

Abstract
Natural products have been a great source of many small molecule drugs for various diseases. In spite of recent advances in biochemical engineering and fermentation technologies that allow us to explore microorganisms and the marine environment as alternative sources of drugs, more than 70% of the current small molecule therapeutics derive their structures from plants used in traditional medicine. Natural-product-based drug discovery relies heavily on advances made in the sciences of biology and chemistry. Whereas biology aims to investigate the mode of action of a natural product, chemistry aims to overcome challenges related to its supply, bioactivity, and target selectivity. This review summarizes the explorations of the caged Garcinia xanthones, a family of plant metabolites that possess a unique chemical structure, potent bioactivities, and a promising pharmacology for drug design and development.
Keywords: cyclic compounds, cycloaddition, domino reactions, natural products, synthesis design
Introduction
The trees of the genus Garcinia belong to the family of Guttiferae (Clusiaceae) and are found mostly in lowland rainforests of India, Indochina, Indonesia, West and Central Africa, and Brazil.[1] They are slow-growing polygamous trees that produce scented flowers and fruits that often contain a fleshy, edible endocarp (Figure 1). Throughout the years Garcinia trees have retained considerable value as sources for medicines, pigments, gums, waxes, resins, foodstuffs (fruit), fuel (wood, seed oil), and lumber.[2,3] Arguably, the most widely cultivated and well-known Garcinia tree is Garcinia mangostana. It yields one of the most highly prized tropical fruits, the mangosteen, valued for its delicious endocarp and rind, both of which are thought to have medicinal potential.[4]
Figure 1.

a) Garcinia mangostana; b) mangosteen; c) gamboge and gambogic acid; and d) an extract from a Japanese painting in which gamboge was used as the yellow colorant.
The utility of the Garcinia trees in the arts and sciences is well documented. In fact, gamboge, the pulverized gold-colored resin collected primarily from Garcinia hanburyi, and to a lesser extent from Garcinia morella, has a particularly long and rich history in the arts and sciences.[5,6] For instance, the yellow colorant used on 8th century artifacts from East Asia is presumed to be a gamboge-based water-color. The importation of gamboge in Europe took place in the 15–16th century where it was used mainly as a coloring material by Flemish painters. In fact, this pigment has been noted on a painting by Rembrandt, currently found in the Staatliche Kunstsammlungen Museum in Dresden.[7] The toxicity of gamboge was also noted early on and several accounts warn against licking brushes containing gamboge. In the recent years, gamboge has been utilized primarily for research on identification of biologically active substances. Nonetheless, it is worth noting that Jean Baptiste Perrin used a colloidal suspension of gamboge particles to investigate Brownian motion and derive a value for the Avogadro number in a series of experiments that gave him the Nobel Prize in physics in 1926.[8]
Family of Caged Garcinia Xanthones
Isolation
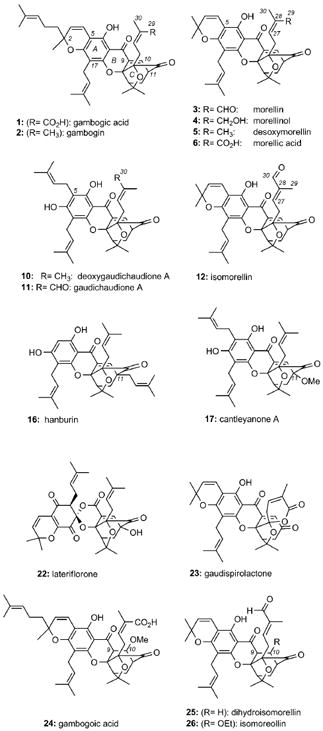
Gamboge (tenghuang in Chinese) has been the subject of analytical and chemical studies for about 200 years.[9] Depending on the originating tree, the resin composition changes but in all cases it contains about 70% of organic constituents that are insoluble in water. Studies with resin extracts of Garcinia hanburyi led to the identification of gambogic acid (1). Although this compound was initially isolated in 1934 as its monoacetyl derivative,[10] its chemical structure was unambiguously determined in 2001 by an X-ray of its pyridine salt.[11] Related investigations from the seeds and the resin of Garcinia morella led in 1937 to the isolation of morellin (3).[12] In 1963 the p-bromobenzenesulfonyl ester of morellin was prepared and its constitution was determined by X-ray crystallography.[13]
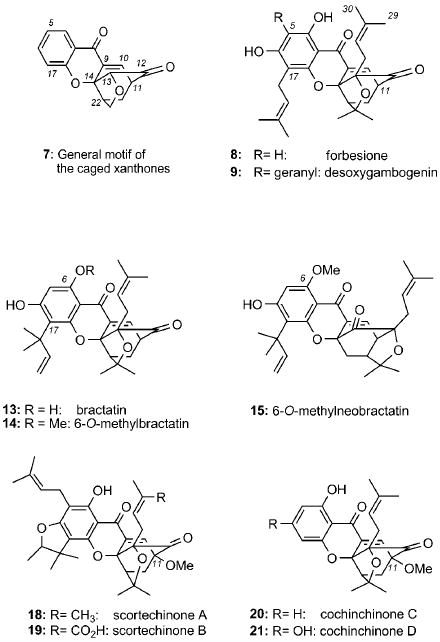
The crystal structures of 1 and 3 defined a new class of natural products that are collectively referred to as caged Garcinia xanthones. Common to their structure is a xanthone backbone in which the C ring has been converted to an unusual 4-oxatricyclo[4.3.1.03,7]dec-8-en-2-one (caged) scaffold.[14] This motif (see structure 7) is further customized through substitutions on the aromatic ring (ring A) and peripheral oxidations to produce a variety of structural subfamilies. This is exemplified by the structure of forbesione (8), a natural product isolated from Garcinia forbesii[15] and Garcinia hanburyi.[16] For example, prenylation at the C5 center of forbesione (gambogic acid numbering) gives access to the gaudichaudione scaffold,[17] represented by deoxygaudichaudione (10).[18] Oxidation at the C30 center could then lead to gaudichaudione A (11).[17a] Alternatively, prenylation of 8 at C5, followed by cyclization with the pendant phenol gives access to the morellin scaffold,[19] represented by desoxymorellin (5).[20] Progressive oxidations at the C29 center of 5 produce morellinol (4), morellin (3), and morellic acid (6).[21] Compounds arising from isomerization around the C27=C28 double bond of morellins have also been isolated. Thus, morellin (3), with the cis configuration about the C27=C28 double bond, is known to isomerize to the trans isomer, isomorellin (12).[22] Similar observations have been reported for 1.[23]
Geranylation at the C5 center of forbesione forms desoxygambogenin (9) and, after formation of the pyran ring, produces the structure of gambogin (2).[24] Further oxidation at C29 leads to the structure of 1.[16a] Isolated from Garcinia bracteata, the bractatin subfamily (13, 14, 15)[25] serves as an example of forbesione-type natural products that contain a reverse prenyl group at the C17 center. Interestingly, 6-O-methylneobractatin (15) is the only natural product known to contain a modified caged structure, referred to as the neo motif.[25] The C11 center is another diversity point. For example, hanburin (16)[24] is a C11 prenylated product of forbesione, while cantleyanone A (17)[26] and scortechinones A (18)[27] and B (19)[28] contain a methoxy group at that position. This motif has also been identified in the structures of cochinchinones C (20) and D (21), two natural products isolated from the roots of Cratoxylum cochinchinense.[29]
In addition to the structures above, there are a number of Garcinia natural products containing a rearranged caged xanthone motif. For instance, in the structure of lateriflorone (22),[30] the caged motif is attached to an unprecedented spiroxalactone core, likely a product of an oxidative rearrangement of the central xanthone ring. Similar rearrangements at the C ring may account for the structure of gaudispirolactone (23).[31, 32] Gambogoic acid (24),[18, 33] dihydroisomorellin (25),[20b] and isomoreollin (26)[34] are a few representative examples of caged xanthones with a modified C9=C10 double bond.
It should be noted that the chemical structures shown do not define the absolute stereochemistry of the caged motif. This is because the vast majority of the caged Garcinia xanthones have been characterized by NMR spectroscopy methods and thus their absolute stereochemistry remains undetermined. It has been suggested that these natural products exist as racemic mixtures and their enantiomeric composition can be enriched by repeated crystallizations.[25] For instance, chiral HPLC analysis of 13 showed that this compound exists as a 6:4 mixture of two enantiomers, the ratio of which varies with different crystallizations.[25] Moreover, gambogic acid (1) occurs in nature as a mixture of epimers at the C2 center that can be separated by modern chromatographic and analytical techniques.[35] The optical rotation recorded for the C2-R epimer is , whereas that recorded for the C2-S epimer, also referred to as epigambogic acid, is .[36] More recently, a combination of these techniques has been used for the identification of new bio-active xanthones from Garcinia plants.[37]
Biogenesis studies
Biosynthetically, the xanthone backbone of the caged Garcinia natural products is presumed to derive from common benzophenone intermediates of a mixed shikimate–acetate pathway.[38] Whereas xanthones produced in fungi have been shown to be wholly acetate derived,[39] those found in higher plants exhibit oxygenation patterns that originate from a combination of the acetate (ring A) and shikimate (ring C) pathways. This proposed biogenetic scenario is illustrated with the synthesis of maclurin (31) and tetrahydroxy xanthone 32 (Scheme 1).[40] An aldol-type condensation of phosphoenolpyruvate (PEP) and d-erythrose 4-phosphate leads to shikimic acid (27) that after oxidation, dehydration, and enolization forms protocatechuic acid (28). Reaction of 28 with coenzyme A (HSCoA) produces activated ester 29 that can then react with three units of malonyl coenzyme A (malonyl-CoA) to produce intermediate 30. An intramolecular Claisen condensation followed by enolization leads to benzophenones such as maclurin (31). Depending upon the benzophenone produced, this may be a branch point in the biogenesis of other benzophenone-type natural products. It is generally accepted that the formation of xanthones such as 1,3,5,6-tetrahydroxyxanthone occurs by means of phenolic coupling of maclurin (31).[40c,d]
Scheme 1.
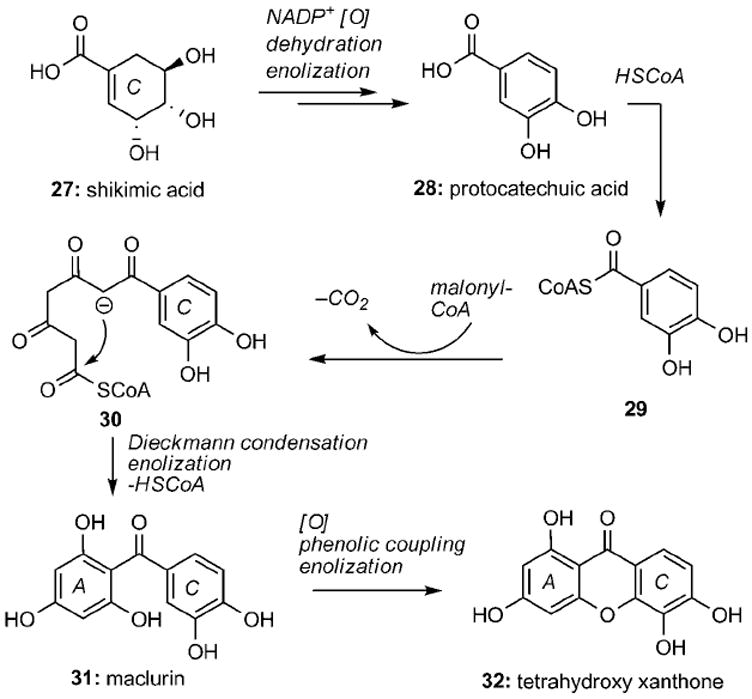
Proposed biosynthesis of benzophenones and xanthones in higher plants. NADP+ = Nicotinamide adenine dinucleotide phosphate.
Different hypotheses have been proposed for the biosynthetic conversion of xanthones, such as 32, to the unusual motif of the caged Garcinia xanthones. The first proposal departed from prenylation of a xanthone, such as 33, at C11 and C13 centers to produce compound 34 (Scheme 2).[13a] Oxidation at the C13 center would then introduce the essential tertiary alcohol, while reduction of the C10=C11 double bond would form compound 35 that can assume the geometry required for cyclization (structure 36). Nucleophilic attack by the C13 tertiary alcohol on the pendant prenyl group was then presumed to initiate the cyclization cascade leading to caged structure 37. The complete caged system 38 could then be formed by oxidation between carbons C9 and C10. The biosynthetic scenarios that followed were variations of this proposal that simply reduced the number of oxidations and reductions.[14a, 19] Unfortunately, both the molecular geometry and the reactivity required for the cascade of nucleophilic attacks from 35 to 37 would make this proposal implausible.
Scheme 2.
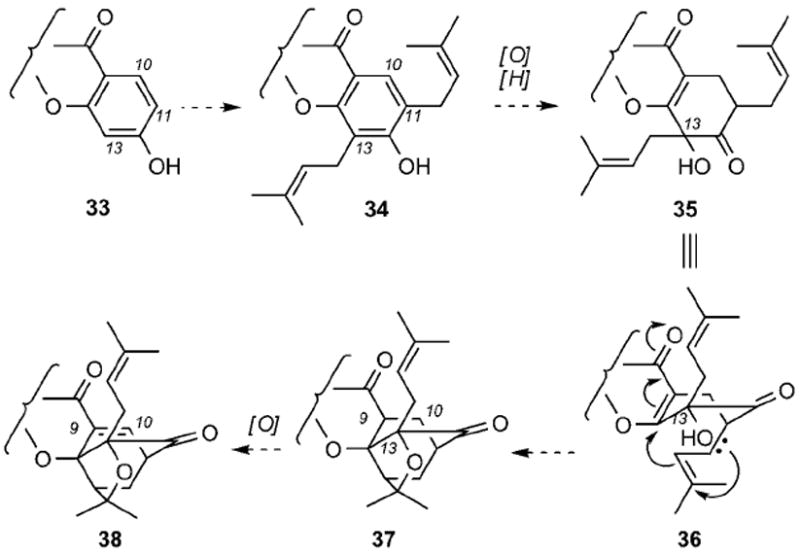
Proposed biosynthesis of the caged xanthone motif through a cascade of nucleophilic attacks.
A more realistic biosynthetic scenario emerged from the pioneering work of Quillinan and Scheinmann.[41] They proposed that “isoprenylation of tetrahydroxyxanthone 32 with four isoprene units can lead to desoxymorellin and related metabolites”. The caged motif of these compounds can thus be formed by a Claisen rearrangement followed by a Diels–Alder reaction on the intermediate dienone. This scenario is highlighted in Scheme 3. It is worth noting that although at that time there was sufficient precedence in support of the Claisen migration,[42] there were no reports on intramolecular Diels–Alder adducts from rearrangements of aryl propargyl ethers. Along these lines, a retro Diels–Alder fragmentation pathway has been detected in mass spectrometry studies of several caged Garcinia xanthones, providing additional support to this biosynthesis proposal.[19e, 36]
Scheme 3.
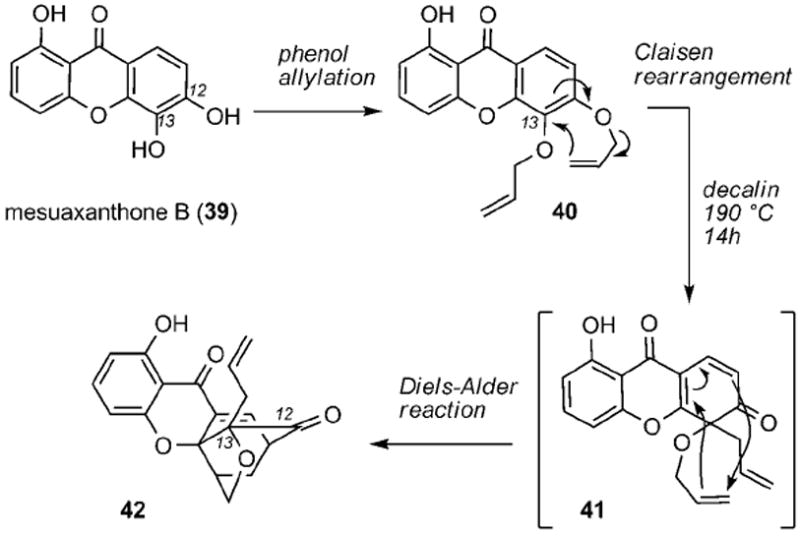
Proposed biosynthesis of the caged xanthone motif by a Claisen/Diels–Alder reaction cascade.
Quillinan and Scheinmann also provided experimental evidence in support of the Claisen/Diels–Alder reaction cascade. Using mesuaxanthone B (39) as the starting material, they successfully prepared and tested their hypothesis on bis(allyloxy) xanthone 40 (Scheme 3). Despite the rather limited experimental details, they showed that heating 40 in boiling decalin (190 °C) for 14 h gave rise to the elusive caged structure, assigned as compound 42, presumably by a Diels–Alder cyclization of Claisen intermediate 41. Compound 42 had similar spectroscopic characteristics as those of morellin, suggesting that this reaction cascade could account for the biogenesis of the caged xanthones.
The lack of details on the synthesis of caged motif 42, together with the high temperature and long reaction times required for the Claisen/Diels–Alder reaction, cast some doubts on the synthetic relevance of this reaction cascade. Likely due to these reasons, the proposal by Quillinan and Scheinmann remained dormant for over 30 years, until Nicolaou and Li masterfully demonstrated its value in a biomimetic synthesis of forbesione (8).[43] Independently, Theodorakis and co-workers explored the site selectivity of the Claisen/Diels–Alder cyclization and further refined the biosyn-thesis scenario for all caged Garcinia xanthones.[44] These studies will be presented in the following sections.
Biological Activities of the Caged Garcinia Xanthones
Antimicrobial and anticancer activities
Aside from their striking chemical structure and biosynthesis, the caged Garcinia natural products exhibit interesting bioactivities and have a documented value in traditional Eastern medicine. In fact, oral and injectable formulations of gamboge have been used in China for the treatment of patients with breast carcinoma and malignant lymphoma.[45] In addition, gamboge has been used topically for treating infected wounds and systemically to alleviate pain and edema. Moreover, in Thai folk medicine, gamboge has been used as a topical anti-infective agent, and internally as a drastic purgative and a vermifuge to treat tapeworm.[23, 46]
Initial biological studies with semipurified gamboge extracts documented its antiprotozoal activities, thus lending support for its indigenous use in the treatment of enteric diseases.[47] Morellin (3) and gambogic acid (1), the major components of semipurified resin extracts of Garcinia morella,[48] exhibited high in vitro specific growth inhibitory effects on Gram-positive bacteria in vitro and a protective action in experimental staphylococcal infections in mice.[49] In particular, acid 1 exhibited high specific inhibitory effect (0.1–1 μg mL−1) on the growth of Gram-positive bacteria, but had little effect against many Gram-negative bacteria, fungi, yeast, and actinomycetes.[50] Further experiments in mice indicated that topical applications of 1 in experimental septic wounds could offer protection against lethal staphylococcal infections at a dose level of 30 mg kg−1 day−1 for 2 days. Interestingly, the susceptibility of a morellin-resistant strain of Staphylococcus aureus to penicillin and erythromycin was unaffected, indicating that there is no cross-resistance between the morellin and the above antibiotics. Both morellin and 1 were well tolerated by rats at a dose of 40 mg kg−1 day−1 for 40 days. However, a dose of 120 mg kg−1 day−1 resulted in side effects including decreased growth and reduced blood cell counts.[50b] More recently, scortechinone B (19) showed antibacterial activity on methicillin-resistant Staphylococcus aureus with an MIC value of 2.0 μg mL−1.[27c]
In addition to their antimicrobial activity, the caged Garcinia xanthones, have received a great deal of attention for their anticancer activity. An ever increasing body of evidence indicates that these compounds are cytotoxic against various cancer cell lines at low μm concentrations.[20c] For instance, desoxymorellin (5) inhibited the growth of HEL (human embryonic lung fibroblasts) and HeLa (Henrietta Lacks cervical cancer) cells with a minimum inhibitory concentration (MIC) of 0.39 μg mL−1.[24, 51] The bractatins (13–15) were cytotoxic against the KB cell line (human epidermoid carcinoma) with 6-O-methylneobractatin (15) showing the lowest IC50 value of 0.20 μg mL−1.[25] The gaudichaudiones (10 and 11) have been tested against a panel of cell lines, and found to be broadly cytotoxic with effective dose (ED50) values between 0.50 and 8.0 μg mL−1.[17a] Selected members of the cantleyanone (17) family displayed significant cytotoxicity against breast cancer (MDA-MB-231 and MCF-7), ovarian cancer (CaOV-3), and HeLa cells with EC50 values ranging from 0.22 to 17.17 μg mL−1.[26] In addition, lateriflorone (22) was cytotoxic against the P388 cancer cell line with an ED50 value of 5.4 μg mL−1.[30] In similar studies, gambogic acid (1) inhibited the proliferation of T47D and DLD-1 breast cancer cells with GI50 values of 0.04 and 0.03 μm, respectively.[52] Similar observations were reported upon screening 1 and related caged Garcinia xanthones against a panel of solid and nonsolid tumor cells.[53]
Of particular interest are the studies on the activity of 1 and related xanthones against various drug-resistant cell lines.[16a,23] For example, compound 1 and its C2 epimer, epigambogic acid, displayed similar cytotoxicity against doxorubicin-sensitive (EC50 1.3 μm) and doxorubicin-resistant (EC50 0.9 μm) human leukemia K562 cells in an MTT assay.[36] More recently, compound 11 was reported to display strong growth inhibitory activity against both parental murine leukemia P388 and P388/doxorubicin-resistant cell lines at low micromolar concentrations.[54] Studies from our own laboratories have confirmed these findings.[53] Specifically, we have found that adriamycin-resistant HL-60 cells have similar sensitivity to the antiproliferative effects of several caged Garcinia xanthones as the parental HL-60 cell line. These findings indicate that the caged Garcinia xanthones are not subjects of the multidrug resistance mechanisms, often associated with overexpression of P glycoprotein that is typical of relapsed cancers. Thus, these compounds represent a pharmacologically promising chemical scaffold.[55]
Mode-of-action studies
The induction of apoptosis has been established as one of the main mechanisms of cytotoxicity exhibited by the caged Garcinia xanthones in several tumor cells.[52, 53] Both 1[52] and 11[54] have been shown to activate caspase 3, a protein that plays a key role in apoptosis.[56] In MGC-803 cells (human gastric carcinoma), apoptosis was induced by 1 after 48 h of treatment with an IC50 of 0.96 μg mL−1.[57] Immunohistochemical studies indicated that 1 regulates the levels of Bax and Bcl-2, a family of proteins that play a crucial role during apoptosis.[58, 59] Several reports have confirmed that 1 increases the expression of bax and decreases expression of bc1-2 genes in a variety of cancer cell lines, including human gastric cancer cells MGC-803,[59] BGC-82,[60] and human malignant melanoma A375 cells.[61] Induction of bax and suppression of bc1-2 is likely to contribute to the apoptosis mechanism in these cell lines. A recent study has reported that 1 competes with BH3 peptides for binding to the Bcl-2 family of proteins thereby inhibiting the antiapoptotic activity of these proteins.[62] In this study, compound 1 inhibited binding of BH3 peptides to 6 members of the Bcl-2 family to various extents with IC50 values of up to 2.0 μm. Importantly, analogues of 1 with reduced ability to compete with BH3 peptides also had lower cytotoxicity. However, compound 1 retained some cytotoxicity in bax−/−/bak−/− cells, suggesting that this compound has additional targets that contribute to its cytotoxicity.
In addition to the regulation of genes directly associated with apoptosis, a collection of work has revealed that multiple other genes are regulated by 1, which may contribute to its anticancer activity. For instance, compound 1 enhanced p53 protein expression, but, interestingly, had no influence on p53 mRNA synthesis.[63] This result is most likely to be due to the down-regulation of mdm2, a negative regulator of p53, at both mRNA and protein levels.
Gambogic acid (1) can affect the cell cycle in multiple ways, ultimately altering cell proliferation.[64] In recent studies, compound 1 suppressed telomerase activity in human gastric carcinoma and lung cancer cells by repressing both the transcriptional activity and posttranslational modification of hTERT.[64a–d] Another study reported that treatment of MCF-7 breast cancer cells with 1 (2.5 μm) caused microtubule cytoskeleton disruption and microtubule depolymerization.[64e] In addition, compound 1 was proposed to inhibit the catalytic activity of human topoisomerase IIα by binding to its ATPase domain.[64f] Other studies have shown that 1 induces G2/M-phase cell-cycle arrest by disturbing the CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma cells.[64g] The proteomic approach has also been used to reveal the molecular targets of 1 in hepatocellular carcinoma. This study showed that the expression of stathmin 1 (STMN1) was significantly down-regulated by 1.[65] This protein plays a major role in the regulation of microtubule dynamics, thus providing additional evidence on the effects of 1 on cell cycle progress.
Steroid receptor coactivator 3 (SRC-3), a member of the p160 family of nuclear receptor coactivators, is an important modulator of cell growth and is often over-expressed in cancer cells. In recent studies, compound 1 was found to decrease the expression of SRC-3 at both the mRNA and protein levels in human lung adenocarcinoma (A549) cells that over-express this receptor coactivator.[66] This action presumably accounts for the observed inhibition of proliferation by 1 in a time- and dose-dependent manner with an IC50 value of (3.17±0.13) μmol L−1 after 24 h of treatment.
Evaluation of the antileukemic effect of 1 led to the observation that nucleoporin Nup88 expression was down-regulated in several leukemia cell lines upon treatment with 1.[67] In addition, the distribution of Nup88 was altered from widely dispersed in both nucleus and cytoplasm to that predominantly localized at the cytoplasmic side of the nuclear membrane. These results suggest that regulation of nucleocytoplasmic transport may be important for the anticancer effects of 1 in leukemia cells.
Of particular significance are the effects of 1 on cancer cell metastasis. The findings indicate that 1 inhibits the adhesion, migration, and invasion of a highly invasive human breast cancer cell line in vitro and in vivo.[68] These effects have been attributed to the ability of 1 to down-regulate the expression of several matrix metalloproteinases. Similar studies in highly metastatic mouse melanoma cells suggest that 1 inhibits cell adhesion and migration by down-regulating α4 integrin expression.[69]
In addition to modulating cellular processes directly affecting cancer cells, compound 1 appears to also have an effect on angiogenesis thereby having the ability to affect tumor cells indirectly. Specifically, compound 1 inhibits angiogenesis by suppressing the activity of vascular endothelial growth factor receptor 2 (VEGFR2) and its downstream protein kinases, c-Src, focal adhesion kinase, and AKT.[70]
The effect of 1 in combination with known anticancer drugs has also been explored. One study showed that 1 could reverse docetaxel resistance in BGC-823/Doc gastric cancer cells.[71] Specifically, treatment of these cells with 1 at concentrations up to 0.2 μm led to a dramatic increase in docetaxel-induced apoptosis. Analysis of apoptosis-associated genes revealed that 1 singly, or in combination with docetaxel, significantly down-regulated the mRNA expression of survivin, a protein associated with resistance to apoptosis.[71] A more recent study showed that the anticancer effect of a simultaneous administration of 5-fluorouridine (5-FU) with 1 was much greater than that of 5-FU or 1 alone.[72] Furthermore, compound 1 was found to regulate the metabolic enzymes involved in 5-FU metabolism. Specifically, compound 1 decreased mRNA levels of thymidine synthetase (TS) and dihydropyrimidine dehydrogenase (DPD), whereas it increased the mRNA level of orotate phosphoribosyltransferase (OPRT). The authors of this study suggest that this mechanism accounts for the observed synergistic effect of 1 used in combination with 5-FU.[72] These studies attest to the potential of 1 in combination therapies.
The combination of published reports on the anticancer activity of 1 clearly indicates that this compound affects several cellular processes and has multiple targets. Studies have indicated that 1 binds to transferrin receptor type 1 (TfR1), a membrane-bound protein involved in iron homeostasis.[73] The authors of this study have proposed that such binding induces a unique signal leading to rapid apoptosis of tumor cells.[74] In this study, the cytotoxicity of 1 and its derivatives correlated with their ability to bind to TfR1. Furthermore, down-regulation of TfR1 in T47D and 293T cells by RNAi, significantly decreased their sensitivity to 1-induced apoptosis. Further evidence for a role of the TfR in the induction of apoptosis by 1 is the finding that 1 potentiated tumor necrosis factor (TNF)-induced apoptosis in human leukemia cells through modulation of the NF-κB signaling pathway, and that this was dependent on the expression of TfR.[75] Down-regulation of TfR by RNAi reversed the effects of 1 on NF-κB signaling and apoptosis. Furthermore, this study demonstrated that 1 enhanced the effect of TNF and chemotherapeutic agents in inhibiting the expression of gene products involved in antiapoptosis, cell proliferation, invasion, and angiogenesis; all of which are known to be regulated by NF-κB. In addition, compound 1 suppressed NF-κB activation induced by various inflammatory agents and carcinogens. Noteworthy, however, is that 1 alone had no effect on NF-κB signaling, in contrast to results of other studies.[76] The authors concluded that 1 inhibits TNF-induced NF-κB signaling and potentiates apoptosis through its interaction with the TfR-1. Although the results of this study indicate that TfR-1 plays a role in potentiating TNF-induced apoptosis, the direct binding of 1 to TfR-1 has not been determined. Hence, it is not clear whether TfR-1 is a primary target of 1 in this case. Interestingly, in another study, 1 was equally cytotoxic against CHO (Chinese hamster ovary) cells deficient in endogenous TfR1 (TRVb-neo) and those expressing exogenous human TfR1 (TRVb-hTfR1), suggesting that the cytotoxicity of 1 is independent of TfR1.[77] These results clearly indicate that 1 has multiple targets, and that binding to TfR1 may or may not be required for the induction of cytotoxic effects; this is likely to depend on the cell context.
More recently, compound 1 was found to induce production of reactive oxygen species (ROS) in human hepatoma SMMC-7721 cells, resulting in the collapse of the mitochondrial membrane potential. This led to the release of cytochrome c and apoptosis-inducing factor from mitochondria, ultimately leading to apoptosis.[78] Moreover, compound 1 elevated the phosphorylation of c-Jun-N-terminal protein kinase (JNK) and p38, downstream effects of ROS accumulation. N-Acetylcysteine, an inhibitor of ROS production, partly reversed the activation of JNK and p38 and the induction of apoptosis in cells treated with 1. These results indicate that 1 induces apoptosis, in part, by activating the cell stress associated MAPK pathway through the production of ROS.
In summary, compound 1 and related molecules appear to have multiple targets and mechanisms accounting for their cytotoxicity against cancer cells. It is likely that the mechanisms involved will depend on the cellular context. Importantly, further work by independent laboratories will be needed to verify the primary target(s) and most relevant key signaling pathways involved in the action of 1.
Pharmacology and animal model studies
Several studies with 1 in animal models have documented its low toxicity and promising chemotherapeutic value. Of note, in a study using a rat glioma model, it was shown that 1 was taken up by brain microvascular endothelial cells (rBMEC) in a time-dependent fashion, indicating that this compound could pass through the blood brain barrier. Furthermore, intravenous injection of 1 once a day for two weeks could significantly reduce tumor volume by inhibiting angiogenesis and inducing apoptosis of glioma cells.[79] As such, this study reveals a possible new therapeutic lead in glioma therapy. In a study using a rat gastric carcinoma model,[57] intravenous (iv) injection of 1 at 6 mg kg−1 (4 doses on alternate days) did not affect the body weight or white blood cell count of healthy rats, but induced apoptosis of MGC-803 gastric carcinoma cells. These findings support the notion that 1 has little toxicity at therapeutic doses and displays significant tumor selectivity.
Further evidence for the tumor selectivity of 1 is provided by multiple studies both in vitro and in vivo.[80] For instance, compound 1 selectively induced apoptosis of human hepatoma SMMC-7721 cells, whereas it had relatively less effect on human normal embryonic hepatic L02 cells and primary rat hepatic cells.[80a] This study showed that treatment of mice with SMMC-7721 tumors with 1 at dosages of 2, 4, 8 mg kg−1 resulted in 33.1, 50.3, and 64.2% inhibition of tumor growth, respectively, compared with vehicle control. Moreover, compound 1 was more potent than the standard agent cyclophosphamide (66% inhibition of tumor growth at 30 mg kg−1). The authors suggest that the tumor selectivity of 1 may be partly due to its longer retention time in grafted tumor than in the liver, renal system, and other organs. Additional evidence for the tumor selectivity and efficacy of 1 in animal tumor models is the finding that 1 activated T lymphocytes to induce cancer cell apoptosis in H22 transplanted mice.[80c] Hence, the anticancer effects of 1 appear to be at two levels, including direct effects on tumor cells as well as activation of immune cells against tumor cells.
In a study examining the acute and chronic toxicity in experimental animals, the LD50 of 1 in albino mice was in the range of 43.18–48.45 mg kg−1.[81] The results from the chronic toxicity studies using beagles demonstrated that the toxicity targets were liver and kidney. The innocuous dose was established to be 4 mg kg−1 after administration to dogs for a total of 13 weeks at a frequency of one injection every other day. This dose was approximately 9.6 (body weight) or 5.1 (body surface area) times the dosage (25 mg/60 kg, every other day) usually recommended for human trials. Similarly, in a chronic toxicity study using Sprague Dawley rats, oral administration of 1 at 120 mg kg−1 for 13 weeks resulted in damage of the kidney and liver.[82] An innocuous dose was established to be 60 mg kg−1 upon oral administration for a total of 13 weeks at a frequency of one administration every other day. This dose was approximately 18.0 (body weight) or 9.6 (body surface area) times higher than that of the dose (200 mg/60 kg, every other day) used for human trials.[82 Additional toxicology studies using beagles revealed that doses of 1 up to 4 mg kg−1 administered intravenously had no effect on blood pressure, heart rate, or rate of respiration.[83] However, higher doses of 1 in mice could reduce motor co-ordination in a dose-dependent manner. The most significant toxicity effects of 1 in this study were the reduction of maternal and fetal body weight as well as inhibition of fetal skeletal development at doses of 15 mg kg−1 and above. Also of note was the analgesic activity of 1, which was hypothesized to be due to the anti-inflammatory properties of 1.[83]
The plasma pharmacokinetics, excretion, and tissue distribution of 1 were investigated in several recent studies.[84] The results showed that 1 was not detected in the urine after iv administration and was rapidly eliminated from the blood and transferred to the tissues. The tissue distribution of 1 was limited with the highest concentrations found in the liver. Moreover, the majority of 1 appeared to be excreted into the bile within 16 h of iv administration. In metabolism studies using rat liver microsomes, compound 1 was rapidly converted to 10-hydroxygambogic acid, 9,10-epoxygambogic acid, and their glucuronyl derivatives.[85] The formation of 10-hydroxygambogic acid via cytochrome P-450 1A2 was found to be crucial for the elimination of 1 in rats. Hence, inhibitors of cytochrome P-450 1A2 can affect the metabolism of 1 and its bioactivity. This finding suggests that possible drug–drug interactions may result in combination therapies if cytochrome P-450 1A2 activity is affected. Moreover, issues related to the stability and tissue distribution of 1 can be addressed by developing appropriate delivery platforms. For instance, gambogic acid (1)-loaded micelles based on chitosan derivatives showed increased stability and decreased acute toxicity and vein irritation than a non formulated delivery of this compound.[86]
In summary, potent anticancer activity, both in vitro and in vivo, and relatively low toxicity indicate that 1 may be an effective chemotherapeutic agent warranting further study in clinical trials. In fact, this compound has entered clinical trials in cancer patients in China.[87] In turn, this suggests that the caged Garcinia xanthones and designed analogues thereof have promising clinical potential.
Synthetic Strategies Toward the Caged Garcinia Xanthones
Due to the impressive combination of unique chemical architecture, intriguing biological activities, and good potential in medicine, the chemical scaffold of the caged Garcinia xanthones received significant attention as synthetic targets. The synthesis efforts rely on two general strategies for the construction of the caged motif: a tandem Wessely oxidation/Diels–Alder reaction and a sequence of Claisen rearrangements and Diels–Alder reaction.
Tandem Wessely oxidation/Diels–Alder reaction
Since Wessely’s first reports in 1950s,[88] lead tetraacetate mediated oxidation of phenols has become a convenient method to generate o-benzoquinones that, in the presence of a dienophile, can participate in a Diels–Alder cycloaddition reaction. If the phenol and the dienophile are covalently linked in the starting material, then the tandem Wessely oxidation/Diels–Alder reaction can produce tricyclic motifs. Yates and co-workers applied this reaction for the synthesis of compounds reminiscent to the structures of the caged Garcinia xanthones (Scheme 4). Thus treatment of phenol 43 with Pb(OAc)4 in acetic acid produced 2,4-cyclohexadienone 44 that, upon heating at 140 °C, formed compound 45 (gambogic acid numbering).[89] In a subsequent study, xanthene 46 was treated with lead tetraacrylate (formed in situ by Pb-(OAc)4 and acrylic acid) to produce dienone 47 and, after an intramolecular Diels–Alder reaction, caged compound 48.[90]
Scheme 4.
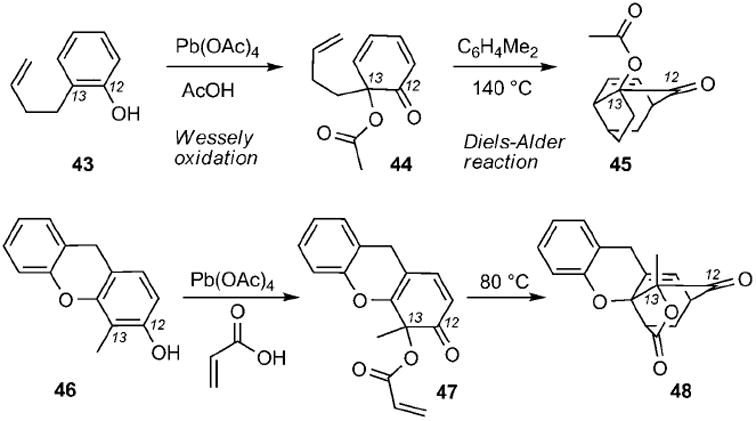
Representative examples of caged structures 45 and 48 formed by a Wessely oxidation/Diels–Alder reaction cascade.
Theodorakis and co-workers[91] investigated the application of this strategy for the synthesis of a more hydroxylated caged motif reminiscent to the structure of lateriflorone (22) (Scheme 5). Treatment of 49 with Pb(OAc)4 in acrylic acid/dichloromethane produced, after heating in benzene at reflux (80 °C), tricyclic lactone 51 in 82% combined yield. Crystallographic studies established that 51 is a constitutional isomer of desired structure 54 and is reminiscent of the so-called neo caged structure. The connectivity of compound 51 suggested that during the Wessely oxidation the acrylate unit was attached exclusively at the more electronically rich C11 center of 49, instead of the desired C13 carbon. In turn, this produced dienone 50 that subsequently underwent an efficient Diels–Alder cycloaddition with the pendant acrylate dienophile. To alter the connectivity of the caged structure, one could have the acetoxy group preinstalled at the C13 center and promote the migration of the prenyl group. Along these lines, heating of allyl ether 52 in m-xylene (140 °C), gave rise exclusively to caged motif 54 through a Claisen rearrangement and Diels–Alder cycloaddition. The selectivity of the Claisen rearrangement at the C13 center can be explained by considering that intermediate 53 has the necessary geometry that allows it to be trapped as the Diels–Alder adduct.
Scheme 5.
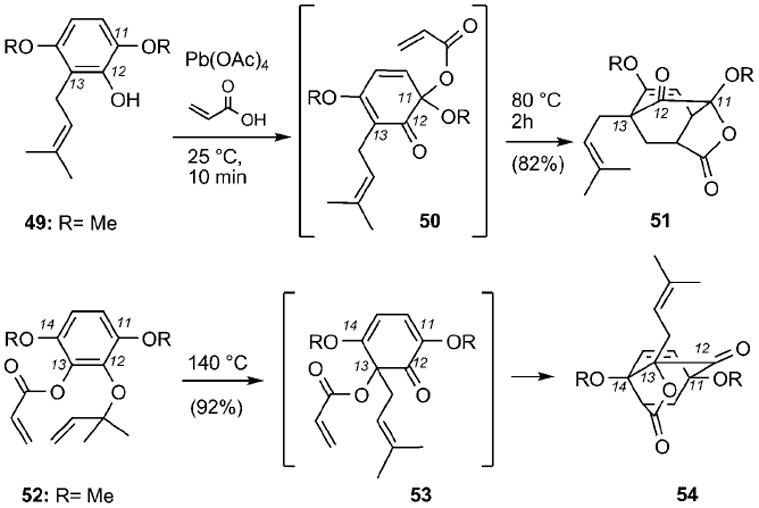
Synthesis of caged structures 51 and 54.
The tandem Wessely oxidation/Diels–Alder sequence was revived recently by Mehta and Maity (Scheme 6).[92] Exposure of phenol 55 to lead tetraacrylate followed by heating in benzene at reflux produced a mixture of two caged structures 56 and 57, reminiscent of the regular and neo caged motifs respectively, in 1:1 ratio and 71% combined yield. In this case, the acrylate addition proceeds at both C11 and C13 centers with low selectivity due to their similar electronic density.
Scheme 6.
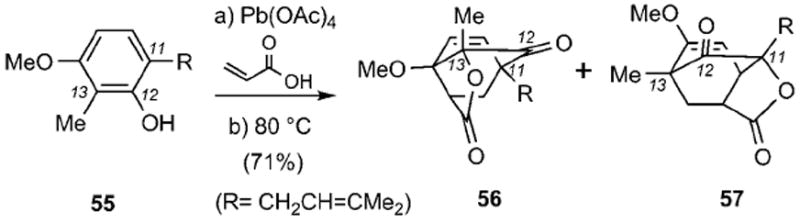
Synthesis of caged structures 56 and 57.
Tandem Claisen/Diels–Alder reaction
Inspired by the Quillinan and Sheinman proposed biosynthesis, Nicolaou and Li evaluated the tandem Claisen/Diels–Alder sequence for the conversion of bis(allylated) coumarin 59 to a caged scaffold (Scheme 7).[43] In principle, two competing migrations can occur during the initial Claisen rearrangement of 59 to form intermediates 60 and 61 (path A and path B, respectively). Each one of these intermediates can participate in two intramolecular Diels–Alder reactions with the coumarin core producing compounds 62–65 (Scheme 7). Indeed, upon heating of 59 at 125 °C, these compounds were isolated in 91% combined yield.
Scheme 7.
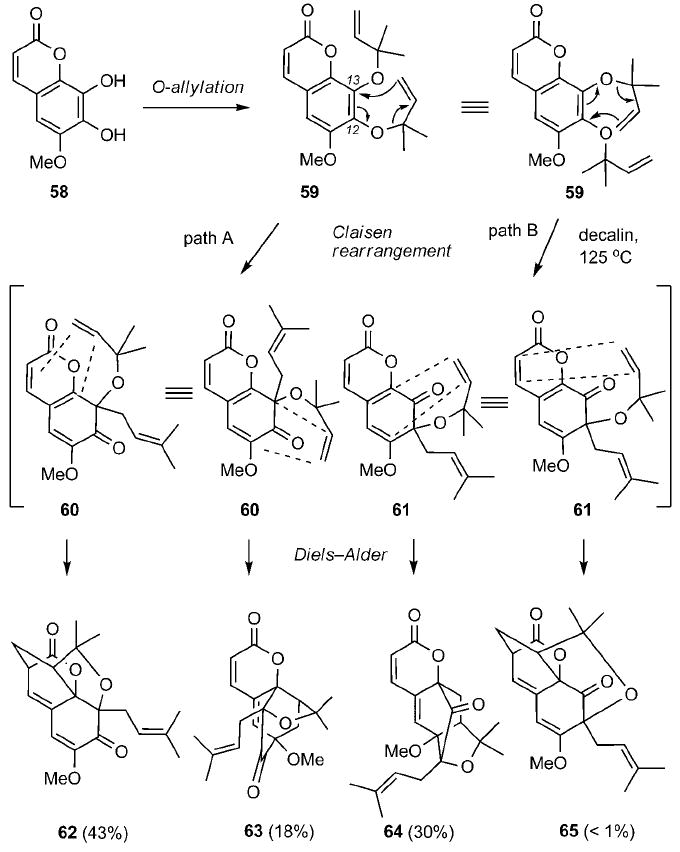
Model studies on the Claisen/Diels–Alder reaction cascade with prenylated coumarin 59.
Application of this approach to the synthesis of forbesione[43] and lateriflorone[93, 94] provided further support for the Quillinan–Scheinmann biosynthetic hypothesis. For example, heating of allyl ether 66 at 120 °C produced a 1:1 mixture of 67 and 68 and in 89% combined yield (Scheme 8).[93] The carbon connectivity of these products corresponds to that of the regular and neo caged scaffolds and indicates that, under these conditions, the Claisen rearrangement proceeds with no significant site selectivity. Interestingly, heating of compound 69 to 110 °C proceeded through a selective Claisen rearrangement to form dienone 70, which after the Diels–Alder cycloaddition afforded exclusively the regular caged structure 71 (85% isolated yield).[94] Comparison of these results suggests that the selectivity of the opening Claisen rearrangement can be controlled by the electronic density of the C8 carbon. It is reasonable to postulate that the C8 carbonyl group, which is para to the C12 allyloxy group, facilitates its Claisen migration at the C13 center.
Scheme 8.
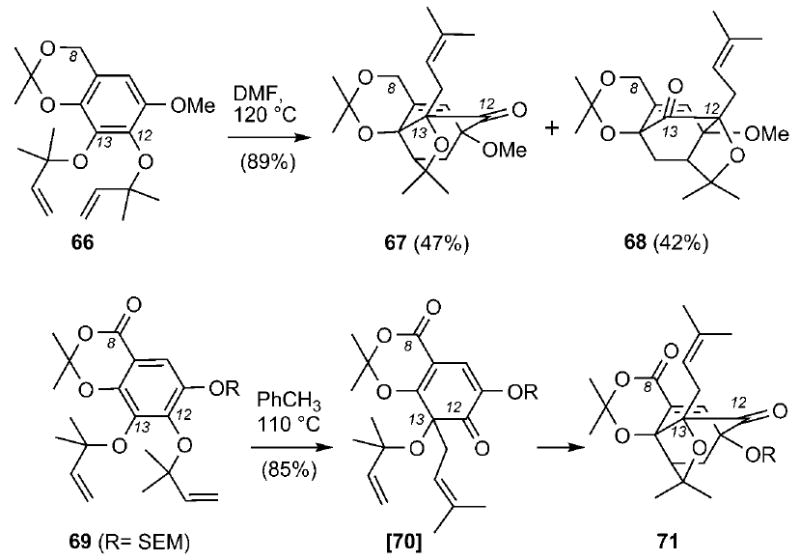
Construction of caged structures 67, 68 and 71 through a biomimetic Claisen/Diels–Alder reaction cascade. SEM = [2-(trimethylsilyl)ethoxy]methyl.
Parallel studies by the Nicolaou[43] and Theodorakis[95] laboratories evaluated the possibility to synthesize forbesione (8) in one pot by using tris(allylated) xanthone 72 as the starting material (Scheme 9). In principle, exposure of such a motif to heat could produce four products arising from a combination of two competing C-ring Claisen/Diels–Alder reactions (producing regular and neo caged structures) and two competing A-ring Claisen migrations (producing C17 and C5 prenylations). Working with methoxy xanthone 72 c (R = Me), the Nicolaou group was the first to describe its conversion to methyl forbesione (73 c) and methyl neoforbesione (74 c) in a 2.4:1 ratio and 89% combined yield.[43] On the other hand, studies by the Theodorakis group showed that heating of xanthone 72 a to 120 °C led only to isolation of forbesione (73 a) and isoforbesione (75 a) in 84% combined yield. The neo C-ring isomers 74 a and 76 a were not detected in this case. More impressively, the O6-acetylated xanthone 72 b afforded, upon heating, solely acetyl forbesione (73 b, 79% isolated yield).[95] Similar observations have recently been reported by other groups.[96] Several model studies were designed to rationalize these findings.[44] The results can be summarized as follows:
The C-ring Claisen/Diels–Alder rearrangement proceeds first and is followed by the A-ring Claisen reaction.
The site selectivity of the A-ring Claisen rearrangement (C5 versus C17 prenylation) is controlled by the steric and electronic effects of the C6 phenolic substituent.
The site-selectivity of the C-ring Claisen/Diels–Alder reaction is attributed to and governed by the electronic density of the C8 carbonyl group. Being para to the C12 allyloxy unit, the electronically deficient C8 carbonyl carbon polarizes selectively the O–C28 bond and facilitates its rupture. In turn, this leads to a site-selective Claisen rearrangement of the C12 allyloxy unit onto the C13 center, thereby producing exclusively the regular caged motif found in the structure of forbesione (73 a).
The substitution of the C6 phenol can regulate the electronic density of the C8 carbonyl group and thus affect the site selectivity of the C-ring Claisen/Diels Alder reaction.
Scheme 9.
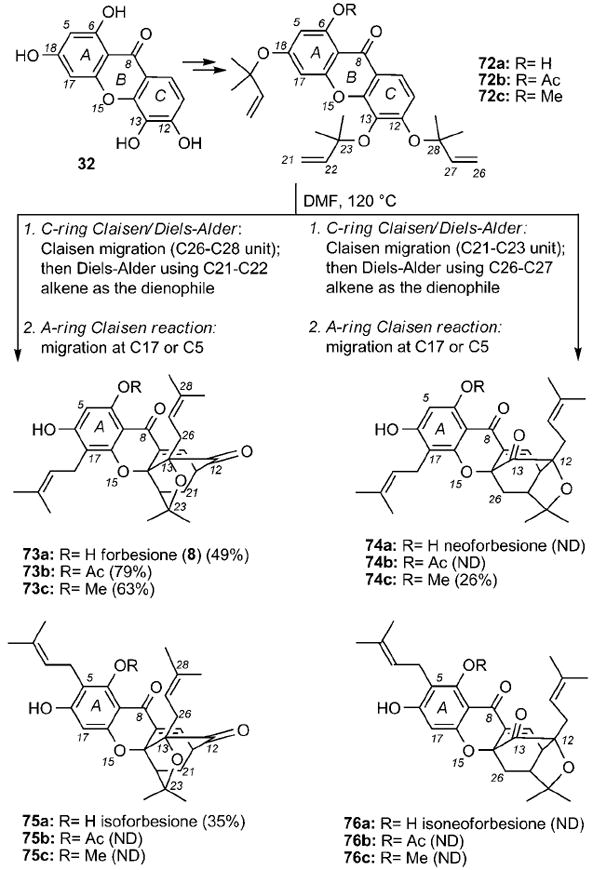
Biomimetic synthesis of forbesione (8) and related structures through a Claisen/Diels–Alder/Claisen reaction cascade. ND = yield not determined.
The experimental findings on the tandem Claisen/Diels–Alder/Claisen reaction cascade provide useful insights regarding the biosynthesis of all known caged Garcinia xanthones.[44] All natural products share a common caged motif, exemplified by structure 7, except 6-O-methylneobractatin (15), which contains the neo caged motif. The remote electronic effects of the seemingly innocuous 6-O-methyl group may explain the concomitant biosynthesis of both 6-O-methylbractatin (14) and 6-O-methylneobractatin (15).
Recent studies by the Nicolaou group have shown that the Claisen/Diels–Alder reaction can be accelerated in the presence of polar solvents.[97] For instance, the conversion of allyl ether 77 to caged structure 78 a and its neo isomer 78b (structure shown in Scheme 13) was dramatically accelerated upon changing the solvent from benzene (100 °C, 4 h, 0%) to DMF (100 °C, 2 h, 75%) to a 1:2 mixture of MeOH/water (100 °C, 0.5 h, 100%) (Scheme 10). It has been proposed that polar aprotic solvents, such as DMF, and more impressively protic solvents, such as water, can accelerate the Claisen rearrangement by stabilizing its polar transition state.[98] The concurrent acceleration of the Diels–Alder component of this cascade is likely due to the hydrophobic effect of water[99] rather than to a polarity or hydrogen-bonding phenomena.[100] Computational studies on the reaction depicted in Scheme 10 have also concluded that the Claisen rearrangement is reversible and the energetics of the irreversible Diels–Alder cyclization can determine the product formation.[101]
Scheme 13.
Biomimetic synthesis of gambogin (2). HMDS = hexamethyldisilazane, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Scheme 10.
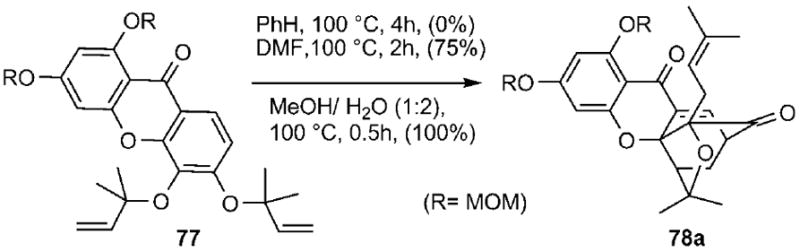
Solvent effect on the rate of the Claisen/Diels–Alder reaction cascade. MOM = methoxymethyl.
Synthesis of selected caged Garcinia xanthones
Biomimetic synthesis of 6-O-methylforbesione
Developed by Nicolaou and Li,[43] the synthetic strategy toward 6-O-methylforbesione is highlighted in Scheme 11. Construction of xanthone 81 proceeded in 5 steps: a) coupling of the lithium salt of 79 with aldehyde 80; b) deprotection of the C14 silyl ether; c) oxidation of the C8 alcohol; d) cyclization of the C14 alkoxy group; and e) deprotection of the C12 and C13 benzyl ethers (78% combined yield). Treatment of 81 with α-bromoisobutyraldehyde (82) and tBuOK followed by Wittig olefination produced a mixture of 83 a and 83b that, after reiteration of the alkylation/olefination steps, gave rise to tris(prenylated) xanthone 72 c in 55% combined yield. Heating of 72 c in DMF at 120 °C for 20 min induced the anticipated Claisen/Diels–Alder/Claisen reaction and produced compound 73 c together with its neo isomer 74 c in 89% combined yield.
Scheme 11.
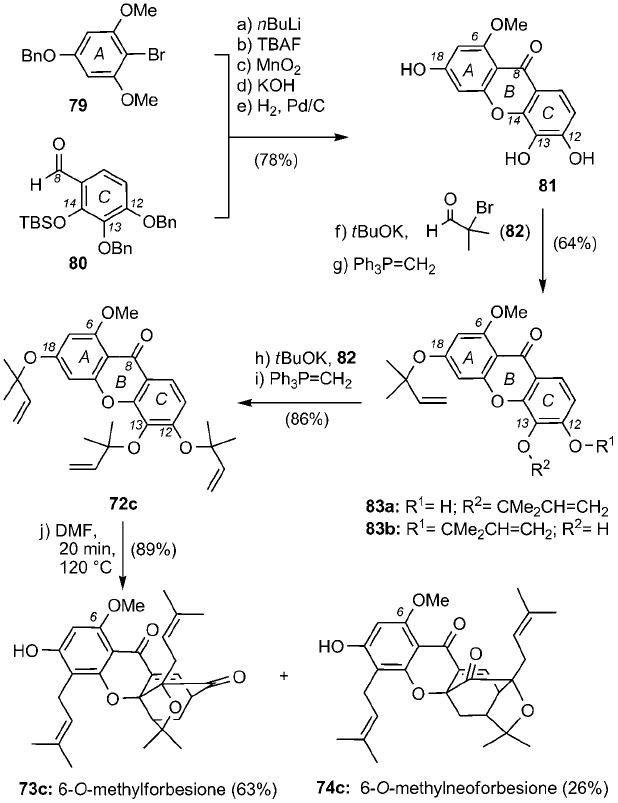
Biomimetic synthesis of 6-O-methylforbesione. TBAF = tetrabutylammonium fluoride, Bn = benzyl, TBS = tert-butyldimethylsilyl.
Unified synthesis of caged Garcinia xanthones
The common structural features of several caged Garcinia natural products suggest that these compounds can be synthesized by decorating the A ring of forbesione (8). Such a strategy, developed by the Theodorakis group, uses forbesione (8) as a node in a unified synthesis of representative members of the gaudichaudiones (10), morellins (5), and gambogins (2).[44] Scheme 12 highlights this plan. ZnCl2-mediated condensation of phloroglucinol (84) with benzoic acid 85 produced xanthone 32 in 46% yield. Propargylation of 32 with propargyl chloride 86 followed by partial reduction of the alkyne units and acetylation of the C6 phenol formed compound 72b in 16% combined yield. The Claisen/Diels–Alder/Claisen reaction cascade gave rise, after deacetylation, forbesione (8) in 72% combined yield. Propargylation of the C18 phenol with chloride 86 afforded, after Lindlar reduction and Claisen rearrangement desoxygaudichaudione A (10) in 34% yield. On the other hand, propargylation and Claisen rearrangement of forbesione formed desoxymorellin (5) in 61% combined yield. In a similar manner, condensation of 8 with citral (88) in Et3N produced gambogin (2) in 75% overall yield.
Scheme 12.
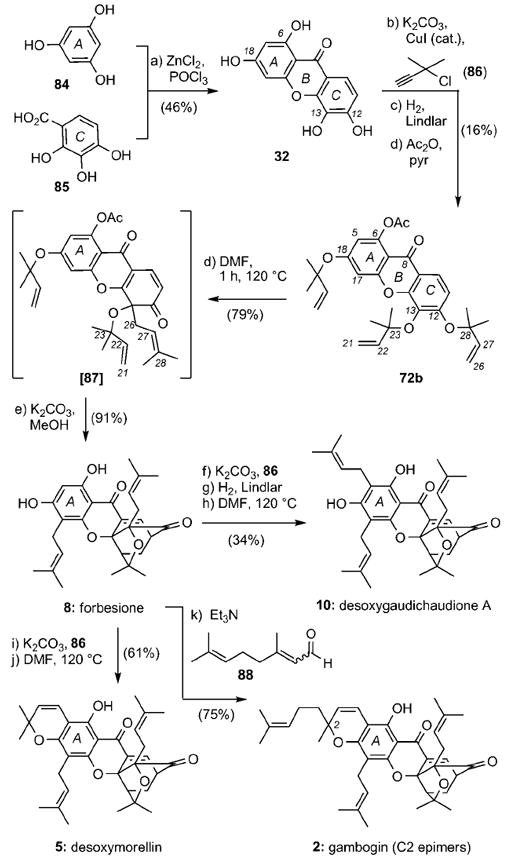
Unified biomimetic synthesis of caged Garcinia xanthones.
Synthesis of 2
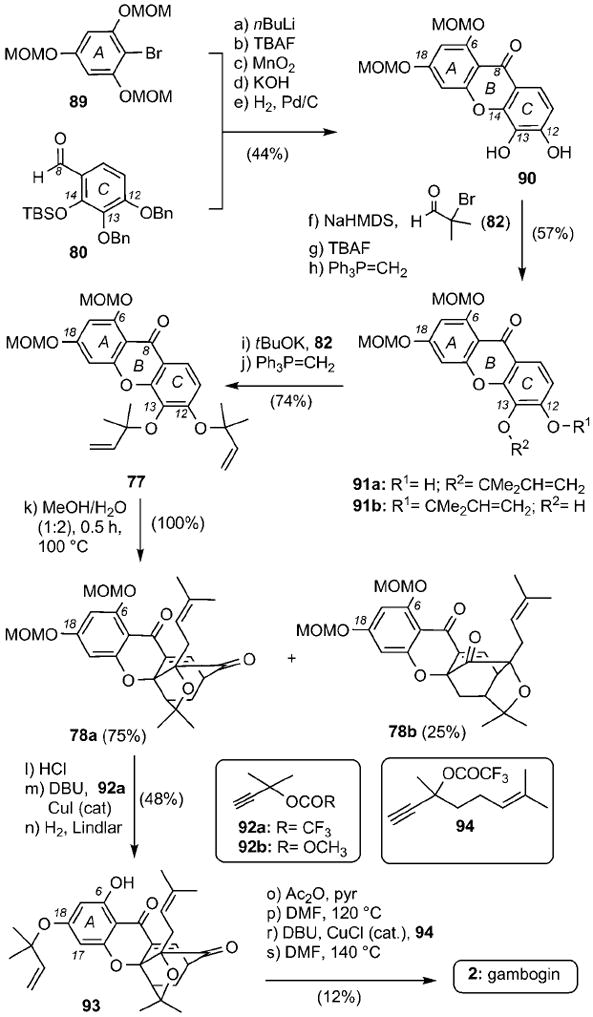
An alternative synthesis of 2 has recently been reported by the Nicolaou group (Scheme 13).[97] The plan involved construction of partially protected xanthone 90, available in 5 steps from coupling of bromide 89 with aldehyde 80 (44% overall yield). Two rounds of alkylation with α-bromoisobutyraldehyde (82) and Wittig olefination produced compound 77 in 42% combined yield. The Claisen/Diels–Alder reaction proceeded in quantitative yield in MeOH/H2O (1:2) at reflux to form regular caged motif 78 a together with the neo isomer 78b in a 3:1 ratio. MOM deprotection of 78 a followed by propargylation with 92 a at the C18 center and Lindlar reduction afforded compound 93 in 48% combined yield. This compound was then converted to 2 by a sequence of steps that involved acetylation of the C6 phenol, Claisen rearrangement to install the prenyl group at the C17 center, propargylation of the resulting phenol with alkyne 94, and Claisen rearrangement to form the dihydropyran ring of the natural product (12% over 4 steps).
Studies toward the synthesis of lateriflorone
It has been proposed that the unprecedented spiroxalactone motif of lateriflorone (22) could be formed by condensation of two fully functionalized fragments 95 and 96 (Scheme 14). An alternative and likely more biosynthetically relevant hypothesis could involve conversion of xanthone 97 into dioxepanone 98 that, upon hydrolysis and spirocyclization at the C16 center, could form the spiroxalactone ring system of lateriflorone.
Scheme 14.
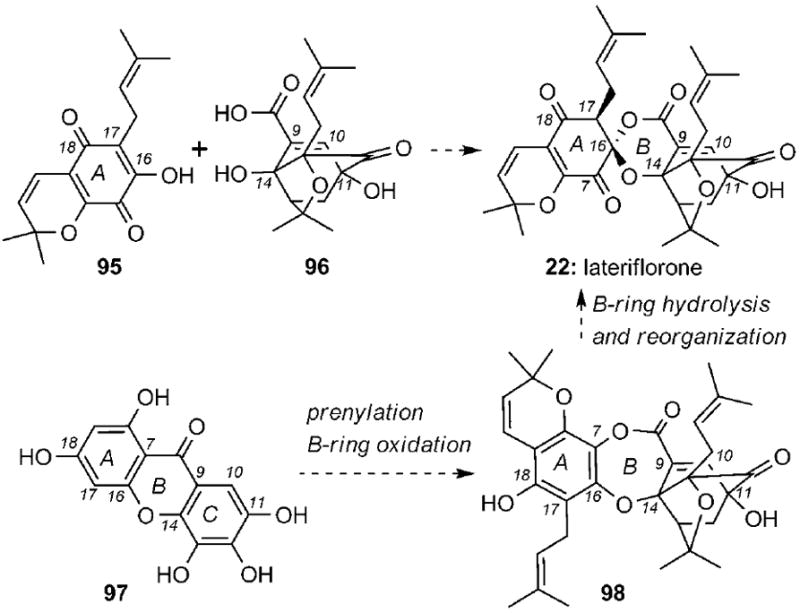
Synthetic plans toward lateriflorone (22) based on biogenetic scenarios.
The Theodorakis group has reported an approach toward the synthesis of quinone 95 representing the A ring of lateriflorone (Scheme 15).[102] A sequence of six steps was used to convert phloroglucinol (84) to chromanol 99, which, upon propargylation with 92b, Lindlar reduction and Claisen rearrangement, gave rise to phenol 100 (32% combined yield). Oxidation of 100 at the C7 center using Fremy’s salt and deprotection of the MEM ether then formed chromenequinone 95 in 66% combined yield.
Scheme 15.
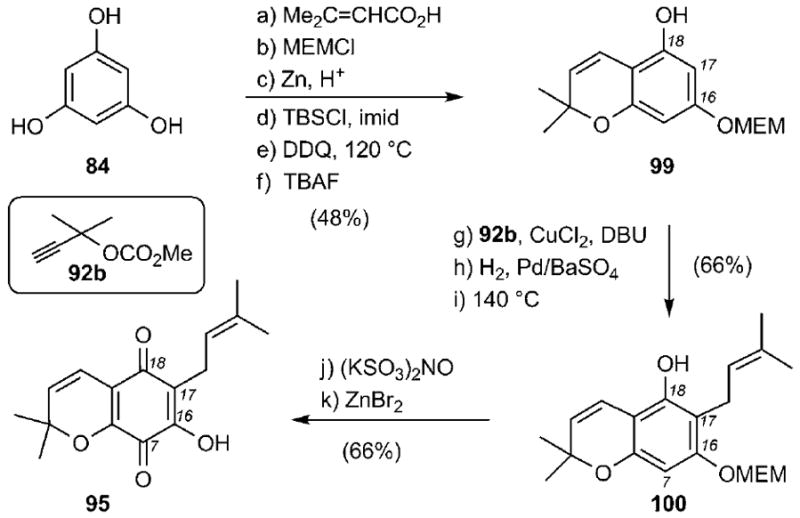
Synthesis of chromenequinone 95. MEMCl = 2-methoxyethoxymethyl chloride, DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
The synthesis of caged motif 96 and its coupling with quinone 95 are summarized in Scheme 16. 4-Hydroxysalicylic acid (101), containing only two of the four hydroxy groups needed, was selectively brominated at the C11 center and then converted to benzopyran 102 (50% combined yield). Oxygenation at the C11 center via the intermediacy of a boronic acid, followed by oxygenation and reverse prenylation at the C13 center, produced compound 69.[94] A site-selective Claisen/Diels–Alder reaction gave rise, after deprotection, to desired fragment 96 (45% combined yield). Coupling of 95 with 96 then produced secolateriflorone (104), which did not undergo the desired spiroxalactonization reaction.
Scheme 16.
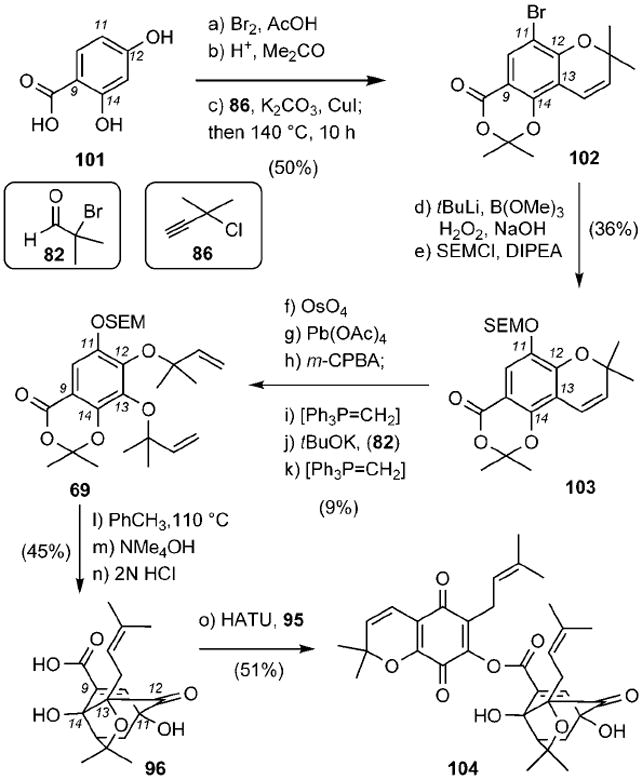
Synthesis of secolateriflorone (104). DIPEA = N,N-diisopropylethylamine, m-CPBA = m-chloroperoxybenzoic acid, HATU = 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate.
More recently, the Nicolaou group has reported a synthesis of C11-methyllateriflorone (110) (Scheme 17).[103] Key to the strategy was the coupling of orthogonally protected hydroquinone 105 with acid 106, which after selective deprotection of the C7 MOM ether produced compound 107 (61% combined yield). Oxidation of 107 in the presence of iodosobenzene bis(trifluoroacetate) in methanol, followed by heating under acidic conditions formed spiroxalactone 109 in 42% combined yield. Acid-catalyzed hydrolysis of 109 gave rise to C11-methyllateriflorone (110) in 66% yield.
Scheme 17.
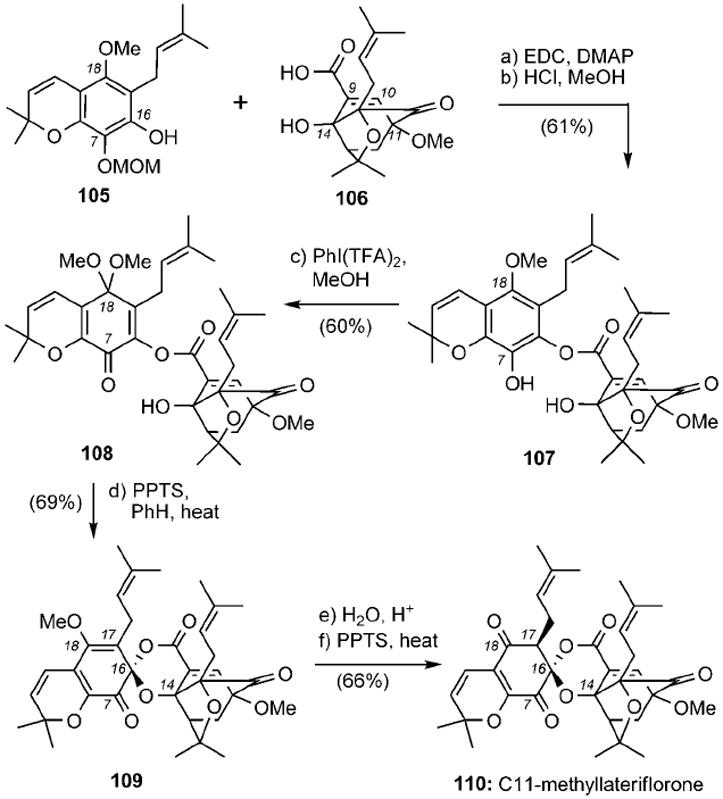
Synthesis of C11-methyllateriflorone (110). EDC = N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide, DMAP = 4-(N,N-dimethylamino)pyridine, TFA = trifluoroacetate, PPTS = pyridinium p-toluenesulfonate.
Structure–Activity Relationship (SAR) Studies
The underexplored chemical structures and promising biological activities of the caged Garcinia xanthones have fuelled several structure-activity relationship studies. The majority of this effort has been focused on the evaluation of derivatives of 1. Early SAR studies showed that the carboxylic acid of 1 can be functionalized with groups that modulate the solubility and selectivity properties of the parent molecule without affecting substantially its bioactivity.[52] For example, 2-ethoxyethyl-gambogamide (111)[104] and glycine conjugate 112[105] exhibited comparable antitumor efficacy, but improved water solubility as compared with 1 (Scheme 18). More recently, studies with neurons showed that gambogic amide 113 can bind to and activate tyrosine kinase A (TrkA) by promoting its dimerization and auto-phosphorylation. Furthermore, this compound prevents neuronal cell death and provokes prominent neurotrophic activity.[106] Under the same conditions, gambogic acid (1) fails to induce TrkA phosphorylation. The carboxylic acid has also been used for the incorporation of biotin and related probes, such as in compound 114, in an effort to identify the cellular target of 1.[52]
Scheme 18.
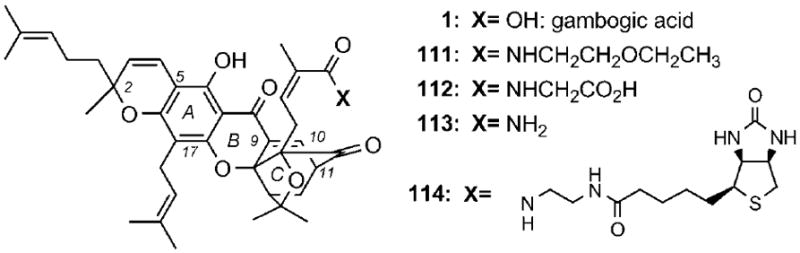
Selected structures of gambogic acid conjugates.
Recent studies have shown that oxidation and/or epoxidation of the prenyl groups of 1 can lead to analogues with improved solubilities and cytotoxicities.[107] On the other hand, the caged core of 1 is needed for bioactivity, since regular xanthones display reduced cytotoxicities.[108] More importantly, the α,β-unsaturated motif of 1 is critical to its bioactivity and metabolic stability. In fact, conjugate reduction or alkylation of the C9=C10 double bond, to form compounds 115 and 116, respectively, decreased the cytotoxicity of the parent molecule by more than two orders of magnitude (Scheme 19).[52] A similar decrease in activity has been recorded for compounds 24 and 117.[20c,33] These compounds have been identified upon prolonged storage (one week at room temperature) of 24 in methanol and ethanol, respectively. Moreover, the C10 hydroxygambogic acid (118) was identified as one of the main metabolites of 1 formed in vivo in rat bile.[85] These findings indicate that 1 and related caged Garcinia xanthones could exert their bioactivities by reacting in cells as conjugate electrophiles across the C9–C10 enone motif.
Scheme 19.
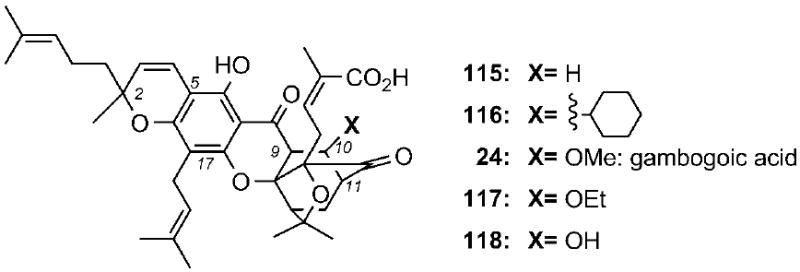
Selected structures of gambogic acid derivatives containing functionalities at the C9–C10 bond.
To evaluate whether the entire xanthone backbone of the caged natural products is essential to the bioactivity, the Theodorakis group synthesized several simplified analogues of the caged xanthone motif, containing progressively smaller fragments of the caged structure.[53, 109] Representative examples of this effort are shown in Scheme 20. Comparison of the cytotoxicity values indicated that compound 119, which contains the central caged xanthone structure but lacks the prenyl groups at the periphery of the A ring, maintains the bioactivity of 1 and related natural products. However, compounds 120, 121, and 122, containing further deletions of the xanthone backbone, have substantially decreased bioactivities. Similar observations have been made for compounds 123 and 124.[110]
Scheme 20.
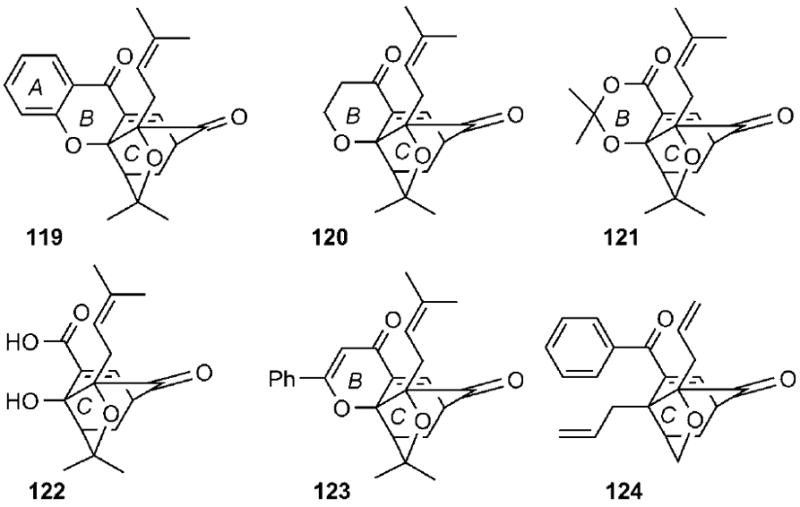
Selected caged structures used to evaluate the minimum pharmacophore of the caged Garcinia xanthones.
Scheme 21 summarizes the SAR studies with 1. The minimum pharmacophoric motif is represented by the red structure and includes the ABC tricyclic xanthone backbone and the C-ring caged structure. Functionalization at the periphery of this motif is not essential to bioactivity, but could be used to address issues related to solubility and target selectivity of the parent molecule.
Scheme 21.
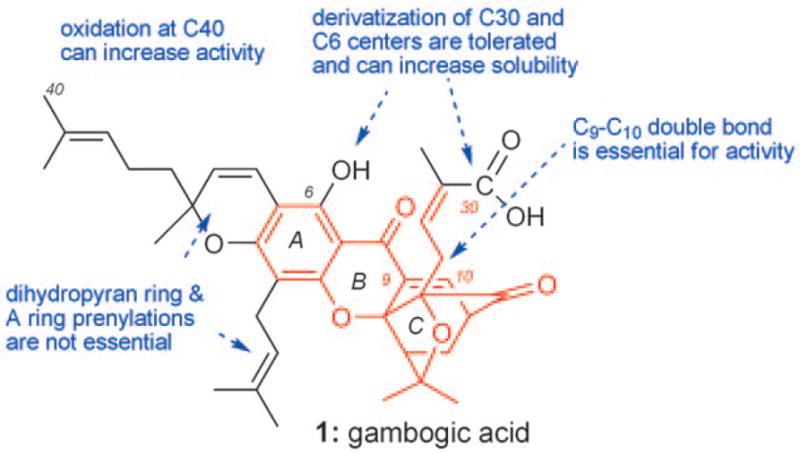
Summary of SAR studies with 1 and related analogues.
An optimized synthesis of 119, referred to as cluvenone, has been recently reported (Scheme 22).[109] Xanthone 128 can be prepared by a Friedel–Crafts acylation of 2-fluorobenzoyl chloride (125) with pyrrogallol (126) followed a base-induced cyclization of the resulting benzophenone 127 (two steps, 34% combined yield). A new palladium-catalyzed reverse prenylation of 128 with 1,1-dimethylpropenyl tert-butyl carbonate formed compound 129, which upon heating at 120 °C gave 119 in 81% yield together with small amounts of the neo isomer 130 (14%). The streamlined synthesis of this compound allows further preclinical investigation of this and related caged Garcinia xanthone analogues.
Scheme 22.
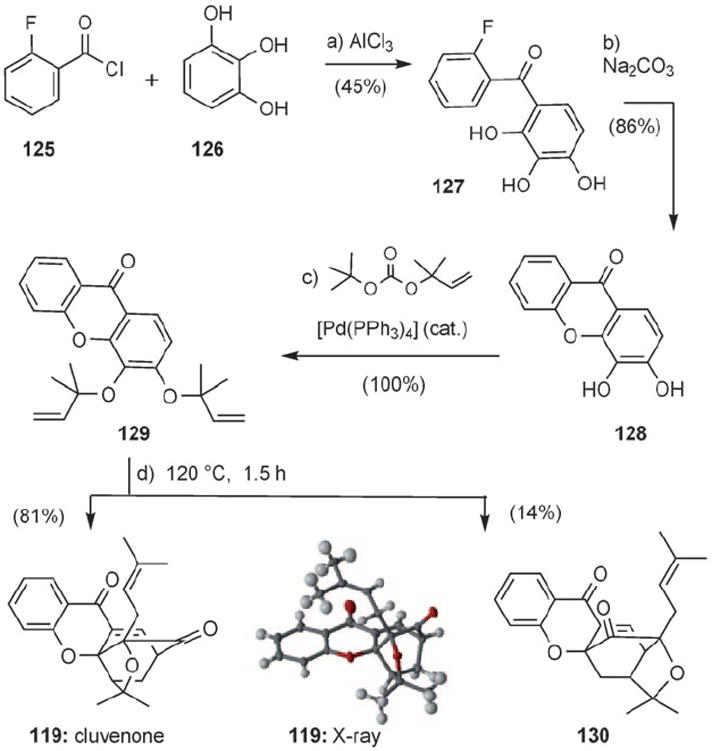
Optimized synthesis of cluvenone (119) by using a Pd0-catalyzed reverse prenylation reaction.
Summary and Outlook
Gamboge, the golden resin of certain Garcinia trees, has a spectacular history in the arts and sciences. Its use in traditional medicine led, in the recent years, to the discovery of a new family of natural products, collectively referred to as caged Garcinia xanthones. The chemical structure, biosynthesis, biology, and medicinal potential of these compounds are remarkable. Their chemical structure is represented by an unusual xanthone backbone in which the C ring has been converted to a 4-oxa-tricyclo[4.3.1.03,7]dec-8-en-2-one (caged) scaffold. Their biosynthesis is proposed to involve a cascade of Claisen and Diels–Alder reactions and has provided the inspiration for the development of efficient laboratory syntheses of the parent molecules and designed analogues. Their biology is fascinating and complex. This is evident by the nearly one hundred recent publications describing just the biology of 1, the most studied member of this family. The biology of other members of the Garcinia family remains largely unexplored. Preclinical and clinical studies with 1 and a few other family members are preliminary, but appear promising, in part due to their low toxicity. With many biological questions relating to mode-of-action as yet unanswered, interest in this family of natural products is likely to increase in the near future. It is conceivable that this family will enrich the armamentarium of anticancer medicines, such as podophyllotoxin and camptothecin, which originate from plant ethnomedicine. Along these lines, chemistry in conjunction with biology can generate analogues of these natural products with the desired pharmacological and clinical profile.
Acknowledgments
Financial support from the National Institutes of Health (CA 133002) is gratefully acknowledged. We also acknowledge partial support to O.C. and W.C. from the Thailand Research Fund in the form of a Royal Golden Jubilee Ph.D. Fellowship (grant no. PHD/0223/2548) and the Center for Petroleum, Petrochemistry and Advanced Materials. We thank the National Science Foundation for instrumentation grants CHE-9709183 and CHE-0741968. We also thank Drs. E. J. Tisdale, C. Chowdhury, B. G. Vong, H. Li, S. H. Kim, I. Slobodov, T. Lam, V. Wascholowski, and Professors A. Giannis and A. L. Yu for significant contributions to various aspects of this project. We are grateful to Teresa Abendroth for assistance with the design of the graphics and frontispiece.
Dedicated with respect and admiration to our mentor Professor K. C. Nicolaou
Biographies

Oraphin Chantarasriwong earned her B.Sc. and M.Sc. in chemistry from Chulalongkorn University in 2006, working with Professor Warinthorn Chavasiri. She is currently pursuing a Ph.D. in organic chemistry under the direction of Professor Warinthorn Chavasiri and Professor Emmanuel A. Theodorakis at Chulalongkorn University (Royal Golden Jubilee Ph.D. Fellowship). Her research interests include synthetic organic chemistry and the development of new synthetic methods and the construction of pharmaceutically important compounds.

Ayse Batova received a B.A. in Chemistry from the College of Wooster (Wooster, OH). She obtained a Ph.D. in 1991 in Biochemistry from the University of South Carolina working on the mechanisms of growth inhibition by retinoic acid in human keratinocytes. Dr. Batova did post-doctoral work at Telios Pharmaceuticals in San Diego, and at the University of California, San Diego (UCSD) where she studied the molecular biology of leukemia and neuroblastoma and investigated novel molecularly targeted therapeutics. She is currently a member of the faculty at UCSD in the department of Pediatrics. Her research focuses on the discovery and development of natural products and analogues as cancer therapeutics.

Warinthorn Chavasiri received his B.Sc. and M.Sc. in chemistry from Chulalongkorn University under the supervision of Professor Udom Kokpol. After that he pursued his Ph.D. in chemistry at Texas A&M University under the supervision of Sir Derek H.R. Barton. Since 1993, he has joined the chemistry department at Chulalongkorn University. His research focuses on the methodologies in organic synthesis, metal organic chemistry, natural product chemistry, and chemical ecology.

Emmanuel A. Theodorakis received his B.S. Degree in Chemistry from the University of Athens and his M.S. Degree (D.E.A.) and Ph.D. Degree in Organic Chemistry from Paris XI University (France). His M.S. studies were performed at the Institute of Chemistry of Natural Products (ICSN-CNRS) under the direction of Professor H.-P. Husson, while his Ph.D. studies were performed at Texas A&M University under the direction of Sir Derek H. R. Barton. Following a postdoctoral appointment at TSRI in the laboratories of Professor K. C. Nicolaou, in 1995 he joined the faculty at UCSD. His research focuses on the synthesis and bioorganic studies of natural products and designed small molecules with biological and medicinal interest.
References
- 1.a) Gustafsson MHG, Bittrich V, Stevens PF. Int J Plant Sci. 2002;163:1045–1054. [Google Scholar]; b) Sultanbawa MUS. Tetrahedron. 1980;36:1465–1506. [Google Scholar]
- 2.Mabberley DJ. The Plant-book: A Portable Dictionary of the Vascular Plants. 2. Cambridge University Press; New York: 1997. p. 293. [Google Scholar]
- 3.Kumar P, Baslas RK. Herba Hung. 1980;19:81–91. [Google Scholar]
- 4.Morton JF. In: Fruits of Warm Climates. Dowling CF Jr, editor. Florida Flair Books; Miami: 1987. pp. 301–304. [Google Scholar]; Howes FN. Vegetable Gums and Resins. Waltham, Mass: 1949. p. 161. [Google Scholar]
- 5.Winter J. In: Artists’ Pigments: A Handbook of their History and Characteristics. Fitzhugh EW, editor. Vol. 3. Oxford University Press; New York: 1997. pp. 143–155. [Google Scholar]
- 6.For selected publications on this topic, see: Eremin K, Stenger J, Green ML. J Raman Spectrosc. 2006;37:1119–1124.; Yamasaki K, Emoto Y. Ars Orientalis. 1979;11:1–14.; Yamasaki K, Nishikawa K. Stud Conserv. 1970;15:278–293.; Eremin K, Stenger J, Huang J-F, Aspuru-Guzik A, Betley T, Vogt L, Kassal I, Speakman S, Khandekar N. J Raman Spectrosc. 2008;39:1057–1065.
- 7.Kuehn H. Maltechnik-Restauro. 1977;83:223–233. [Google Scholar]
- 8.a) Perrin J. Ann Chim Phys. 1909;18:1–114. [Google Scholar]; b) Perrin J. Compt Rend. 1909;147:530–532. [Google Scholar]; c) Oseen CW. Nobel Prize in Physics, 1926—Presentation Speech. [12.28.2009]; http://nobelprize.org/nobel_prizes/physics/laureates/1926/press.html.
- 9.For selected reviews on this topic, see: Sousa ME, Pinto MMM. Curr Med Chem. 2005;12:2447–2479. doi: 10.2174/092986705774370736.; Pouli N, Marakos P. Anti-Cancer Agents Med Chem. 2009;9:77–98. doi: 10.2174/187152009787047699.; Han Q-B, Xu H-X. Curr Med Chem. 2009;16:3775–3796. doi: 10.2174/092986709789104993.; Xu H-X. Curr Med Chem. 2009;16:3775–3796. doi: 10.2174/092986709789104993.; El-Seedi HR, El-Ghorab DMH, El-Barbary MA, Zayed MF, Goransson U, Larsson S, Verpoorte R. Curr Med Chem. 2009;16:2581–2626. doi: 10.2174/092986709788682056.
- 10.Yates P, Karmarkar SS, Rosenthal D, Stout GH, Stout VF. Tetrahedron Lett. 1963;4:1623–1629.. According to the authors, acetyl gambogic acid was initially reported by Furrer M, editor. Dissertation. University of Basel (Switzerland); 1934.
- 11.a) Anthony A, Desiraju GR. Supramol Chem. 2001;13:11–23. [Google Scholar]; b) Weakley TJR, Cai SX, Zhang H-Z, Keana JFW. J Chem Crystallogr. 2001;31:501–505. [Google Scholar]
- 12.Rao BS. J Chem Soc. 1937:853–855. [Google Scholar]
- 13.Kartha G, Ramachandran GN, Bhat HB, Nair PM, Raghavan VKV, Venkataraman K. Tetrahedron Lett. 1963;4:459–472.. Previous efforts to determine the chemical structure of morellin led to incomplete structural assignments: Kartha G. Curr Sci. 1954;23:8.; Rao RVGS, Padmanabhan VM, Kartha G. Curr Sci. 1954;23:216.; Krishna Murthy DV, Rao PLN. Experientia. 1961;17:445–446.; Dayal B, Mathur BSC. Acta Crystallogr. 1960;13:269.; Rao PLN, Murthy DVK, Verma SCL. Naturwissenschaften. 1954;41:66–67.
- 14.a) Ollis WD, Ramsay MVJ, Sutherland IO, Mongolsuk S. Tetrahedron. 1965;21:1453–1470. [Google Scholar]; b) Ahmad SA, Rigby W, Taylor RB. J Chem Soc C. 1966:772–779. [Google Scholar]
- 15.Leong YW, Harrison LJ, Bennett GJ, Tan HTW. J Chem Res Synop. 1996:392–393. [Google Scholar]
- 16.a) Wang LL, Li ZL, Xu YP, Liu XQ, Pei YH, Jing YK, Hua HM. Chin Chem Lett. 2008;19:1221–1223. [Google Scholar]; Xu YP, Liu XQ, Pei YH, Jing YK, Hua HM. Chin Chem Lett. 2008;19:1221–1223. [Google Scholar]; b) Li S-L, Song J-Z, Han Q-B, Qiao C-F, Xu H-X. Biomed Chromatogr. 2008;22:637–644. doi: 10.1002/bmc.981. [DOI] [PubMed] [Google Scholar]
- 17.a) Cao SG, Sng VHL, Wu XH, Sim KY, Tan BHK, Pereira JT, Goh SH. Tetrahedron. 1998;54:10915–10924. [Google Scholar]; b) Cao SG, Wu XH, Sim KY, Tan BKH, Pereira JT, Wong WH, Hew NF, Goh SH. Tetrahedron Lett. 1998;39:3353–3356. [Google Scholar]; c) Xu YJ, Yip SC, Kosela S, Fitri E, Hana M, Goh SH, Sim KY. Org Lett. 2000;2:3945–3948. doi: 10.1021/ol006730t. [DOI] [PubMed] [Google Scholar]; d) Wu XH, Tan BKH, Cao SG, Sim KY, Goh SH. Nat Prod Lett. 2000;12:453–458. [Google Scholar]
- 18.Han Q-B, Wang YL, Yang L, Tso TF, Qiao C-F, Song JZ, Xu LJ, Chen SL, Yang DJ, Xu HX. Chem Pharm Bull. 2006;54:265–267. doi: 10.1248/cpb.54.265. [DOI] [PubMed] [Google Scholar]
- 19.a) Venkataraman K. Proc Indian Nat Sci Acad Part A. 1974;39:365–381. [Google Scholar]; b) Adawadkar PD, Srinivasan R, Yemul SS. Indian J Chem Sect B. 1976;14:19–21. [Google Scholar]; c) Feng F, Liu WY, Chen YS, Guo QL, You QD. J Asian Nat Prod Res. 2007;9:735–741. doi: 10.1080/10286020701189146. [DOI] [PubMed] [Google Scholar]; d) Karanjgaonkar CG, Nair PM, Venkataraman K. Tetrahedron Lett. 1966;7:687–691. [Google Scholar]; e) Yemul SS, Rama Rao AV. Org Mass Spectrom. 1974;9:1063–1072. [Google Scholar]
- 20.a) Bhat HB, Nair PM, Venkataraman K. Indian J Chem. 1964;2:405–410. [Google Scholar]; b) Reutrakul V, Anantachoke N, Pohmakot M, Jaipetch T, Sophasan S, Yoosook C, Kasisit J, Napaswat C, Santisuk T, Tuchinda P. Planta Med. 2007;73:33–40. doi: 10.1055/s-2006-951748. [DOI] [PubMed] [Google Scholar]; c) Tao S-J, Guan SH, Wang W, Lu Z-Q, Chen GT, Sha N, Yue Q-X, Liu X, Guo DA. J Nat Prod. 2009;72:117–124. doi: 10.1021/np800460b. [DOI] [PubMed] [Google Scholar]
- 21.Sukpondma Y, Rukachaisirikul V, Phongpaichit S. Chem Pharm Bull. 2005;53:850–852. doi: 10.1248/cpb.53.850. [DOI] [PubMed] [Google Scholar]
- 22.a) Rao PLN, Verma SCL, Murthy DVK. Curr Sci. 1951;20:234. [Google Scholar]; b) Nair PM, Venkataraman K. Indian J Chem. 1964;2:402–404. [Google Scholar]
- 23.Lin L-J, Lin LZ, Pezzuto JM, Cordell GA, Ruangrungsi N. Magn Reson Chem. 1993;31:340–347. [Google Scholar]
- 24.Asano J, Chiba K, Tada M, Yoshii T. Phytochemistry. 1996;41:815–820. doi: 10.1016/0031-9422(95)00682-6. [DOI] [PubMed] [Google Scholar]
- 25.a) Thoison O, Fahy J, Dumontet V, Chiaroni A, Riche C, Tri MV, Sevenet T. J Nat Prod. 2000;63:441–446. doi: 10.1021/np9903088. [DOI] [PubMed] [Google Scholar]; b) Thoison O, Cuong DD, Gramain A, Chiaroni A, Hung NV, Sevenet T. Tetrahedron. 2005;61:8529–8535. [Google Scholar]
- 26.Shadida KA, Shaari K, Abas F, Israf DA, Hamzah AS, Syakroni N, Saha K, Lajis NH. Phytochemistry. 2007;68:2537–2544. doi: 10.1016/j.phytochem.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 27.a) Rukachaisirikul V, Kaewnok W, Koysomboon S, Phongpaichit S, Taylor WC. Tetrahedron. 2000;56:8539–8543. [Google Scholar]; b) Sukpondma Y, Rukachaisirikul V, Phongpaichit S. J Nat Prod. 2005;68:1010–1017. doi: 10.1021/np0580098. [DOI] [PubMed] [Google Scholar]; c) Rukachaisirikul V, Phainuphong P, Sukpondma Y, Phongpaichit S, Taylor SWC. Planta Med. 2005;71:165–170. doi: 10.1055/s-2005-837785. [DOI] [PubMed] [Google Scholar]
- 28.Rukachaisirikul V, Painuphong P, Sukpondma Y, Koysomboon S, Sawangchote P, Taylor WC. J Nat Prod. 2003;66:933–938. doi: 10.1021/np030080v. [DOI] [PubMed] [Google Scholar]
- 29.Mahabusarakam W, Nuangnaowarat W, Taylor WC. Phytochemistry. 2006;67:470–474. doi: 10.1016/j.phytochem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Kosela S, Cao SG, Wu X-H, Vittal JJ, Sukri T, Masdianto, Goh S-H, Kim K-Y. Tetrahedron Lett. 1999;40:157–160. [Google Scholar]
- 31.Wu J, Xu YJ, Cheng XF, Harrison LJ, Sim KY, Goh SH. Tetrahedron Lett. 2001;42:727–729. [Google Scholar]
- 32.a) Hunt BJ, Rigby W. J Chem Soc C. 1970:2459–2462. [Google Scholar]; b) Rao DR, Gurudutt KN, Mamatha S, Rao LJM. Magn Reson Chem. 2007;45:578–582. doi: 10.1002/mrc.2011. [DOI] [PubMed] [Google Scholar]
- 33.Han QB, Cheung S, Tai J, Qiao CF, Song JZ, Xu HX. Biol Pharm Bull. 2005;28:2335–2337. doi: 10.1248/bpb.28.2335. [DOI] [PubMed] [Google Scholar]
- 34.a) Rao GSRS, Rathnamala S, Sivaramakrishnan R. Proc Indian Acad Sci Sect A. 1978;87:75–86. [Google Scholar]; b) Rao GSRS, Mala SR, Surendranath V, Gupta VS, Rao PLN. Tetrahedron Lett. 1974;15:1259–1262. [Google Scholar]
- 35.a) Han QB, Song JZ, Qiao JZ, Wong L, Xu HX. J Chromatogr A. 2006;1127:298–301. doi: 10.1016/j.chroma.2006.07.044. [DOI] [PubMed] [Google Scholar]; b) Cardillo G, Merlini L. Tetrahedron Lett. 1967;8:2529–2530. [Google Scholar]
- 36.Han Q, Yang L, Liu Y, Wang Y, Qiao C, Song J, Xu L, Yang D, Chen S, Xu H. Planta Med. 2006;72:281–284. doi: 10.1055/s-2005-916193. [DOI] [PubMed] [Google Scholar]
- 37.a) Zhou Y, Liu X, Yang J, Han Q-B, Song J-Z, Li S-L, Qiao C-F, Ding L-S, Xu H-X. Anal Chim Acta. 2008;629:104–118. doi: 10.1016/j.aca.2008.09.044. [DOI] [PubMed] [Google Scholar]; b) Han Q-B, Zhou Y, Feng C, Xu G, Huang S-X, Li S-L, Qiao C-F, Song J-Z, Chang DC, Luo KQ, Xu H-X. J Chromatogr B. 2009;877:401–407. doi: 10.1016/j.jchromb.2008.12.046. [DOI] [PubMed] [Google Scholar]
- 38.For selected reviews and monographs on this topic, see: Dewick PM. Nat Prod Rep. 1998;15:17–58. doi: 10.1039/a815017y.; Knaggs AR. Nat Prod Rep. 2003;20:119–136. doi: 10.1039/b100399m.; Herrmann KM, Weaver LM. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:473–503. doi: 10.1146/annurev.arplant.50.1.473.; Beerhues L, Liu B. Phytochemistry. 2009;70:1719–1727. doi: 10.1016/j.phytochem.2009.06.017.; Dewick PM, P M. Medicinal Natural Products: a Biosynthetic Approach. 2. Wiley; New York: 2002. pp. 121–164.; Gottlieb OR. Phytochemistry. 1968;7:411–421.
- 39.a) Birch AJ, Baldas J, Hlubucek JR, Simpson TJ, Westerman PW. J Chem Soc Perkin Trans. 1976;1:898–904. [Google Scholar]; b) Holker JSE, Lapper RD, Simpson TJ. J Chem Soc Perkin Trans. 1974;1:2135–2140. [Google Scholar]; c) Hill JG, Nakashima TT, Vederas JC. J Am Chem Soc. 1982;104:1745–1748. [Google Scholar]
- 40.a) Locksley HD, Moore I, Scheinmann F. Tetrahedron. 1967;23:2229–2234. [Google Scholar]; b) Carpenter I, Locksley HD, Scheinmann F. Phytochemistry. 1969;8:2013–2025. [Google Scholar]; c) Bennett GJ, Lee HH. J Chem Soc Chem Commun. 1988:619–620. [Google Scholar]; d) Bennett GJ, Lee HH, Das N. J Chem Soc Perkin Trans. 1990;1:2671–2676. [Google Scholar]
- 41.Quillinan AJ, Scheinmann F. J Chem Soc Chem Commun. 1971:966–967. [Google Scholar]
- 42.a) Jackson B, Locksley HD, Scheinmann F. J Chem Soc C. 1967:785–796. [Google Scholar]; b) Jefferson A, Scheinmann F. Q Rev Chem Soc. 1968;22:391–420. [Google Scholar]; c) Locksley HD, Quillinan AJ, Scheinmann F. J Chem Soc C. 1971:3804–3814. [Google Scholar]
- 43.Nicolaou KC, Li J. Angew Chem. 2001;113:4394–4398. [Google Scholar]; Angew Chem Int Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 44.Tisdale EJ, Slobodov I, Theodorakis EA. Proc Natl Acad Sci USA. 2004;101:12030–12035. doi: 10.1073/pnas.0401932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Dictionary of Chinese Traditional Medicines. Shanghai Scientific and Technical Publishers; Shanghai: 1977. pp. 3805–3807. [Google Scholar]; b) the anticancer gamboge research team. Jiangxi Med J. 1982;17:1–5. [Google Scholar]
- 46.a) Panthong A, Norkaew P, Kanjanapothi D, Taesotikul T, Anantachoke N, Reutrakul V. J Ethnopharmacol. 2007;111:335–340. doi: 10.1016/j.jep.2006.11.038. [DOI] [PubMed] [Google Scholar]; b) Evan WC. Trease and Evans Pharmacognosy. 14. Saunders Company Limited; Nottingham: 1996. p. 290. [Google Scholar]
- 47.a) Rao PLN, Verma SCL. J Scient Ind Res. 1951;10B:184–185. [Google Scholar]; b) Gupta VS, Rao AVSP, Rao PLN. Ind J Exper Biol. 1963;1:146–147. [Google Scholar]; c) Sani BP, Rao PLN. Ind J Exper Biol. 1966;4:27–28. [PubMed] [Google Scholar]; d) Santhaman K, Rao PLN. Ind J Exper Biol. 1968;6:158–159. [Google Scholar]; e) Puttanna CR, Rao PLN. Ind J Exper Biol. 1968;6:150–152. [PubMed] [Google Scholar]
- 48.In the early chemical literature, Garcinia natural products were commonly named as guttiferins and 1 was also known as β-guttiferin; for selected references on this issue, see: Gupta VS, Rao PLN, Vaidya SN, Ramaseshan S. Chem Ind. 1962:1469–1470.; Rao KVN, Rao PLN. Experientia. 1961;17:213–214.
- 49.a) Santhanam K, Rao PLN. Ind J Exper Biol. 1969;7:34–36. [PubMed] [Google Scholar]; b) Sani BP, Rao PLN. Ind J Chem. 1969;7:680–684. [Google Scholar]; c) Verma SCL, Rao PLN. Ind J Exper Biol. 1967;5:106–109. [PubMed] [Google Scholar]; d) Rao DR, Gupta TR, Gupta VS, Rao KVN, Rao PLN. Ind J Chem. 1963;1:276–277. [Google Scholar]
- 50.a) Rao KVN, Rao PLN. Ind J Exper Biol. 1967;5:101–105. [PubMed] [Google Scholar]; b) Rao KVN, Rao PLN. Ind J Exper Biol. 1967;5:96–100. [Google Scholar]
- 51.Khalid RM, Jabit ML, Abas F, Stanslas J, Shaari K, Lajis NH. Nat Prod Commun. 2007;2:271–276. [Google Scholar]
- 52.Zhang H-Z, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, Drewe J, Cai S-X. Bioorg Med Chem. 2004;12:309–317. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 53.Batova A, Lam T, Wascholowski V, Yu AL, Giannis A, Theodorakis EA. Org Biomol Chem. 2007;5:494–500. doi: 10.1039/b612903j. [DOI] [PubMed] [Google Scholar]
- 54.Wu X, Cao S, Goh S, Hsu A, Tan BKH. Planta Med. 2002;68:198–203. doi: 10.1055/s-2002-23142. [DOI] [PubMed] [Google Scholar]
- 55.For selected reviews, see: La Porta CAM. Curr Med Chem. 2007;14:387–391. doi: 10.2174/092986707779941078.; Larsen AK, Escargeuil AE, Skladanowski A. Pharmacol Ther. 2000;85:217–229. doi: 10.1016/s0163-7258(99)00073-x.
- 56.For selected reviews, see: Earnshaw WC, Martins LM, Kaufmann SH. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383.; Thornberry NA, Lazebnic Y. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312.; Taylor RC, Cullen SP, Martin SJ. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312.; Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9.; Porter AG, Janicke RU. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476.
- 57.Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Biol Pharm Bull. 2004;27:998–1003. doi: 10.1248/bpb.27.998. [DOI] [PubMed] [Google Scholar]
- 58.For selected reviews on this topic, see: Yip KW, Reed JC. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307.; Reed JC, Pellecchia M. Blood. 2005;106:408–418. doi: 10.1182/blood-2004-07-2761.; Cory S, Adams JM. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883.; Reed JC. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6.; Kroemer G, Reed JC. Nat Med. 2000;6:513–519. doi: 10.1038/74994.; Gupta S, Kass GEN, Svegezdi E, Joseph B. J Cell Mol Med. 2009;13:1004–1033. doi: 10.1111/j.1582-4934.2009.00697.x.
- 59.For selected reviews on the therapeutic value of Bcl-2 inhibitors, see: Doshi JM, Xing C. Mini-Rev Org Chem. 2008;5:171–178.; Zhai D, Jin C, Satterthwait AC, Reed JC. Cell Death Differ. 2006;13:1419–1421. doi: 10.1038/sj.cdd.4401937.; Vogler M, Dinsdale D, Dyer MJS, Cohen GM. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137.
- 60.a) Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. World J Gastroenterol. 2005;11:3655–3659. doi: 10.3748/wjg.v11.i24.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gu HY, Rao SY, Zhao J, Wang J, Mu R, Rong JJ, Tao L, Qi Q, You QD, Guo QL. J Cancer Res Clin Oncol. 2009;135:1777–1782. doi: 10.1007/s00432-009-0624-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu X, Liu Y, Wang L, He J, Zhang H, Chen X, Li Y, Yang J, Tao J. Int J Dermatol. 2009;48:186–192. doi: 10.1111/j.1365-4632.2009.03946.x. [DOI] [PubMed] [Google Scholar]
- 62.Zhai D, Jin C, Shiau C-W, Kitada S, Satterthwait AC, Reed JC. Mol Cancer Ther. 2008;7:1639–1646. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.a) Gu H, Wang X, Rao S, Wang J, Zhao J, Ren FL, Mu R, Yang Y, Qi Q, Liu W, Lu N, Ling H, You Q, Guo Q. Mol Cancer Ther. 2008;7:3298–3305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]; b) Rong JJ, Hu R, Qi Q, Gu HY, Zhao Q, Wang J, Mu R, You QD, Guo QL. Cancer Lett. 2009;284:102–112. doi: 10.1016/j.canlet.2009.04.011. [DOI] [PubMed] [Google Scholar]; c) Gu HY, Guo QL, You QD, Liu W, Qi Q, Zhao L, Yuan ST, Zhang K. Chin J Nat Med. 2005;3:168–172. [Google Scholar]
- 64.a) Wu ZQ, Guo QL, You QD, Zhao L, Gu HY. Biol Pharm Bull. 2004;27:1769–1774. doi: 10.1248/bpb.27.1769. [DOI] [PubMed] [Google Scholar]; b) Yu J, Guo Q-L, You Q-D, Lin S-S, Li Z, Gu H-Y, Zhang H-W, Tan Z, Wang X. Cancer Chemother Pharmacol. 2006;58:434–443. doi: 10.1007/s00280-005-0177-2. [DOI] [PubMed] [Google Scholar]; c) Zhao Q, Yang Y, Yu J, You QD, Zeng S, Gu HY, Lu N, Qi Q, Liu W, Wang XT, Guo QL. Cancer Lett. 2008;262:223–231. doi: 10.1016/j.canlet.2007.12.002. [DOI] [PubMed] [Google Scholar]; d) Guo QL, Lin SS, You QD, Yu J, Zhao L, Qi Q, Liang F, Tan Z, Wang XT. Life Sci. 2006;78:1238–1245. doi: 10.1016/j.lfs.2005.06.046. [DOI] [PubMed] [Google Scholar]; e) Chen J, Gu H-Y, Lu N, Yang Y, Liu W, Qi Q, Rong J-J, Wang X-T, You Q-D, Guo Q-L. Life Sci. 2008;83:103–109. doi: 10.1016/j.lfs.2008.05.003. [DOI] [PubMed] [Google Scholar]; f) Qin Y, Meng L, Hu C, Duan W, Zuo Z, Lin L, Zhang X, Ding J. Mol Cancer Ther. 2007;6:2429–2440. doi: 10.1158/1535-7163.MCT-07-0147. [DOI] [PubMed] [Google Scholar]; g) Yu J, Guo Q-L, You Q-D, Zhao L, Gu H-Y, Yang Y, Zhang H-w, Tan Z, Wang X. Carcinogenesis. 2006;28:632–638. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Chen Y, Han Q-b, Chan C-y, Wang H, Liu Z, Cheng CH-k, Yew DT, Lin MCM, He M-l, Xu H-x, Sung JJY, Kung H-f. Proteomics. 2009;9:242–253. doi: 10.1002/pmic.200800155. [DOI] [PubMed] [Google Scholar]
- 66.a) Li R, Chen Y, Shu WX, Zhao F, Liu Y, Wen L. Chin J Cancer Res. 2009;21:68–73. [Google Scholar]; b) Li R, Chen Y, Zeng L-I, Shu W-x, Zhao F, Wen L, Liu Y. Toxicology. 2009;262:98–105. doi: 10.1016/j.tox.2009.04.059. [DOI] [PubMed] [Google Scholar]
- 67.a) Shu W-X, Chen Y, Li R, Wu Q, Cui G, Ke W, Chen Z. Basic Clin Pharmacol Toxicol. 2008;103:530–537. doi: 10.1111/j.1742-7843.2008.00292.x. [DOI] [PubMed] [Google Scholar]; b Shu WX, Chen Y, He J. Chin J Oncol. 2008;30:484–489. [PubMed] [Google Scholar]
- 68.a) Qi Q, Gu HY, Yang Y, Lu N, Zhao J, Liu W, Ling H, You QD, Wang XT, Guo QL. J Mol Med. 2008;86:1367–1377. doi: 10.1007/s00109-008-0398-z. [DOI] [PubMed] [Google Scholar]; b) Qi Q, Lu N, Wang XT, Gu HY, Yang Y, Liu W, Li CL, You QD, Guo QL. Biochem Cell Biol. 2008;86:386–395. doi: 10.1139/o08-104. [DOI] [PubMed] [Google Scholar]
- 69.Zhao J, Qi Q, Yang Y, Gu HY, Lu N, Liu W, Wang W, Qiang L, Zhang LB, You QD, Guo QL. Eur J Pharmacol. 2008;589:127–131. doi: 10.1016/j.ejphar.2008.04.063. [DOI] [PubMed] [Google Scholar]
- 70.a) Yi T, Yi Z, Cho S-G, Luo J, Pandley MK, Aggarwal BB, Liu M. Cancer Res. 2008;68:1843–1850. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu N, Yang Y, You QD, Ling Y, Gao Y, Gu H-Y, Zhao L, Wang X-T, Guo Q-L. Cancer Lett. 2007;258:80–89. doi: 10.1016/j.canlet.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 71.Wang T, Wei J, Qian X, Ding Y, Yu L, Liu B. Cancer Lett. 2008;262:214–222. doi: 10.1016/j.canlet.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Liu W, Zhao Q, Qi Q, Lu N, Yang Y, Nei F-F, Rong J-J, You QD, Guo QL. Toxicology. 2009;256:135–140. doi: 10.1016/j.tox.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 73.For selected reviews on the role of TfR-1 in cancer, see: Aisen P. Int J Biochem Cell Biol. 2004;36:2137–2143. doi: 10.1016/j.biocel.2004.02.007.; Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I, Zarkovic N. Cancer Lett. 2006;238:188–196. doi: 10.1016/j.canlet.2005.07.001.; Ryschich E, Huszty G, Knaebel HP, Hartel M, Buechler MW, Schmidt J. Eur J Cancer. 2004;40:1418–1422. doi: 10.1016/j.ejca.2004.01.036.
- 74.Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma N, Jiang S, Zhang H-Z, Gehlsen KR, Cai SX, Green DR, Tsang B. Proc Natl Acad Sci USA. 2005;102:12095–12100. doi: 10.1073/pnas.0406731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.a) Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB. Blood. 2007;110:3517–3525. [Google Scholar]; b) Zhu XL, Zhang HM, Lin Y, Chen PS, Min J, Wang ZZ, Xiao W, Chen BA. J Chemother. 2009;21:666–672. doi: 10.1179/joc.2009.21.6.666. [DOI] [PubMed] [Google Scholar]
- 76.a) Wang Y, Chen Y, Chen Z, Wu Q, Ke WJ, Wu QL. Acta Phamacol Sin. 2008;29:349–354. doi: 10.1111/j.1745-7254.2008.00762.x. [DOI] [PubMed] [Google Scholar]; b) Palempalli UD, Gandhi U, Kalantari P, Vunta H, Arner RJ, Narayan V, Ravindran A, Prabhu KS. Biochem J. 2009;419:401–409. doi: 10.1042/BJ20081482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ortiz-Sanchez E, Daniels TR, Helguera G, Martinez-Maza O, Bonavida B, Penichet ML. Leukemia. 2009;23:59–70. doi: 10.1038/leu.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu W, Lu N, Wu Z, You Q, Guo Q. Toxicology. 2009;260:60–67. doi: 10.1016/j.tox.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 79.Qiang L, Yang Y, You Q-D, Ma Y-J, Yang L, Nie F-F, Gu H-Y, Zhao L, Lu N, Qi Q, Liu W, Wang X-T, Guo Q-L. Biochem Pharmacol. 2008;75:1083–1092. doi: 10.1016/j.bcp.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 80.a) Yang Y, Yang L, You QD, Nie FF, Gu HY, Zhao L, Wang XT, Guo QL. Cancer Lett. 2007;256:259–266. doi: 10.1016/j.canlet.2007.06.014. [DOI] [PubMed] [Google Scholar]; b) Guo Q-L, You Q-D, Wu Z-Q, Yuan S-T, Zhao L. Acta Pharmacol Sin. 2004;25:769–774. [PubMed] [Google Scholar]; c) Gu H, You Q, Liu W, Yang Y, Zhao L, Qi Q, Zhao J, Wang J, Lu N, Ling H, Guo Q, Wang X. Intern Immunopharmacol. 2008;8:1493–1502. doi: 10.1016/j.intimp.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 81.a) Guo Q, Qi Q, Gu H, Wu Z. Basic Clin Pharmacol Toxicol. 2006;99:178–184. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]; b) Hao K, Zhao XP, Liu XQ, Wang GJ. Biomed Chromatogr. 2007;21:279–283. doi: 10.1002/bmc.752. [DOI] [PubMed] [Google Scholar]
- 82.Qi Q, You Q, Gu H, Zhao L, Liu WL, Lu N, Guo QL. J Ethnopharmacol. 2008;117:433–438. doi: 10.1016/j.jep.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 83.Zhao L, Zhen C, Wu Z, Hu R, Zhou C, Guo Q. Drug Chem Toxicol. 2010;33:88–96. doi: 10.3109/01480540903173534. [DOI] [PubMed] [Google Scholar]
- 84.Hao K, Liu XQ, Wang GJ, Zhao XP. Eur J Drug Metab Pharmacokinet. 2007;32:63–68. doi: 10.1007/BF03190993. [DOI] [PubMed] [Google Scholar]
- 85.a) Liu Y-T, Hao K, Liu X-Q, Wang G-J. Acta Pharmacol Sin. 2006;27:1253–1258. doi: 10.1111/j.1745-7254.2006.00369.x. [DOI] [PubMed] [Google Scholar]; b) Feng F, Liu W, Wang Y, Guo Q, You Q. J Chromatogr B. 2007;860:218–226. doi: 10.1016/j.jchromb.2007.10.037. [DOI] [PubMed] [Google Scholar]; c) Ding L, Huang D, Wang JW, Li ST. J Chromatogr B. 2007;846:112–118. doi: 10.1016/j.jchromb.2006.08.034. [DOI] [PubMed] [Google Scholar]; d) Yang J, Ding L, Jin SH, Liu XX, Liu WY, Wang ZZ. J Chromatogr B. 2010;878:659–666. doi: 10.1016/j.jchromb.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 86.a) Qu G, Zhu X, Zhang C, Ping Q. Drug Delivery. 2009;16:363–370. doi: 10.1080/10717540903075545. [DOI] [PubMed] [Google Scholar]; b) Zhu X, Zhang C, Wu XL, Tang XY, Ping QN. Drug Dev Ind Pharm. 2008;34:2–9. doi: 10.1080/03639040701378001. [DOI] [PubMed] [Google Scholar]
- 87.Zhou ZT, Wang JW. Chin J New Drugs. 2007;16:79–82. [Google Scholar]
- 88.a) Wessely F, Lauterbach-Keil G, Sinwel F. Monatsh Chem. 1950;81:811–818. [Google Scholar]; b) Wessely F, Sinwel F. Monatsh Chem. 1950;81:1055–1070. [Google Scholar]; c) Wessely F, Kotlan J, Sinwel F. Monatsh Chem. 1952;83:902–914. [Google Scholar]; d) Metlesics W, Wessely F, Budzikiewicz H. Monatsh Chem. 1958;89:102–109. [Google Scholar]
- 89.Bhamare NK, Granger T, John CR, Yates P. Tetrahedron Lett. 1991;32:4439–4442. [Google Scholar]
- 90.Bichan DJ, Yates P. J Am Chem Soc. 1972;94:4773–4774. [Google Scholar]
- 91.Tisdale EJ, Chowdhury C, Vong BG, Li H, Theodorakis EA. Org Lett. 2002;4:909–912. doi: 10.1021/ol017278w. [DOI] [PubMed] [Google Scholar]
- 92.Mehta G, Maity P. Tetrahedron Lett. 2008;49:318–322. [Google Scholar]
- 93.Nicolaou KC, Sasmal PK, Xu H, Namoto K, Ritzen A. Angew Chem. 2003;115:4357–4361. doi: 10.1002/anie.200351805. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:4225–4229. doi: 10.1002/anie.200351805. [DOI] [PubMed] [Google Scholar]
- 94.Tisdale EJ, Li H, Vong BG, Kim SH, Theodorakis EA. Org Lett. 2003;5:1491–1494. doi: 10.1021/ol034276y. [DOI] [PubMed] [Google Scholar]
- 95.Tisdale EJ, Slobodov I, Theodorakis EA. Org Biomol Chem. 2003;1:4418–4422. doi: 10.1039/b311833a. [DOI] [PubMed] [Google Scholar]
- 96.Li NG, Wang J-J, Liu X-R, Lin C-J, You Q-D, Guo Q-L. Tetrahedron Lett. 2007;48:6586–6589. [Google Scholar]
- 97.Nicolaou KC, Xu H, Wartmann M. Angew Chem. 2005;117:766–771. doi: 10.1002/anie.200462211. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:756–761. doi: 10.1002/anie.200462211. [DOI] [PubMed] [Google Scholar]
- 98.For selected accounts on this topic, see: Gajewski JJ. Acc Chem Res. 1980;13:142–148.; Ganem B. Angew Chem. 1996;108:1014–1023.; Angew Chem Int Ed Engl. 1996;35:936–945.; Severance DL, Jorgensen WL. J Am Chem Soc. 1992;114:10966–10968.; Gajewski JJ. Acc Chem Res. 1997;30:219–225.
- 99.For a review of hydrophobic effects, see: Tanford C. The Hydrophobic Effect. 2. Wiley; New York: 1980.
- 100.For selected accounts on Diels–Alder reactions accelerated in water, see: Breslow R. Acc Chem Res. 2004;37:471–478. doi: 10.1021/ar040001m.; Lindstroem U. Chem Rev. 2002;102:2751–2771. doi: 10.1021/cr010122p.; Grieco PA, Kaufman MD. J Org Chem. 1999;64:6041–6048.
- 101.Hayden AE, Xu H, Nicolaou KC, Houk KN. Org Lett. 2006;8:2989–2992. doi: 10.1021/ol060917o. [DOI] [PubMed] [Google Scholar]
- 102.Tisdale EJ, Vong BG, Li H, Kim SH, Chowdhury C, Theodorakis EA. Tetrahedron. 2003;59:6873–6887. [Google Scholar]
- 103.Nicolaou KC, Sasmal PK, Xu H. J Am Chem Soc. 2004;126:5493–5501. doi: 10.1021/ja040037+. [DOI] [PubMed] [Google Scholar]
- 104.Tao Z, Zhou Y, Lu J, Duan W, Qin Y, He X, Lin L, Ding J. Cancer Biol Ther. 2007;6:691–696. doi: 10.4161/cbt.6.5.3960. [DOI] [PubMed] [Google Scholar]
- 105.Xie H, Qin Y-X, Zhou Y-L, Tong L-J, Lin L-P, Geng M-Y, Duan W-H, Ding J. Acta Pharmacol Sin. 2009;30:346–354. doi: 10.1038/aps.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jang S-W, Okada M, Sayeed I, Xiao G, Stein D, Jin P, Ye K. Proc Natl Acad Sci USA. 2007;104:16329–16334. doi: 10.1073/pnas.0706662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.a) Wang J, Zhao L, Hu Y, Guo Q, Zhang L, Wang X, Li N, You Q. Eur J Med Chem. 2009;44:2611–2620. doi: 10.1016/j.ejmech.2008.09.034. [DOI] [PubMed] [Google Scholar]; b) Mu R, Lu N, Wang J, Yin YH, Ding Y, Zhang XX, Gui H, Sun Q, Duan HQ, Zhang L, Zhang YC, Ke X, Guo QL. Eur J Cancer Prev. 2010;19:61–67. doi: 10.1097/CEJ.0b013e328333fb22. [DOI] [PubMed] [Google Scholar]; c) Li QL, Cheng H, Zhu GQ, Yang L, Zhou A, Wang XS, Fang NB, Xia LZ, Su JJ, Wang M, Peng DY, Xu Q. Biol Pharm Bull. 2010;33:415–420. doi: 10.1248/bpb.33.415. [DOI] [PubMed] [Google Scholar]
- 108.a) Li NG, You QD, Huang XF, Wang JX, Guo QL, Chen XG, Li Y, Li HY. Chin Chem Lett. 2007;18:659–662. [Google Scholar]; b) Han QB, Yang NY, Tian HL, Qiao C-F, Song J-Z, Chang DC, Chen S-L, Luo KQ, Xu H-X. Phytochemistry. 2008;69:2187–2192. doi: 10.1016/j.phytochem.2008.05.019. [DOI] [PubMed] [Google Scholar]; c) Sousa EP, Silva AMS, Pinto MMM, Pedro MM, Cerqueira FAM, Nascimento MSJ. Helv Chim Acta. 2002;85:2862–2876. [Google Scholar]
- 109.Chantarasriwong O, Cho WC, Batova A, Chavasiri W, Moore C, Rheingold AL, Theodorakis EA. Org Biomol Chem. 2009;7:4886–4894. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuemmerle J, Jiang S, Tseng B, Kasibhatla S, Drewe J, Cai SX. Bioorg Med Chem. 2008;16:4233–4241. doi: 10.1016/j.bmc.2008.02.084. [DOI] [PubMed] [Google Scholar]