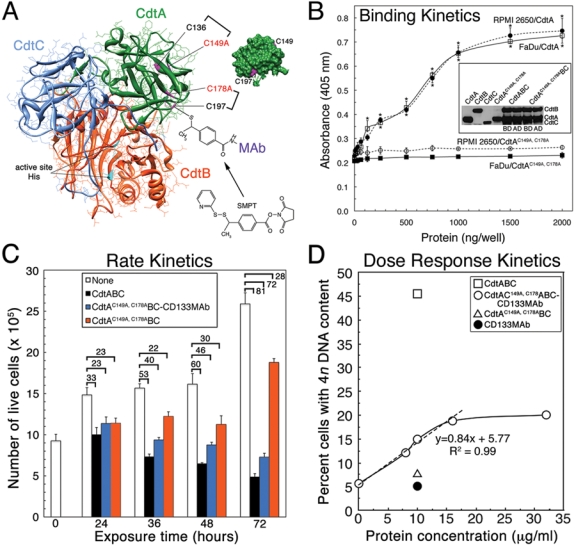
Construction and characterization of a genetically modified Cdt for targeting CD133-expressing epithelial cells. (A) Crystal structure of the A. actinomycetemcomitans Y4 Cdt (Yamada et al., 2006) modeled with UCSF Chimera (Pettersen et al., 2004) and Protein Data Bank (PDB) file 2F2F. Essential active-site residues H160 and H274 in CdtB are shown in cyan, and cysteine residues in CdtA are displayed in magenta. Insert shows a surface model of CdtA, rotated 90° counterclockwise relative to that in the ribbon structure, to show surface-exposed C149 and C197. Brackets mark two predicted disulfides, C136/C149 and C178/C197 (Cao et al., 2006), and the two mutations, C149A and C178A. Mouse anti-human AC133.1 MAb was attached to C197 in CdtAC149A, C178A-His6 via SMPT. (B) Kinetics of CdtAC149A, C178A-His6 and CdtA-His6 binding to RPMI 2650 and FaDu cells. Bound protein was detected with anti-His•Tag MAb (1:3000) and goat anti-mouse HPR-conjugated IgG (1:3000). Data are represented as mean values ± SD (n = 3) in each group. Statistically significant differences were found between the binding of the wild-type and mutated CdtA to RPMI 2650 (top asterisks) and FaDu (bottom asterisks). P ≤ 0.05. Inset shows a Western blot depicting the ability of CdtAC149A, C178A-His6 to form a heterotrimer with wild-type CdtB and CdtC. Aliquots of reconstituted samples were taken before (BD) and after (AD) dialysis. (C) Rate kinetics of RPMI 2650 cell viability following exposure to CdtAC149A, C178ABC-CD133MAb, CdtABC, or CdtAC149A, C178ABC for 24-72 hrs. Live cells were counted after staining with trypan blue. Data are presented as the mean values ± SD (n = 3). Values above the brackets represent the percent decrease between the number of untreated and treated cells at each exposure time. (D) Following treatment of RPMI 2650 with increasing concentrations of CdtAC149A, C178ABC-CD133MAb for 36 hrs, the percentage of cells having a 4n DNA content (diploid G2) were quantified by flow cytometry and plotted against the input concentration of toxin. Regression analysis showed that the dose response was linear (dashed line), with an R-squared value (square of the correlation coefficient) of 0.99, up to 16 µg/mL of protein/1 × 106 cells (at time of toxin addition). Saturation kinetics showed that the effect on cell cycle was specific and indicated that CD133+ cells comprised approximately 18% of the total cell culture. Kinetic experiments were performed twice.
Selective Inhibition of CD133+ Cells in Head and Neck Squamous Cell Carcinoma Cell Lines
RPMI 2650 was exposed to CdtABC, CdtAC149A, C178ABC-CD133MAb, or CdtAC149A, C178ABC and stained to detect live cells. There was a 23 to 72% decrease in the number of live cells following exposure to CdtAC149A, C178ABC-CD133MAb over a period of 24-72 hrs (Fig. 2C; blue bars). We observed a similar 33 to 81% decrease (white bars) in viability over the same time period of exposure to CdtABC. However, there was no specific decrease (orange bars) in the number of live cells after treatment with CdtAC149A, C178ABC.
RPMI 2650 was cell-cycle-arrested in a dose-dependent fashion when treated with CdtAC149A, C178ABC-CD133 MAb (Fig. 2D). The response was linear over the range 0 to 16 µg/mL of protein (R-squared = 0.99). Saturation kinetics was observed, and maximum cell cycle arrest (18% of the total cell population) was attained with 22 µg/mL of protein. This response was well below that of RPMI 2650 cells treated with the wild-type Cdt under the same conditions. Analysis of these data supported the results of the immunofluorescence experiments and cell sorting (data not shown), which indicated that the CD133+ cells comprised a subset (approximately 16-20%) of the total cell population. On average, 26.4 ± 0.6% (P = 0.002) of the total RPMI 2650 cell population had a 4n DNA content following a 36-hour exposure to a TD50 of CdtAC149A, C178ABC-CD133MAb. In comparison, 54.0 ± 1.6% (P = 0.001) and 8.5 ± 0.7% of the total cells had a 4n DNA content after exposure to the same concentration of CdtABC and no toxin, respectively.
Following treatment with CdtAC149A, C178ABC-CD133MAb for 36 hrs, RPMI 2650 and JHU012 contained a subpopulation of cells with a 4n DNA content indicative of cell-cycle arrest at G2/M (Fig. 3). On average, 15, 84, and 19% of the total RPMI 2650, JH012, and Caco-2 cell populations (3.5 × 106 cells), respectively, had a 4n DNA content after treatment with 10 µg/mL of toxin (Table). Cultures treated with either the CD133 MAb (AC133.1) alone or with toxin reconstituted with the unconjugated CdtA mutant (CdtAC149A, C178ABC) did not exhibit cell-cycle arrest. Both primary and immortalized HGEC were not affected by CdtAC149A, C178ABC-CD133MAb, even though > 90% of these cells arrested at G2/M when treated with the wild-type Cdt.
Figure 3.
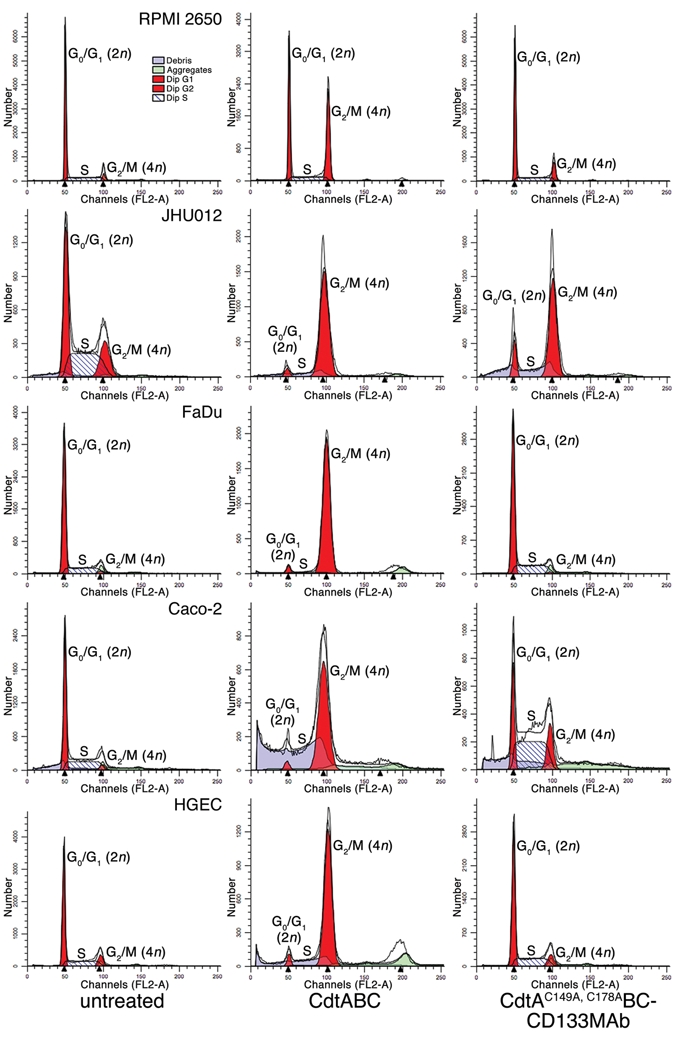
Cell-cycle analysis of HNSCC cell lines and HGEC treated with anti-human CD133 MAb conjugated to the Cdt. Cultures (3.5 × 106 cells) of RPMI 2650 and JHU012 were left untreated or treated with 10 or 2.5 µg/mL of CdtABC or CdtAC149A, C178ABC-CD133MAb for 36 or 24 hrs, respectively. Caco-2 (positive control), FaDu (negative control), and HGEC (normal oral keratinocytes) were treated with 10 µg/mL of protein for 36 hrs. Propidium-iodide-stained nuclei were examined by flow cytometry. ModFit was used to generate the plots. The stained DNA profile is shown as the heavy black line. Experiments were performed a minimum of three times.
Table.
Cell Cycle Analysis of HNSCC Cell Lines and HGEC Treated with the Toxin-Monoclonal Antibody Complex
| Cell Line | Treatment | Diploid G1 (2n) % of Cell Population | Diploid G2 (4n) % of Cell Population | Diploid S % of Cell Population | Coefficient of Variance (%) |
|---|---|---|---|---|---|
| RPMI 2650 | None | 68.55 | 5.63 | 25.82 | 1.78 |
| CdtABC | 37.72 | 45.49 | 16.78 | 2.05 | |
| CdtAC149A, C178ABC-CD133MAb | 59.38 | 14.95* | 25.67 | 1.96 | |
| CdtAC149A, C178ABC | 71.96 | 7.56 | 20.48 | 3.89 | |
| CD133MAb | 69.42 | 5.08 | 25.50 | 2.14 | |
| JHU012 | None | 39.69 | 19.09 | 41.22 | 6.00 |
| CdtABC | 3.92 | 96.08 | 0 | 5.80 | |
| CdtAC149A, C178ABC-CD133MAb | 16.04 | 83.96* | 0 | 4.83 | |
| CdtAC149A, C178ABC | 44.17 | 26.38 | 29.45 | 7.35 | |
| CD133MAb | 49.15 | 17.22 | 33.63 | 6.50 | |
| CAL-27 | None | 24.80 | 1.54 | 73.65 | 3.99 |
| CdtABC | 9.28 | 21.27 | 69.45 | 4.76 | |
| CdtAC149A, C178ABC-CD133MAb | 42.80 | 0 | 57.20 | 4.34 | |
| CdtAC149A, C178ABC | NT | NT | NT | NT | |
| CD133MAb | 31.64 | 0 | 68.36 | 2.30 | |
| FaDu | None | 71.07 | 2.47 | 26.47 | 4.93 |
| CdtABC | 2.71 | 94.61 | 2.68 | 4.78 | |
| CdtAC149A, C178ABC-CD133MAb | 65.97 | 1.35 | 32.68 | 4.11 | |
| CdtAC149A, C178ABC | NT | NT | NT | NT | |
| CD133MAb | 66.73 | 3.90 | 29.37 | 4.29 | |
| Caco-2 | None | 57.04 | 4.10 | 38.87 | 3.41 |
| CdtABC | 3.97 | 96.03 | 0 | 6.69 | |
| CdtAC149A, C178ABC-CD133MAb | 23.03 | 19.37* | 57.60 | 3.99 | |
| CdtAC149A, C178ABC | 45.47 | 10.46 | 44.07 | 5.49 | |
| CD133MAb | 55.99 | 4.68 | 39.33 | 3.77 | |
| HGEC | None | 59.96 | 11.05 | 28.99 | 3.23 |
| CdtABC | 4.26 | 94.22 | 1.52 | 4.78 | |
| CdtAC149A, C178ABC-CD133MAb | 58.85 | 9.74 | 31.42 | 3.98 | |
| CdtAC149A, C178ABC | NT | NT | NT | NT | |
| CD133MAb | NT | NT | NT | NT |
NT, not tested.
The increase in the diploid G2 population was statistically significant (P < 0.005) when compared with that for untreated cells.
Discussion
There is evidence that HNSCC may be maintained by a subpopulation of cells with acquired stem-cell-like properties (Hamburger and Salmon, 1977). These cells form “cancer spheroids” in culture and tumors when transplanted into mice (Harper et al., 2007; Prince et al., 2007). CD133, a 120-kDa cell-surface glycoprotein (Shmelkov et al., 2005), appears to be uniquely expressed by these cells (Harper et al., 2007; Okamoto et al., 2009). Less than 0.2% of cells in cultures of the cell lines CA1, CaLH2, Cal-27, and FaDu exhibited high expression of CD133 (Harper et al., 2007; Cameron et al., 2010). Zhang et al. (2009) found that 3 HNSCC cell lines and 10 biopsy samples contained up to 2% and 3% CD133+ cells, respectively. Approximately 60% of the cells with stem-cell-like properties in the cell lines SAS and OECM1 bound a CD133 antibody (Chiou et al., 2008). Two of 5 HNSCC cell lines (RPMI 2650 and JHU012) cultured in our study had relatively high numbers of CD133+ cells, and 2 cell lines (CAL-27 and FaDu) failed to express the CD133 gene at levels sufficient for detection of the glycoprotein on the cell surface. This differential expression may have been a consequence of cell passage in vitro. Our analysis of 10 HNSCC biopsies suggests that CD133+ cells may be more prevalent, but in relatively low numbers, in vivo, where conditions for tumor growth and metastasis are optimal. Cells with acquired stem-cell-like properties isolated from HNSCC, based on the expression of CD133, were resistant to standard chemotherapy in vitro and in vivo (Zhang et al., 2009). These cells use ATP-binding cassette membrane transporters (Goodell et al., 1996) or high expression of aldehyde dehydrogenase (Storms et al., 1999) to rapidly export or degrade xenobiotics. Targeting the subpopulation of cells with acquired stem-cell-like properties and development of treatment procedures to overcome their potential drug resistance seems vital for both tumor growth inhibition and prevention of tumor recurrence (Tu et al., 2009).
Our study appears to be the first proof-of-concept to demonstrate the selective inhibition of proliferation of cells with acquired stem-cell-like properties (CD133+) in heterogeneous HNSCC cell populations. The A. actinomycetemcomitans Cdt was chosen as the inhibitory component due to the sensitivity of epithelioid-like cells to the wild-type Cdt. Various human epithelial and keratinocyte cell lines are cell-cycle-arrested at G2/M when exposed to the Cdt (reviewed in Whitehouse et al., 1998). Likewise, we found that GMSM-K, an immortalized orolabial cell line, is very sensitive to the toxin (Kanno et al., 2005). To the best of our knowledge, the susceptibility of primary human epithelial cells to the Cdt had not been previously examined. Here, we report that proliferation of HGEC is inhibited by the native Cdt, and that these cells do not express the CD133 gene. Based on these findings, we mutated the primary receptor binding subunit (CdtA) of the toxin to eliminate intoxication of normal cells. Sonoporation has been used to deliver a plasmid containing the A. actinomycetemcomitans cdtB gene into Ca9-22, a human gingival squamous cell carcinoma cell line (Iwanaga et al., 2007). Transfected cells underwent apoptosis when treated in vitro and in mice. However, specific cell delivery mechanisms have not been tested. We conjugated an anti-human CD133 MAb to Cdt, containing the mutated CdtA subunit, to specifically deliver the toxin to CD133-expressing cells. The CdtAC149A, C178ABC-CD133MAb inhibited proliferation only in cells that displayed detectable levels of CD133 on the surface. The relative disparities among the HNSCC cell lines in susceptibility to CdtAC149A, C178ABC-CD133MAb could be attributed to variable levels of expression of the CD133 gene.
Molecular targeted therapy in which the products of selectively expressed genes that contribute to the neoplastic phenotype are exploited as targets of antibodies, small molecules, or genetic constructs is a promising therapeutic strategy (Choi and Myers, 2008). Targeted therapy should have a higher therapeutic index and, therefore, be less toxic than cytotoxic drugs. Unfortunately, recombinant immunotoxins and immunoreagents have had a poor clinical track record, primarily due to problems with immunogenicity, selectivity, penetration into solid tumors (Schrama et al., 2006; Pastan et al., 2007), and, possibly, failure to target tumor-forming cells. Subsequent studies are required to evaluate targeted approaches for the inhibition of proliferation of CD133+ cells that could lead to impaired development of primary and secondary tumors in vivo. The toxin-monoclonal antibody complex may have utility, in a clinical setting, as a topical or locally delivered reagent for treating dysplastic lesions and very-early-stage masses that have the potential to develop into mature tumors.
Acknowledgments
We thank Dr. Aaron Weinberg, DMD, PhD, Case Western Reserve University, for advice on isolating HGEC. This work was supported by USPHS Research Grants DE012593 and DE017679 from the National Institute of Dental and Craniofacial Research. The authors have no conflicting financial interests.
References
- Alby F, Mazars R, de Rycke J, Guillou E, Baldin V, Darbon JM, et al. (2001). Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett 491:261-265 [DOI] [PubMed] [Google Scholar]
- Braakhuis BJ, Leemans CR, Brakenhoff RH. (2004). A genetic progression model of oral cancer: current evidence and clinical implications. J Oral Pathol Med 33:317-322 [DOI] [PubMed] [Google Scholar]
- Cameron SR, Dahler AL, Endo-Munoz LB, Jabar I, Thomas GP, Leo PJ, et al. (2010). Tumor-initiating activity and tumor morphology of HNSCC is modulated by interactions between clonal variants within the tumor. Lab Invest 90:1594-1603. [DOI] [PubMed] [Google Scholar]
- Cao L, Volgina A, Huang CM, Korostoff J, DiRienzo JM. (2005). Characterization of point mutations in the cdtA gene of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans . Mol Microbiol 58:1303-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Volgina A, Korostoff J, DiRienzo JM. (2006). Role of intrachain disulfides in the activities of the CdtA and CdtC subunits of the cyto-lethal distending toxin of Actinobacillus actinomycetemcomitans. Infect Immun 74:4990-5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, et al. (2008). Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res 14:4085-4095 [DOI] [PubMed] [Google Scholar]
- Choi S, Myers JN. (2008). Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res 87:14-32 [DOI] [PubMed] [Google Scholar]
- Corbeil D, Roper K, Hellwig A, Tavian M, Miraglia S, Watt SM, et al. (2000). The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem 275:5512-5520 [DOI] [PubMed] [Google Scholar]
- Elwell CA, Dreyfus LA. (2000). DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol 37:952-963 [DOI] [PubMed] [Google Scholar]
- Fiala S. (1968). The cancer cell as a stem cell unable to differentiate. A theory of carcinogenesis. Neoplasma 15:607-622 [PubMed] [Google Scholar]
- Ghetie V, Thorpe P, Ghetie MA, Knowles P, Uhr JW, Vitetta ES. (1991). The GLP large scale preparation of immunotoxins containing deglycosylated ricin A chain and a hindered disulfide bond. J Immunol Methods 142:223-230 [DOI] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. (1996). Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183:1797-1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger AW, Salmon SE. (1977). Primary bioassay of human tumor stem cells. Science 197:461-463 [DOI] [PubMed] [Google Scholar]
- Harper LJ, Piper K, Common J, Fortune F, Mackenzie IC. (2007). Stem cell patterns in cell lines derived from head and neck squamous cell carcinoma. J Oral Pathol Med 36:594-603 [DOI] [PubMed] [Google Scholar]
- Hofmann K, Tomiuk S, Wolff G, Stoffel W. (2000). Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc Natl Acad Sci USA 97:5895-5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga K, Tominaga K, Yamamoto K, Habu M, Maida H, Akifusa S, et al. (2007). Local delivery system of cytotoxic agents to tumors by focused sonoporation. Cancer Gene Ther 14:354-363 [DOI] [PubMed] [Google Scholar]
- Jones RJ, Matsui WH, Smith BD. (2004). Cancer stem cells: are we missing the target? J Natl Cancer Inst 96:583-585 [DOI] [PubMed] [Google Scholar]
- Kanno F, Korostoff J, Volgina A, DiRienzo JM. (2005). Resistance of human periodontal ligament fibroblasts to the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. J Periodontol 76:1189-1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K, Araki K, Wang D, Li G, Li X, Zhang J, et al. (2010). Head and neck cancer radiosensitization by the novel poly(ADP-ribose) polymerase inhibitor GPI-15427. Head Neck 32:381-391 [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. (2000). A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354-357 [DOI] [PubMed] [Google Scholar]
- Locke M, Heywood M, Fawell S, Mackenzie IC. (2005). Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res 65:8944-8950 [DOI] [PubMed] [Google Scholar]
- Mackenzie IC. (2004). Growth of malignant oral epithelial stem cells after seeding into organotypical cultures of normal mucosa. J Oral Pathol Med 33:71-78 [DOI] [PubMed] [Google Scholar]
- Mao X, DiRienzo JM. (2002). Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell Microbiol 4:245-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bueno LC, Hansen EJ, DiRienzo JM. (1999). Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun 67:1227-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda D, Watson E. (1990). Human oral epithelial cell culture I. Improved conditions for reproducible culture in serum-free medium. In Vitro Cell Dev Biol 26:589-595 [DOI] [PubMed] [Google Scholar]
- Okamoto A, Chikamatsu K, Sakakura K, Hatsushika K, Takahashi G, Masuyama K. (2009). Expansion and characterization of cancer stem-like cells in squamous cell carcinoma of the head and neck. Oral Oncol 45:633-639 [DOI] [PubMed] [Google Scholar]
- Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. (2007). Immunotoxin treatment of cancer. Annu Rev Med 58:221-237 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GM, Greenblatt DM, Meng EC, et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605-1612 [DOI] [PubMed] [Google Scholar]
- Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba B, et al. (2007). Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 104:973-978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrama D, Reisfeld RA, Becker JC. (2006). Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 5:147-159 [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, St Clair R, Lyden D, Rafii S.(2005). AC133/CD133/Prominin-1. Int J Biochem Cell Biol 37:715-719 [DOI] [PubMed] [Google Scholar]
- Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, et al. (1999). Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA 96:9118-9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LC, Foltz G, Lin E, Hood L, Tian Q. (2009). Targeting stem cells—clinical implications for cancer therapy. Curr Stem Cell Res Ther 4:147-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CA, Balbo PB, Pesci EC, Cottle DL, Mirabito PM, Pickett CL. (1998). Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect Immun 66:1934-1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Komoto J, Saiki K, Konishi K, Takusagawa F. (2006). Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci 15:362-372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. (2009). A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett 289:151-160 [DOI] [PubMed] [Google Scholar]