

Abstract
Histone deacetylase inhibitors (HDACi) are an emerging class of novel anti-cancer drugs that cause growth arrest, differentiation, and apoptosis of tumor cells. In addition, they have shown promise as anti-parasitic, anti-neurodegenerative, anti-rheumatologic and immunosuppressant agents. To date, several structurally distinct small molecule HDACi have been reported including aryl hydroxamates, benzamides, short-chain fatty acids, electrophilic ketones, and macrocyclic peptides. Macrocyclic HDACi possess the most complex cap-groups which interact with HDAC enzyme’s outer rim and have demonstrated excellent HDAC inhibition potency and isoform selectivity. This review focuses on the recent progress and current state of macrocyclic HDACi.
Keywords: Cyclic tetrapeptide, depsipeptide, peptide mimetic, macrolide, ketolide, macrocycles, histone deacetylase inhibitors, HDAC
1. INTRODUCTION
Epigenetic control of DNA transcription is a powerful means to regulate cell function [1]. The main epigenetic mechanisms in humans are DNA methylation and post-translational modifications of histones, including methylation, acetylation, phosphorylation, ubiquitination, sumoylation, and ADP-ribosylation [2–6]. Histone acetylation state, specifically, is linked with transcriptional activation and is controlled by two enzymes with opposing functions: histone acetyltransferase (HAT) and histone deacetylase (HDAC) [7–9] HAT acetylates ε-NH3 groups of histone lysines, disrupting the charge interaction between the negatively-charged DNA backbone and positively-charged histone lysines. This causes nucleosomes to adopt a more relaxed conformation, accessible to transcription factors. HDAC, on the other hand, removes acetyl groups from histone lysines, ultimately resulting in transcriptional suppression via nucleosomal inaccessibility [7].
HDAC has 18 known isoforms, organized into four different classes. Class I, II, and IV depend on a catalytic zinc ion and differ by cellular localization, while Class III HDACs require NAD+ [2]. Altered expression levels of HDAC enzymes have been observed in many diseases, most notably in cancer [2, 10–12]. HDAC inhibitors (HDACi) have been shown to cause growth arrest, differentiation, and apoptosis in a variety of cancer cell lines [13–15]. In addition to cancer, HDACi are being investigated as a potential treatment option for infectious diseases, such as malaria and leishmania [16–21], as well as in CNS disorders [22–24].
Early evidence of small molecule-induced histone hy-peracetylation came in the 1970s with the revelation that butyric acid caused histone acetylation in HeLa and Friend erythroleukemia cell cultures [25, 26]. Other compounds were subsequently shown to cause anticancer activity in cell culture with similar association of histone acetylation [27]. In 1990, the natural product trichostatin A was shown to potently inhibit HDAC activity at low nanomolar ranges [28]. However, it was not until the cyclic tetrapeptide trapoxin was used as an affinity tag to isolate the first HDAC that the enzymes were recognized as the specific molecular targets for HDACi [29]. Since then, HDACi have drawn much interest and research efforts have culminated in the validation of HDAC inhibition as a clinically viable approach for cancer treatment with the approval of SAHA for the treatment of cutaneous T-cell lymphoma [30–33].
HDACi generally conform to a three-motif pharmacophoric model consisting of a zinc-binding group (ZBG), a hydrophobic linker chain, and a cap group [34]. The recognized mode of inhibition involves ZBG interaction with the catalytic zinc ion at the base of the active site while the linker efficiently positions the cap group to make interactions with amino acid residues on the surface of the enzyme [35]. Most HDACi chelate the active site Zn using the hydroxamate moiety as a ZBG [35–37]. However, because of the poor bioavailability and the possibility of undesirable side reactions, there is significant interest in finding better ZBGs [38, 39]. Notable nonhydroxamate inhibitors include depsipeptide FK-228, a cyclic peptide containing a disulfide bond, which is thought to be reduced in the cell to a thiol moiety which serves to bind the zinc ion. Other examples include azumamides, apicidins, trapoxins, the benzamide derivative MS-275, and the recently discovered natural product largazole [13, 40–42]. It should be noted that in spite of the common pharmacophoric model, there is significant structural diversity among HDACi (Fig. 1).
Fig. (1).

Selected Examples of HDAC Inhibitors: (a) Linear HDACi, (b) Macrocyclic HDACi.
Macrocyclic HDACi, including cyclic tetrapeptides and depsipeptides, possess the most complex cap groups, capable of making numerous interactions with the enzyme surface. Because the amino acid composition of the enzyme surface differs between HDAC isoforms, these macrocyclic HDACi have the potential for isoform-selective inhibition [42]. It is possible that macrocyclic HDACi could be very useful as agents customized and targeted against specific cancer types since it has been shown that certain HDAC isoforms have altered expression in specific cancers [43–46]. Also, with the lack of crystal structures for all isoforms except HDAC 8 and the catalytic domain of HDAC 7, an isoform-selective inhibitor could enhance both the structural knowledge of HDAC isoforms and elucidate the distinct cellular functions of structurally similar isoenzymes [47, 48]. Initally, the progress of macrocyclic HDACi lagged behind that of their non-cyclic counterpart. This is mainly due to the peptidyl nature of these macrocyclic templates which offered only a limited number of side-chain modifications, in addition to the difficulty in the synthesis of strained ring architectures [49–51]. These problems have been partially addressed through substitution of amino acids within common macrocyclic frameworks, scaffold attachments, and, most recently, with the search for nonpeptide macrocyclic HDACi [52–56]. The goal of this review is to provide a current view on the progress of macrocyclic HDACi. The examples contained are by no means representative of the complete structural diversity of HDACi, but our intent is to cover relevant macrocyclic HDACi in order to provide an accurate perspective of the current state of this field of HDACi [57–64].
2. REVIEW OF MACROCYCLIC HDACI
2.a. Peptide-Based Macrocyclic HDACi
Cyclic peptide based HDACi are among the most potent and the most structurally complex class of HDACi. They generally fit the overall pharmacophoric model of all HDACi and have been suggested to bind in similar manner as long chain hydroxamate HDACi [34]. This realization has inspired the identification of synthetic and semi-synthetic analogs, in addition to other natural products. Variations within the cap group moiety of these HDACi result in modulation of biological activity with enhanced isoform selectivity among different HDAC isoforms [65]. Based on their macrocyclic moieties, these HDACi can be subdivided into two families. The first class, as exemplified by trapoxin, apicidin, azumamide and HC-toxin, are cyclic tetrapeptides (Fig. 2) containing a mixture of L- and D-amino acids and cyclic amino acids such as proline or pipecolic acid. Both of these features reduce the ring strain associated with cyclic tetrapeptides. The second family comprises bicyclic depsipeptides [66] (Fig. 3) which are isolated from bacterial fermentation extracts. These bicyclic depsipeptides are less stereochemically constrained because of larger macrocycles compared to the cyclic tetrapeptides [67]. Unlike the tetrapeptide analogs, most of the bicyclic depsipeptides are prodrugs, which must undergo intracellular reactions to unmask thiol-containing side chains which then serve as the ZBGs. Even though thiol has weaker affinity for zinc compared to a hydroxamic acid, interaction between the macrocyclic scaffold and the amino acid side chains within enzyme hydrophobic outer rings that lead to the active site is thought to result in the significantly higher overall potency observed, compared to SAHA. As the enzyme rim residues are less conserved than those of the active site [68], these depsipeptides discriminate well between the eleven HDAC isoforms in classes I, II and IV. They have pronounced selectivity [42] for the Class I HDACs (HDAC1, HDAC2, HDAC3 and HDAC8), which happen to be the isoforms most heavily implicated [65, 69, 70] in cancer and other proliferative disease conditions.
Fig. (2).

Examples of cyclic tetrapeptide HDACi.
Fig. (3).

Examples of depsipeptide HDACi.
The development of synthetic analogs based on these natural products is hampered by complex and low yielding routes necessary to access these macrocycles. In addition, the peptidyl nature of the macrocycles offer limited opportunities for semi-synthetic modification. Many of these challenges have been overcome and several synthetic analogs have been reported in the past decades. A detailed discussion of the structure activity relationship (SAR) of cyclic tetrapeptide and depsipeptide HDACi is presented in section 2. a. i. below.
2.a.i. Cyclic Tetrapeptide HDACi
Early examples of cyclic tetrapeptides include HC-toxins [71–74], Cyl-1/2 [75–77], WF-3161 [78, 79] chlamydocin [80] and trapoxins A and B [81] (Fig. 2). They belong to the family of hydrophobic cyclotetrapeptides containing a unique amino acid 2-amino-8-oxo-9,10-epoxydecanoic acid (Aoe). These HDACi inhibit HDAC through their epoxy-ketone functionality which reacts with the target enzyme in an irreversible manner [29]. Replacement of the epoxy-ketone moiety with CH2=CH2, CH2CH3, or CH2 CH2OH has resulted in reduction of activity, whereas reduction of ketone to alcohol leads to loss of activity in the case of chlamydocin [80]. Similarly, the 9,10-diol analogue of HC-toxin shows no significant activity against HDAC [82]. HC-toxin shows HDAC class I selectivity of greater than 1000-fold with IC50=10 nM against rat liver HDAC proteins, which are comprised of predominantly HDAC 1–3 [83]. Chlamydocin, Cyl-2, trapoxin A and B showed 640 to 57000-fold selectivity for HDAC1 versus HDAC6 with IC50 = 110–820 pM against HDAC1 [84].
Apicidin, a fungal metabolite from two fusarium species (ATCC 74289 and ATCC 74322) [85], belongs to the same class of cyclic tetrapeptide HDACi as the trapoxins. Unlike the trapoxins however, apicidin lacks the terminal α-keto-epoxide and it is no surprise that it is a reversible HDACi. Apicidin inhibits HDAC1 with single digit nanomolar IC50 (IC50 = 1 nM), suggesting that the epoxy-ketone group is not necessary for exertion of biological activity. Moreover, apicidin exhibits low nanomolar activity against the apicomplexan family of parasites by inhibiting apicomplexan HDAC enzyme [85]. Successively, it was evaluated as an antiproliferative agent by Han et al. [86] who demonstrated that apicidin inhibits proliferation of tumor cells via induction of p21WAF1/CIP1 in a similar manner to other HDACi. A follow-up study by the same group revealed that apicidin induces apoptosis by induction of Fas/Fas ligand expression [87].
In 2002, Singh et al. [88] reported isolation of five new analogs of apicidin namely, apicidin A, C, D1, D2 and D3, together with the known apicidin isolated from Fusarium pallidoroseum [89]. These apicidins differ essentially by the nature of the side-chain at C-2 (Fig. 2). The anti-HDAC activity of these apicidin analogs were evaluated using extracts from human HeLa cells and E. tenella protozoa cells. Along with apicidin, apicidin A and apicidin D1 were among the most active compounds with IC50 in the 1–4 nM range. In contrast, compounds such as apicidin D2 lacking the C=O group at C-8, or having the C=O reduced to alcohol, converted into oxime, and to CH2, have considerably lower anti-HDAC activity, with IC50 greater than 400 nM. This suggests that, in addition to the cyclic nature of the peptide and the presence of a tryptophan residue, the C-8 keto group is critical for the biological activity. This result conforms with the early observation by Colletti and coworkers [90] who conducted SAR studies on the side chain of apicidin. They found that reduction of the C-8 keto group to alcohol or CH2 group or conversion to olefin or epoxide resulted in the attenuation of the anti-HDAC activity of apicidin. However, substitution of the ethyl ketone moiety of apicidin side chain with hydroxamic acid, epimeric epoxide, and hydroxyketone resulted in analogs with improved HDAC activity relative to apicidin. Interestingly, the methyl ester derivative was also found to be active, emphasizing the importance of the macrocyclic ring in enzyme-inhibitor interaction. Altogether, they identified several apicidin analogs with picomolar cytotoxic activity against human HeLa cells and E. tenella protozoa cells.
Similarly, Komatsu and co-workers [84, 91] explored the idea of replacing the epoxy-ketone of trapoxins, WF-3161, chlamydocin and HC-toxin with hydroxamic acid as the ZBG. These synthetic hybrids of TSA and cyclic tetrapeptides are termed cyclic hydroxamic-acid containing peptides (CHAPs). Detailed SAR study on amino acid constituents of the ring structure, chirality of amino acids, and side chain structure have resulted in potent HDACi within this class of molecules. All CHAPs synthesized in this study were potent inhibitors of HDAC1 with chlamydocin-type CHAP showing the highest inhibition of HDAC1 (IC50 = 0.44 nM) among all of them. Interestingly, CHAP carboxylic acid analogs exhibit good HDAC activity (IC50 = 100 nM). Isoform selectivity for HDAC1 over HDAC6 varied with the structure of cyclic tetrapeptide with chlamydocin-type CHAP showing 86-fold higher selectivity for HDAC1 versus HDAC6. However, the epoxy-ketone group-containing parent natural products show strikingly large isoform selectivity (640–57000-fold), indicating importance of the epoxy-ketone group in isoform selectivity. Although parent natural products show potent in vitro activity along with better isoform selectivity, they behave poorly in in vivo studies, partly because of the chemical instability of the epoxy-ketone group in blood. CHAP compounds may circumvent this problem because of their superior half-life when compared to Aoe-containing parent natural products [84].
Recently, Ghadiri’s group [92] has probed the bioactive conformation of apicidin utilizing the 1,4 and 1,5 triazole-modified cyclic tetrapeptide constructs 1 and 2, to mimic a trans and cis amide bond, respectively. They provided evidence that supports cis-trans-trans-trans (c-t-t-t) conformation of the cyclic peptide backbone as the most potent conformation of apicidins, and not the all-trans (t-t-t-t) that predominates in solution. Against HDAC1 and HDAC3, the 1,5-disubstituted triazole 2 (IC50 = 7 nM and 9 nM) and apicidin (IC50 = 3 nM and 11 nM), were more potent than the 1,4-disubstitited, 1 (IC50 = 25 nM and 16 nM), while against HDAC6 and HDAC8, the 1,5-disubstituted 2 (IC50 = 6100 nM and 105 nM) is more active than apicidin (IC50 = >10,000 nM and 750 nM). This study provides strong evidence on the influence of the conformational state of the cyclic tetrapeptide moiety of apicidin on its affinity for HDAC, a fact that hitherto remained unclear.
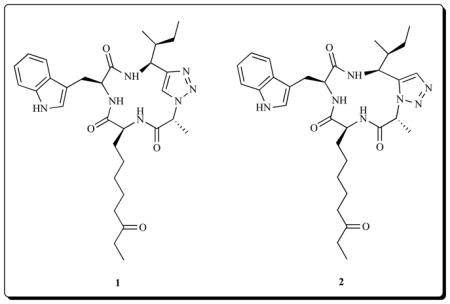
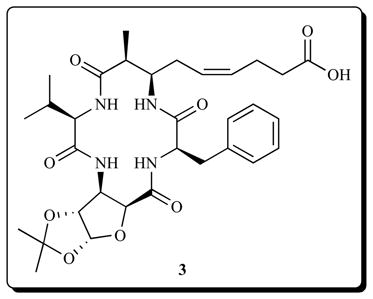
Structurally similar to the apicidins are the azumamides A-E which were isolated from the marine sponge Mycale izuensis [93]. They belong to a rare class of HDACi, incorporating four nonribosomal amino acid residues; three of which are R-amino acids (D-series), while the fourth one is a unique β-amino acid [(Z)-(2S,3R)-3-amino-2-methyl-5-nonenedioic acid-9-amide (Amnaa)] in azumamide A, B, and D and a free acid in C and E. The azumamides have potent HDAC inhibitory activity with an IC50 range of 0.045 – 1.3 μM. Azumamide E, one of the most potent of the azumamides, also showed roughly 100-fold selectivity for class I over class II HDACs (HDAC1–3, IC50 = 50–100 nM versus HDAC4–7, 9, IC50 = 9.7–28 μM) [94]. Chandrasekhar et al. [95] reported the total synthesis of azumamide E and sugar amino acid-containing analog (azumamide E-SAA) 3. A near total inhibition of HDAC activity in a cell-free system was achieved by 20 μM of compound 3. However, whole cell activity of this compound was not reported in these studies. Ganesan and coworkers [96] converted the azumamide E zinc binding group to an unnatural hydroxamic acid. This unnatural inhibitor shows activity similar to FK228 (IC50 7.0 ± 2.5 nM), indicating the shift from depsipeptide to peptide can be overcome by increasing the affinity of the zinc binding group. This result is in contrast with earlier findings where substitution of the epoxy-ketone group in chlamydocin to hydroxamic acid actually reduced its biological activity [97].
2.a.ii. Depsipeptide HDACi
FK-228 is the most well studied depsipeptide HDACi to date. FK-228 and a closely related FR901375, are naturally occurring depsipeptides isolated from Chromobacterium violaceum [98, 99] FK-228 has a unique structural feature which includes a 16-membered cyclic depsipeptide bridged by a 15-membered macrocyclic disulfide ring (Fig. 3). FK-228 is a stable prodrug which is activated by glutathione reductase in vivo by reduction of the disulfide bond to reveal a butenyl thiol moiety which then interacts with zinc at the HDAC active site [42]. It shows high selectivity toward class I HDACs compared to HDAC4 and HDAC6. Ganesan and co-workers [49], in their pursuit of a SAR study on FK228, demonstrated that the dehydrobutyrine side chain is not essential for activity. In addition, cleavage of the macrocyclic ester bond to give a linear peptide resulted in loss of activity, an observation which underscores the importance of conformational restriction of the macrocycle. This study also clarified the role of the disulfide moiety in these compounds: the cysteine free thiol resulting from disulfide cleavage facilitates cellular uptake. The potent activity of FK228 against a range of murine and human solid tumor cells, coupled with ability to target epigenetic silencing mechanisms crucial to cancer formation, facilitated its advancement into clinical trials as an anticancer agent [100, 101].
Spiruchostatin A and B, somewhat structurally simpler depsipeptides relative to FK228, were isolated from a culture broth of Pseudomonas sp. by Shinya and co-workers in 2001 [102]. These are 15-membered bicyclic depsipeptides containing (3S,4R)-statine, D-cysteine, D-alanine as amino acid residues along with disulfide bond linkage (Fig. 3). They operate by a similar prodrug strategy as FK228 and display low nanomolar activity against HDAC1. Several total syntheses of spiruchostatins have appeared in the literature [103–107]. Relatively recently, Narita et al. [108] reported total synthesis and biological activities of FK-228, spiruchostatin A and B along with 5″-epi-spiruchostatin B. These cyclic tetrapeptides all show potent activity (IC50 = 2 – 4 nM) against HDAC1 with ~500 fold selectivity over HDAC6. The unnatural 5″-epi-spiruchostatin B analog showed the highest isoform selectivity (1625-fold) for HDAC1 (IC50 = 2.4 nM) over HDAC6 (IC50 = 3900 nM) while maintaining its cytotoxic activity. This isoform selectivity is 15 fold higher than isoform selectivity of FK228, making it the highest level of class I/II selectivity among all natural as well as synthetic analogs of depsipeptides known to date [108].
Largazole, (Fig. 3), a structurally unique depsipeptide, was recently isolated from the marine cyanobacterium Symploca sp. by Luesch and co-workers [40]. It is a 16-membered macrocyclic depsipeptide that exhibits potent biological activity as well as high HDAC1 selectivity [40, 109]. Several research groups have completed the total synthesis of largazole and further investigated biochemical potency of the natural product and assessed key derivatives with enhanced potency against human HDACs [109–114]. These studies collectively established largazole as a thioester prodrug HDACi, which is likely activated by metabolic hydrolysis of the octanoyl residue. Bowers et al. [109, 112] have prepared the thiol derivative (largazole thiol) and reported potent biochemical inhibition against class I HDACs (IC50 = 0.1 nM against HDAC1). In order to study conformation activity relationship, peptide isosteres of FK228 and largazole thiol were synthesized and it was found that the FK228 isostere was 50-fold less active, while the largazole thiol isostere was 9-fold less active than the natural product itself. This reduced activity was attributed to difference in accessible conformational orientation due to subtle changes in chemical structure of the macrocyclic cap group of the isosteres relative to the respective natural product [112]. Cramer and co-workers [113] have shown that the free thiol, after rapid hydrolysis of the thioester under physiological conditions, displayed slightly lower potency (GI50=126 nM compared to GI50=49 nM against human epithelial cancer cell line A431) but higher selectivity against wild type cells using the MTT assay. Interestingly, an extension of the thioester side chain by one or two methylene groups was found not to be compatible with anti-HDAC activity, underscoring the importance of maintaining appropriate distance between the core macrocyclic head group and the thiol ZBG [113]. Further SAR studies have revealed the importance of the C-7 methyl group as demethylated derivatives showed ~1000-fold decrease in activity. Also (R)-configuration at C-17 position was found to be inactive, underscoring the importance of (S)-configuration at the C-17 position for biological activity [110, 115]. Substitution of the macrocycle valine with alanine is well tolerated [115, 116]. Epimerization of the C-2 carbon, valine to proline substitution, and oxidized and strained thiazolethiazole derivatives have all resulted in diminished activity compared to largazole [114]. Replacement of natural 3-hydroxy-7-mercaptohept-4-enoic acid ZBG with benzamide and thioamide has not resulted in more potent analogs [114]. However the use of hydroxamic acid as ZBG was not investigated. Interestingly, substitution of thiazole ring with pyridine resulted in a largazole analog with a more potent HDAC inhibition than largazole. This observation necessitates further study on the effect of substitution of thiazole ring by other six membered heterocycles on the biological activity of larga-zole [114].
Much still needs to be done to decipher the intricacy of the SAR of cyclic tetrapeptide HDACi. However, it is abundantly clear that the nature of the ring substituents and the macrocycle conformations are major determinants of the strength of association of these HDACi with various HDAC isoforms.
2.b. Macrocyclic Peptide Mimetic HDACi
Although cyclic peptide HDACi possess potent HDAC inhibition activity (as discussed in the previous sections), their development has been hampered mainly due to lack of a steady source of natural products and development problems characteristic of large peptides, most especially poor oral bioavailability [49, 51]. Efforts by Curtin and coworkers [117] to prepare less peptidic macrocyclic HDAC inhibitors led to the synthesis of succinimide-based hydroxamic acid macrocycle 4 and reaction byproduct 5 (Fig. 4). It turned out that the byproduct 5 was a more potent HDAC inhibitor (IC50 = 38 nM) than the target compound 4 (IC50 = 2.1 μM) with antiproliferative activity of IC50 = 250 nM and 150 nM against human HT1080 fibrosarcoma and MDA435 breast carcinoma cells, respectively.
Fig. (4).

Structure of Macrocyclic Succinimide Hydroxamic Acids 4 and 5.
Recently, Etzkorn and co-workers [118] reported a cyclic peptide mimic hydroxamate HDACi (6) demonstrating potent anti-HDAC activity in nuclear HDACs, with an IC50 value of 46 nM and a 4-fold selectivity of HDAC1 (IC50 = 57 nM) over HDAC8 (IC50 = 231 nM).
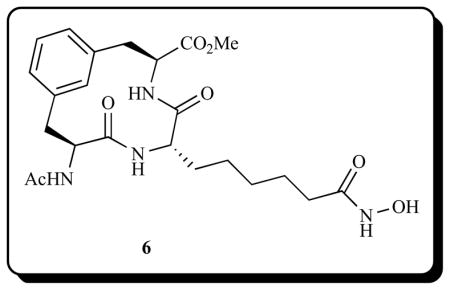
The value of introduction of non-hydrolyzable peptide isosteres in the design of new macrocyclic peptide HDACi head groups has further been validated by Pirali et al. [119]. Using ammonium chloride-catalyzed three-component reaction between various alkynyl-α-isocyanoacetamides, amino-azido amides, and aldehyde derivatives, followed by Cu(I) catalyzed intramolecular [3 + 2] cycloaddition reaction, they identified two peptidomimetic macro-cyclic hydroxamate HDACi 7a–b and 8a–b (Fig. 5).
Fig. (5).

a) Macrocyclic peptide mimetic class of HDAC inhibitors; b) Intermediate of the final macrocyclic hydroxamic acid products 7 and 8.
The cytotoxicity of these compounds against SHSY-5Y cells was reported to be in the high micromolar to low millimolar range. Compounds 7a and 7b have modest cytotoxic activity (IC50 = 100 μM) while 8a and 8b, are more potent with IC50 = 6.2 ± 0.5 μM and 6.6 ± 1.4 μM, respectively, compared to SAHA’s IC50 of 1.4 μM. Additionally, the HDAC inhibition activity of compounds 8a and 8b paralleled their cytotoxicity with IC50 = 4.3 μM and 6.61 μM respectively, compared to SAHA IC50 = 3.4 μM. Molecular modeling studies using the hydrophobic head group of intermediate 9 revealed that the three phenyl rings extended their hydrophobic contacts to three different enzymatic pockets while the macrocyclic portion interacted with the hydrophobic rim of the tubular pocket. This orientation facilitated favorable interaction of the linear C7 hydroxamate moiety-containing chain with the zinc ion at the base of the pocket.
2.c. Non-Peptide Macrocyclic HDACi
Inspired by the peptidomimetic nature of macrolide antibiotics [120], we recently reported the discovery of a new class of non-peptide macrocyclic HDACi [55]. Our molecular design relies on our hypothesis that an appropriate substitution of the cyclic peptide moiety of a prototypical cyclic peptide HDACi with macrolide skeletons will generate a new class of potent non-peptide macrocyclic HDACi [55]. We indeed found that macrocyclic skeletons derived from 14- and 15-membered macrolides are suitable as surrogates for the cap-groups of macrocyclic HDACi. We showed that incorporation of a hydroxamate ZBG, through hydrophobic SAHA-like and aryltriazolyl [121] linker groups, to the desosamine sugar moiety of 15-membered azithromycin and 14-membered clarithromycin resulted in a new class of non-peptide macrocyclic HDACi (Fig. 6). Our initial SAR studies focused on the effects of the modification of the linker-cap group connection moiety (amide and 1,2,3-triazole ring group), macrolide skeletons, and linker length (Fig. 6) on anti-HDAC activity of these non-peptide macrocyclic HDACi [55]. We found that these compounds displayed both linker-length and macrolide-type dependent anti-HDAC activities with IC50 in the low nanomolar range (Table 1).
Fig. (6).

Analogs of (a) 15-membered azithromycin and (b) 14-membered clarithromycin classes of HDAC inhibitors.
Table 1.
In VitroAnti-HDAC Activity (IC50) and Isoform Selectivity of Non-Peptide Macrocyclic HDACi
| Compound | HDAC 1/2 (nM) | HDAC 8 (μM) | Isoform Selectivity |
|---|---|---|---|
| 10 | 107.1 | 6.68 | 62 |
| 11 | 109.8 | 2.32 | 21 |
| 12a | 91.6 | 4.73 | 51.63 |
| 12b | 13.9 | 0.99 | 71.77 |
| 12c | 58.9 | 7.13 | 121 |
| 12d | 145.5 | 11.05 | 38 |
| 12e | 226.7 | ND | c |
| 13a | 88.8 | 3.74 | 42.11 |
| 13b | 10.6 | 1.02 | 96.59 |
| 13c | 72.4 | 6.78 | 94 |
| 14a | 37.0 | 3.99 | 107.90 |
| 14b | 4.1 | 1.89 | 462.10 |
| 14c | 55.6 | 5.88 | 106 |
| 14d | 169.8 | 10.55 | 56 |
| 14e | 223.4 | ND | c |
| 15a | 44.3 | 4.75 | 107.32 |
| 15b | 1.9 | 1.39 | 743.32 |
| 15c | 123.0 | 4.42 | 36 |
| SAHA | 65 | 1.86 | 28.61 |
Specifically, the HDAC inhibition activity was nearly identical between triazole-linked compounds (12a, IC50 = 91.6 nM) and amide-based compounds (10, IC50 = 107.1 nM) (Table 1). This result is in contrast to our previous studies [121] where we found that incorporation of triazole ring on simple aliphatic hydroxamates resulted in enhanced anti-HDAC activity. Optimization of the methylene linker length resulted in analogs with enhanced HDAC affinity, specifically C7-linker compounds (12b, IC50 = 13.9 nM; 13b, IC50 = 10.6 nM; 14b, IC50 = 4.1 nM; 15b, IC50 = 1.9 nM; compare to SAHA, IC50 = 65 nM). A further increase in linker length did not improve HDAC inhibition activity (Table 1). This result was confirmed through interrogation of the interaction between Histone Deacetylase-Like Protein (HDLP) and these macrocyclic HDACi using Auto-Dock [55]. We found that the hydroxamic acid moiety of C7-linker compounds maintained optimal interaction with the catalytic Zn2+ ion at the enzyme’s active site. Additionally, interesting molecular surface complementarities between the macrolide skeleton of C7-linker compounds and the HDAC outer rim further enhanced their binding association. These compounds adopt docked structures whereby the macrolide rings optimally interact with the hydrophobic residues in enzyme outer rim while their hydrophilic hydroxyl groups are oriented away from the pocket’s hydrophobic residues (Fig. 7a). Interestingly, 14-membered compounds 14b and 15b demonstrated a 2- to 5-fold better HDAC inhibition potency relative to their 15-membered congeners 12b and 13b. This may be due to the fact that the larger 15-membered ring is about 0.5 Å closer to the phenyl ring of Phe 338 that defines one of the hydrophobic pockets at the enzyme outer rim compared to that of the 14-membered compound (Fig. 7b). This might compromise the binding affinity of the 15-membered compounds relative to the 14-membered analogs [55].
Fig. (7).

Docked structures of 14- and 15-membered macrolide-derived HDACi at the active site of HDLP. (a) Superposition of the low energy conformation of 13a (blue) and SAHA (yellow) revealed the pocket binding preferences of inhibitors at the HDLP surface. (b) Relative orientation of the macrocyclic rings of 13b (orange) and 15b (yellow) with respect to Phe 338 at the HDLP surface.
Consistent with literature reports [65] on isoform selectivity arising from modifications of the cap group, these non-peptide macrocyclic HDACi are more selective against HDACs 1 and 2 relative to HDAC 8, another class I HDAC isoform, and hence have subclass HDAC isoform selectivity. Particularly 14-membered C7-linker compounds 14b and 15b displayed several hundred-fold preference for HDAC 1/2 over HDAC 8 (14b, HDAC 1/2 IC50 = 4.1 nM, HDAC 8 = 1890 nM; 15b, HDAC 1/2 IC50 = 1.9 nM, HDAC 8 IC50 = 1390 nm) (Table 1). Furthermore, these compounds also displayed class I selectivity over class II when tested against HDAC 6 (15b, HDAC 6 IC50 = 148.5 nM), unlike SAHA which equally inhibited HDAC 1/2 and 6 [55].
To screen for the whole cell activity of these macrocyclic HDACi, we studied their effect on the viability of SKMES-1 (human NSCLC cell line), NCI-H69 (human SCLC cell line), DU-145 (human prostate cancer cell line), and non-transformed human primary lung fibroblasts and mammary epithelial cell lines. These compounds inhibited the proliferation of all transformed cells with EC50 values close to or better than those obtained for SAHA while remaining non-toxic to the non-transformed human primary lung fibroblasts and mammary epithelial cell lines. Specifically, compounds 13a, 14b, and 15b are at least twice as potent as SAHA in DU-145 cells (Table 2). We subsequently validated HDACs as potential intracellular targets of these compounds by monitoring the drug induced changes in the expression levels of p21WAF1/CIP1 protein, a biomarker whose expression is upregulated upon HDAC inhibition [55].
Table 2.
Cell Growth Inhibition Data for Non-Peptide Macrocyclic HDACi
| Compound | SKMES 1 (μM) | NCI-H69 (μM) | DU-145 (μM) | Lung Fibroblast (μM) | HMEC (μM) |
|---|---|---|---|---|---|
| 10 | NT | NT | >25 | NT | NT |
| 11 | NT | NT | <25 | NT | NT |
| 12a | 1.79 | 1.92 | 1.45 | >10 | >10 |
| 12b | 2.33 | 3.45 | 1.88 | >10 | >10 |
| 12c | 4.89 | 4.56 | 5.89 | >10 | >10 |
| 12d | 7.54 | 8.45 | >10 | >10 | >10 |
| 13a | 1.68 | 1.77 | 1.24 | >10 | >10 |
| 13b | 2.56 | 3.01 | 1.97 | >10 | >10 |
| 13c | 4.67 | 3.99 | 5.68 | >10 | >10 |
| 14a | 2.15 | 2.67 | 2.98 | >10 | >10 |
| 14b | 1.33 | 1.45 | 1.12 | >10 | >10 |
| 14c | 4.89 | 5.67 | 6.97 | >10 | >10 |
| 14d | 7.12 | 7.29 | 8.14 | >10 | >10 |
| 15a | 1.95 | 1.92 | 3.29 | >10 | >10 |
| 15b | 1.28 | 1.49 | 1.05 | >10 | >10 |
| 15c | 4.45 | 5.09 | 5.78 | >10 | >10 |
| SAHA | 2.42 | 2.06 | 2.12 | >10 | >10 |
In a follow up investigation, we sought to further explore the SAR of this class of macrocyclic HDACi. Specifically, we have probed the roles played by the hydrophobic pocket bounded by the phenyl ring of Phe 338 in the interaction of the enzyme with the macrocyclic moieties of these HDACi [122]. In this study, we incorporated a tricyclic ketolide (TE-802) scaffold as an alternative macrocyclic surface recognition domain. The resulting second generation macrocyclic non-peptide hydroxamates (Fig. 8) are potent HDACi. Many of these compounds have better anti-HDAC activities relative to their macrolide counterparts. These compounds also showed selectivity for class I over class II HDAC enzymes (Table 3), as well as low nanomolar cytotoxicity against various cancer cell lines with relatively low cytotoxicity on the non-transformed cell lines (Table 4). Molecular docking analysis revealed the structural basis of the enhanced anti-HDAC activity of these ketolide based HDACi. The enhanced hydrophobicity and/or rigidity of the TE-802 moiety of the C5- and C6-linker compounds 16a and 16b, respectively, forced their ring to adopt a docked orientation that is distinct from the corresponding 14-membered clarithromycin template (Fig. 9a). The ketolide ring of 16b fit optimally into the Phe 338 pocket (pocket 1) while the C6-linker methylene group efficiently presented the hydroxamate ZBG to the active site zinc. To accommodate the loss of one methylene group and consequently optimally interact with the active site zinc, the ketolide ring of 16a has to be slightly lifted out of the Phe-338 pocket and be more solvent exposed relative to that of 16b. Concomitantly, a potentially energetically costly sharp kink is introduced into the linker methylene group of 16a (Fig. 9b). This result may explain the 7-fold enhanced anti-HDAC activity of 16b relative to 16a.
Fig. (8).

Non-peptide HDAC inhibitors based on tricyclic ketolide scaffold.
Table 3.
Anti-HDAC Activity and Isoform Selectivity of Tricyclic Ketolide-HDAC Inhibitors
| Compd | n | Nuclear Extract (nM) | HDAC 8 (nM) | HDAC8:Nuclear Extract Isoform Selectivity | HDAC 6 (nM) | HDAC6:Nuclear Extract Isoform Selectivity |
|---|---|---|---|---|---|---|
| 16a | 1 | 7.77 | 796.2 | 102.5 | 1180.1 | 151.9 |
| 16b | 2 | 1.03 | 544.6 | 528.7 | 728.7 | 707.5 |
| 16c | 3 | 104.2 | 1909.3 | 18.3 | 1709.8 | 16.4 |
| 16d | 4 | 163.6 | 2859.9 | 17.5 | 1916.9 | 11.7 |
| 16e | 5 | 208.2 | 4557.8 | 21.9 | 3203.1 | 15.4 |
Table 4.
Antiproliferative Activity of Tricyclic Ketolide-HDAC Inhibitors
| Compd | n | DU-145 (uM) | A549 (uM) | MCF-7 (uM) | Hs1.Lu Normal Lung Fibroblast (uM) |
|---|---|---|---|---|---|
| 16a | 1 | 1.64 | 1.17 | 1.26 | >10 |
| 16b | 2 | 0.82 | 0.66 | 0.75 | >10 |
| 16c | 3 | 3.70 | 1.64 | 1.35 | >10 |
| 16d | 4 | 3.89 | 2.48 | 2.56 | >10 |
| 16e | 5 | 4.83 | 2.81 | 3.68 | >10 |
Fig. (9).

Docked structures of ketolide-derived HDACi at the active site of HDLP. (a) Superposition of the low energy conformation of 13b (grey) and 16b (blue) revealed the pocket binding preferences of inhibitors at the HDLP surface. (b) Relative orientation of the macrocyclic rings of 16a (yellow) and 16b (green) within the Phe 338 pocket. (c) Comparison of the orientation of the macrocyclic rings of the C6-linker compound 16b (grey) and C9-linker compound 16e (light-blue) within the Phe 338 pocket revealed the structural basis for the chain length dependence of the anti-HDAC activities of ketolide-derived HDACi.
Further analysis of the docked orientations of analogs with longer methylene linker shed more light on the chain length dependence of the anti-HDAC activities of these ketolide-based HDACi. An illustrative example, comparing the docked structures of C6-linker 16b with that of C9-linker 16e, is shown in Fig. 9c. In general, increase in linker lengths above C6 progressively extrudes the ketolide ring from the Phe 338 pocket into solvent exposed regions, thereby compromising the compounds’ affinity for the enzyme and resulting in the attenuation of their HDAC inhibition activity.
The non-peptide macrocyclic HDACi described in this section are a relatively new class of HDACi and the breath of their biological activities is yet to be fully explored. However, because of the selective tissue distribution that may be conferred by the appended macrolide moiety, some of these HDACi are anticipated to have targeted anticancer activity. Specifically, compounds incorporating azithromycin skeleton could be selectively accumulated in the lungs, thereby possessing lung-selective anticancer activity. The prospect of tissue-specific HDACi delivery is a particularly enticing alternative to isoform selective HDACi and could lead to the identification of new chemotherapeutic agents for use in targeted cancer therapy applications [55].
2.d. Miscellaneous Macrocyclic HDACi
Fusetani and co-workers [123, 124] recently found that cyclostellettamines, a group of structurally related macrocyclic bispyridinium alkaloids (Fig. 10) possess modest anti-HDAC activity with IC50 between 17 – 80 μM (Table 5). Compared to their anti-HDAC activity, cyclostellettamines possess enhanced cytotoxic activity against HeLa human cervix carcinoma, P388 mouse leukemia and 3Y1 rat fibroblast cancer cell lines (Table 5, IC50 range between 0.6 –11.0 μM). The discrepancy between the anti-HDAC and whole cell activities of cyclostellettamines may indicate additional or alternative mechanism(s) of antiproliferative activity other than intracellular HDAC inhibition. Nevertheless, the allure of the HDAC inhibitory activity of cyclostellettamines is the fact that they are structurally unrelated to other naturally occurring macrocyclic HDACi currently known. It remains to be seen if the HDAC inhibitory potency of this class of macrocyclic HDACi could be enhanced to the level of those discussed in this review.
Fig. (10).

Cyclostellettamine A and its congeners.
Table 5.
Anti-HDAC and Anti-Proliferate Activity of Cyclostellettamines 17 – 20
| Compound | HDAC μM | P388 μM | HeLa μM | 3Y1 μM |
|---|---|---|---|---|
| 17 | 80 | 2.7 | 2.8 | 11 |
| 18 | 17 | 1.3 | 0.6 | 4.3 |
| 19 | 30 | 1.3 | 1.8 | 3.2 |
| 20 | 61 | 2.1 | 1.8 | 7.2 |
3. CLINICAL EVALUATIONS
To date, FK-228 (Romidepsin) is the only macrocyclic HDAC inhibitor to progress through clinical trials. FK-228 was initially assessed for responses against a range of solid tumors, and results from these studies showed promising responses among tested patients [125]. Initially two Phase I dosing trials were carried out. The first trial incorporated two doses at days 1 and 5, repeating every three weeks, while the second trial administered three doses on days 1, 8, and 15, repeating every four weeks. The maximum tolerated dose for each study was determined to be 17.8 mg/m2 and 14 mg/m2, respectively. Most patients showed limited activity, although promising responses against cutaneous T-cell lymphoma (CTCL) were observed [125]. It should be noted that in an additional cohort, included after completion of the first trial, a patient with peripheral T-cell lymphoma experienced complete remission in response to FK-228. Following this success and similar results obtained in patients with cutaneous T-cell lymphomas, a Phase II trial was launched with a recommended dose of 17.8 mg/m2, administered on days 1 and 5 of a 21 day rotation [100].
While responses against CTCL were promising, none of the patients experienced a complete remission [100]. Because of this, other Phase II trials are currently ongoing with the focus on combination therapies of FK-228 and cytotoxic anticancer agents such as the proteasome inhibitor bortezomib and the nucleoside analogue gemcitabine [126, 127]. Additionally, a Phase II trial involving FK-228 as a single agent against previously-treated colorectal cancer has been completed, but no objective responses were observed [128].
Since the publication of the initial clinical results, FK-228 has entered numerous clinical trials against other forms of cancer including lung cancer and various subtypes of leukemia [129, 130]. In trials against acute myeloid leukemia (AML), apoptosis has been observed at concentrations suggesting HDAC inhibition mechanisms [129]. After completion of this study, several patients showed measurable antitumor responses, but no complete responses [128]. Contrasting with this result, another clinical trial using FK-228 against AML, showed no significant change in chromatin acetylation, specifically histone H3 and H4, but did result in one complete remission and six stable disease conditions [131]. In a trial against lung cancer, FK-228 produced no objective responses, but did confirm in vitro findings, namely resulting in histone H4 acetylation, p21 expression, and a general shift of gene expression towards those observed in normal lung epithelia [130].
There has been some concern over preclinical data that suggests that FK-228 could result in cardiac toxicity. One trial reported serious cardiac complications in tested patients and had to be terminated due to the death of one patient [132]. The cause of death could not be determined, but was assumed to be due to a ventricular arrhythmia that may have been related to several factors, FK-228 being one. On the basis of this, the trial was terminated. Also, two patients were observed with asymptomatic ventricular tachycardia and three experienced QTc prolongations, but these resolved between doses. It should be noted that the results of this trial contrast greatly with subsequent clinical results. To date, no other clinical observations of significant cardiac toxicity have been observed in studies using FK-228 [128, 130, 131, 133] however it is important to underscore that the serious adverse cardiac effects seen with FK-228 treatment could be cause for concern [134]. Also, it is important to emphasize that Shah et al. noted that the cause of death in their trial could not be determined to be linked with FK-228 treatment [132]. With the exception of this mar, FK-228 has shown great promise in the clinic, specifically against T-cell lymphoma. Of particular note is the remarkable length of response to FK-228, even after treatment has ceased, extending over 3 years in some cases [135]. Due to the successes of the clinical trials, in November of 2009, FK-228 (ISTODAX®) was approved by the FDA for treatment of CTLC [136]. With this approval, the future looks promising for the clinical use of FK-228, both as a single agent and in combination with other chemotherapeutic agents.
4. FUTURE PROSPECT
The outlook for macrocyclic HDACi is promising. The advent of several new macrocyclic HDACi has enabled an undertaking of one of the first steps toward understanding the SAR of these compounds. It is expected that these compounds will continue to elicit medicinal chemistry interest for many years to come. Identification of better synthetic routes to macrocyclic ring construction will no doubt play a crucial role in laying the groundwork for exploring the wealth of bioactivities of this promising HDACi. Specifically, the flashes of success of FK-228 in clinical trials and its subsequent FDA approval by the FDA, as well as the in vitro potency of other macrocyclic inhibitors, will continue to stimulate interest in the use of this class of HDACi in cancer therapy and could open the doors for other macrocyclic HDACi as clinical agents.
Acknowledgments
This work was financially supported by Georgia Institute of Technology, by the Blanchard fellowship and by NIH Grant R01CA131217 (A.K.O.). W.G. is a recipient of a GAANN predoctoral fellowship from the Georgia Tech Center for Drug Design, Development and Delivery.
ABBREVIATIONS
- HDAC
Histone Deacetylase
- HAT
Histone Acetyltransferase
- HDACi
Histone Deacetylase Inhibitors
- HDLP
Histone Deacetylase-Like Protein
- NAD+
Nicotinamide Adenine Dinucleotide
- ZBG
Zinc Binding Group
- SAR
Structure Activity Relationship
- AML
Acute Myeloid Leukemia
- CTCL
Cutaneous T-cell Lymphoma
- SAHA
Suberoylanilide Hydroxamic Acid
- TSA
Trichostatin A
- SCLC
Small Cell Lung Cancer
- NSLC
Non-Small Cell Lung Cancer
References
- 1.Carey N, La Thangue NB. Histone deacetyalse inhibitors: Gathering pace. Curr Opin Pharmacol. 2006;6:369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Althaus FR, Höfferer L, Kleczkowska HE, Malanga M, Naegeli H, Panzeter PL, Realini CA. Histone Shuttling by Poly ADP-Ribosylation. Mol Cell Biochem. 1994;138:53–59. doi: 10.1007/BF00928443. [DOI] [PubMed] [Google Scholar]
- 6.Shiio Y, Eisenman RN. Histone Sumoylation is Associated with Transcriptional Repression. Proc Natl Acad Sci USA. 2003;100:13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 9.Davie JR, Hendzel MJ. Multiple functions of dynamic histone acetylation. J Cell Biochem. 1994;55:98–105. doi: 10.1002/jcb.240550112. [DOI] [PubMed] [Google Scholar]
- 10.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 11.Gray SG, Dangond F. Rationale for the use of histone deacetylase inhibitors as dual therapeutic modality in multiple schlerosis. Epigenetics. 2006;1:67–75. doi: 10.4161/epi.1.2.2678. [DOI] [PubMed] [Google Scholar]
- 12.Grabiec AM, Tak PP, Reedquist KA. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep Our HATs on? Arthritis Res Ther. 2008;10:226. doi: 10.1186/ar2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-275, with marked in vivo anti-tumor activity against human tumors. Proc Natl Acad Sci USA. 1999;96:4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glick RD, Swindemen SL, Coffey DC, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59:4392–4399. [PubMed] [Google Scholar]
- 15.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- 16.Andrews KT, Tran TN, Lucke AJ, Kahnberg P, Le GT, Boyle GM, Gardiner DL, Skinner-Adams TS, Fairlie DP. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob Agents Chemother. 2008;52:1454–1461. doi: 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agbor-Enoh S, Seudieu C, Davidson E, Dritschilo A, Jung M. Novel inhibitor of plasmodium histone deacetylase that cures P. berghei-infected Mice. Antimicrob Agents Chemother. 2009;53:1727–1734. doi: 10.1128/AAC.00729-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dow GS, Chen Y, Andrews KT, Caridha D, Gerena L, Gettayacamin M, Johnson J, Melendez V, Obaldia N, III, Tran TN, Kozikowski AP. Antimalarial activity of phenylthiazolyly-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob Agents Chemother. 2008;52:3467–3477. doi: 10.1128/AAC.00439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mai A, Cerbara I, Valente S, Massa S, Walker LA, Tekwani BL. Antimalarial and antileishmanial activities of aroyl-pyrroyl-hydroxyamides, a new class of histone deacetylase inhibitor. Antimicrob Agents Chemother. 2004;48:1435–1436. doi: 10.1128/AAC.48.4.1435-1436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergnes B, Vanhille L, Ouaissi A, Sereno D. Stage-specific antileishmanial activity of an inhibitor of SIR2 histone deacetylase. Acta Trop. 2005;94:107–115. doi: 10.1016/j.actatropica.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Patil V, Guerrant W, Chen PC, Gryder B, Benicewicz DB, Khan SI, Tekwani BL, Oyelere AK. Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorg Med Chem. 2010;18(1):415–425. doi: 10.1016/j.bmc.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- 23.Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral schlerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riggs MG, Whittaker RG, Neumann JR, Ingram VR. n-Butyrate causes histone modification in hela and friend erythroleukemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 26.Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem. 1978;253:3364–3366. [PubMed] [Google Scholar]
- 27.Witt O, Lindemann R. HDAC inhibitors: Magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 2009;280:123–124. doi: 10.1016/j.canlet.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 29.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 30.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 31.Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009;280:192–200. doi: 10.1016/j.canlet.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 32.http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm094952.htm
- 33.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA Approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 34.Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46:5097–5116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 35.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetlase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Matsuura A, Kouketsu A, Nakagawa H, Miyata N. Identification of a potent non-hydroxamate histone deacetylase inhibitor by mechanism-based drug design. Bioorg Med Chem Lett. 2005;15:331–335. doi: 10.1016/j.bmcl.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 37.Samoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Mulder GJ, Meerman JH. Sulfonation and glucoridation as competing pathways in the metabolism of hydroxamic acids: the role of n, o-sulfonation in chemical carcinogenesis of aromatic amines. Envir Health Perspect. 1983;49:27–38. doi: 10.1289/ehp.834927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vassilou S, Mucha A, Cuniasse P, Georgiadis D, Lucet-Levannier K, Beau F, Kannan R, Murphy G, Knäuper V, Rio MC, Basset P, Yiotakis A, Dive V. Phosphinic pseudotripeptides as potent inhibitors of matrix metalloproteinases: A structure-activity study. J Med Chem. 1999;42:2610–2620. doi: 10.1021/jm9900164. [DOI] [PubMed] [Google Scholar]
- 40.Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent the floridian marine cyanobacterium symploca Sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida M, Matsuyama A, Komatsu A, Nishino N. From discover to the current generation of histone deacetylase inhibitors. Curr Med Chem. 2003;10:2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]
- 42.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 43.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 44.Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, Kim DY. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson EM, Rotwein P. Control of MyoD function during initiation of muscle differentiation by an autocrine signalling pathway activated by insulin-like growth factor-II. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M605445200. [DOI] [PubMed] [Google Scholar]
- 46.Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, Nam SW. Increased expression of histone deacetylase 2 in human gastric cancer. APMIS. 2005;113:264–268. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 47.Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Human HDAC7 harbors a class IIA histone deacetylase-specific zinc-binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yurek-George A, Cecil AR, Mo AH, Wen S, Rogers H, Habens F, Maeda S, Yoshida M, Packham G, Ganesan A. The first biologically active synthetic analogues of FK228, the depsipeptide histone deacetylase inhibitor. J Med Chem. 2007;50:5720–5726. doi: 10.1021/jm0703800. [DOI] [PubMed] [Google Scholar]
- 50.Shute RE, Megumi K, Rich DH. Conformationally constrained biologically active peptides: tentative identification of the antimitogenic bioactive conformer of the naturally occurring cyclic tetrapeptides. Tetrahedron. 1988;44:685–695. [Google Scholar]
- 51.Loiseau N, Gomis JM, Santolini J, Delaforge M, André F. Predicting the conformational states of cyclic tetrapeptides. Biopolymers. 2003;69(3):363–385. doi: 10.1002/bip.10339. [DOI] [PubMed] [Google Scholar]
- 52.Meutermans WD, Bourne GT, Golding SW, Horton DA, Campitelli MR, Craik D, Scanlon M, Smythe ML. Difficult macrocyclizations: new strategies for synthesizing highly strained cyclic tetrapeptides. Org Lett. 2003;5:2711–2714. doi: 10.1021/ol034907o. [DOI] [PubMed] [Google Scholar]
- 53.Glenn MP, Kelso MJ, Tyndall JD, Fairlie DP. Conformationally homogenous cyclic tetrapeptides: useful new three-dimensional scaffolds. J Am Chem Soc. 2003;125:640–641. doi: 10.1021/ja029205t. [DOI] [PubMed] [Google Scholar]
- 54.Shivashimpi GM, Amagai S, Kato T, Nishino N, Maeda S, Nishino TG, Yoshida M. molecular design of histone deacetylase inhibitors by aromatic ring shifting in chlamydomycin framework. Bioorg Med Chem. 2007;15:7830–7839. doi: 10.1016/j.bmc.2007.08.041. [DOI] [PubMed] [Google Scholar]
- 55.Oyelere AK, Chen PC, Guerrant W, Mwakwari SC, Hood R, Zhang Y, Fan Y. Non-peptide macrocyclic histone deacetylase inhibitors. J Med Chem. 2009;52(2):456–468. doi: 10.1021/jm801128g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deshmukh PH, Schulz-Fademrecht C, Procopiou PA, Vigushin DA, Coombes RC, Barrett AGM. Ring-Closing metathesis in the synthesis of biologically active peptidomimetics of apicidin A. Adv Synth Catal. 2007;349:175–183. [Google Scholar]
- 57.McLaughlin F, La Thangue NB. Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 58.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug, Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 59.Drummond DC, Noble CO, Kirpotin DM, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 60.Marson CM. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med Chem. 2009;9(6):661–692. doi: 10.2174/187152009788679976. [DOI] [PubMed] [Google Scholar]
- 61.Newkirk TL, Bowers AA, Williams RM. Discovery, biological activity, synthesis and potential therapeutic utility of naturally occurring histone deacetylase inhibitors. Nat Prod Rep. 2009;26(10):1293–1320. doi: 10.1039/b817886k. [DOI] [PubMed] [Google Scholar]
- 62.Kalin JH, Butler KV, Kozikowski AP. Creating zinc monkey wrenches in the treatment of epigenetic disorders. Curr Opin Chem Biol. 2009;13(3):263–271. doi: 10.1016/j.cbpa.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 63.Moradeia O, Vaisburg A, Martell RE. Histone Deacetylase Inhibitors in Cancer Therapy: New Compounds and Clinical Update of Benzamide-Type Agents. Curr Top Med Chem. 2008;8(10):841–858. doi: 10.2174/156802608784911581. [DOI] [PubMed] [Google Scholar]
- 64.Jones P, Steinkühler C. From natural products to small molecule ketone histone deacetylase inhibitors: development of new class specific agents. Curr Pharm Des. 2008;14(6):545–561. doi: 10.2174/138161208783885317. [DOI] [PubMed] [Google Scholar]
- 65.Bieliauskas AV, Pflum MKH. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Townsend PA, Crabb SJ, Davidson SM, Johnson PWM, Packham G, Ganesan A. The Bicyclic Depsipeptide Family of Histone Deacetylase Inhibitors. In: Schreiber SL, Kapoor TM, Wess G, editors. Chemical Biology From Small Molecules to System Biology and Drug Design. Vol. 2. Wiley-VCH; Weinheim: 2007. pp. 693–720. [Google Scholar]
- 67.Chorev M, Goodman M. A dozen years of retro-inverso peptidomimetics. Acc Chem Res. 1993;26(5):266–273. [Google Scholar]
- 68.Nielsen TK, Hildmann C, Dickmanns A, Schwienhorst A, Ficner R. Crystal structure of a bacterial class 2 histone deacetylase homologue. J Mol Biol. 2005;354(1):107–120. doi: 10.1016/j.jmb.2005.09.065. [DOI] [PubMed] [Google Scholar]
- 69.Kozikowski AP, Butler KV. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki T, Itoh Y, Miyata N. Isoform-selective histone deacetylase inhibitors. Curr Pharm Des. 2008;14:529–544. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- 71.Gross ML, McCrery D, Crow F, Tomer KB. The structure of the toxin from. Helminthosporium carbonum Tetrahedron Lett. 1982;23:5381–5384. [Google Scholar]
- 72.Liesch JM, Sweeley CC, Staffeld GD, Anderson MS, Weber DJ, Scheffer RP. Structure of HC-toxin, a cyclic tetrapeptide from. Helminthosporium carbonum Tetrahedron Lett. 1982;38:45–48. [Google Scholar]
- 73.Kim SD, Knoche HW. Structure of an amino acid analog of the host-specific toxin from. Helminthosporium carbonum Tetrahedron Lett. 1985;26:969–972. [Google Scholar]
- 74.Tanis SP, Horenstein BA, Scheffer RP, Rasmussen JB. A new host specific toxin from. Helminthosporium carbonum Heterocycles. 1986;24:3423–3431. [Google Scholar]
- 75.Hirota A, Suzuki A, Suzuki H, Aizawa K, Tamura S. Structure of Cyl-2, a novel cyclotetrapeptide from. Cylindrocladium scoparium Agric Biol Chem. 1973;37:955–956. [Google Scholar]
- 76.Hirota A, Suzuki A, Suzuki H, Tamura S. Isolation and biological activity of Cyl-2, a metabolite of. Cylindrocladium scoparium Agric Biol Chem. 1973;37:643–647. [Google Scholar]
- 77.Hirota A, Suzuki A, Suzuki H, Tamura S. Characterization of four amino acids constituting Cyl-2, a metabolite from. Cylindrocladium scoparium Agric Biol Chem. 1973;37:1185–1189. [Google Scholar]
- 78.Kawai M, Pottorf RS, Rich DH. Structure and solution conformation of the cytostatic cyclic tetrapeptide WF-3161, cyclo-[L-leucyl-L-pipecolyl-L-(2-amino-8-oxo-9,10-epoxydecanoyl)-D-phenylalanyl] J Med Chem. 1986;29:2409–2411. doi: 10.1021/jm00161a046. [DOI] [PubMed] [Google Scholar]
- 79.Umehara K, Nakahara K, Kiyoto S, Iwami M, Okamoto M, Tanaka H, Kohsaka M, Aoki H, Imanaka H. Studies on WF-3161, a new antitumor antibiotic. J Antibiot. 1983;36:478–483. doi: 10.7164/antibiotics.36.478. [DOI] [PubMed] [Google Scholar]
- 80.Closse A, Huguenin R. Isolation and structural clarification of chlamydocin. Helv Chim Acta. 1974;57(3):533–545. doi: 10.1002/hlca.19740570306. [DOI] [PubMed] [Google Scholar]
- 81.Itazaki H, Nagashima K, Sugita K, Yoshida H, Kawamura Y, Yasuda Y, Matsumoto K, Ishii K, Uotani N, Nakai H, Terui A, Yoshimatsu S, Ikenishi Y, Nakawaga Y. Isolation and structural elucidation of new cyclotetrapeptides, trapoxin A and B, having detransformation activities as antitumor agents. J Antibiot. 1990;63:1524–1532. doi: 10.7164/antibiotics.43.1524. [DOI] [PubMed] [Google Scholar]
- 82.Ciuffetti LM, Pope MR, Dunkle LD, Daly JM, Knoche HW. Isolation and structure of an inactive product derived from the host-specific toxin produced by Helminthosporium carbonum. Biochemistry. 1983;22(14):3507–3510. [Google Scholar]
- 83.Hildmann C, Wegener D, Riester D, Hempel R, Schober A, Merana J, Giurato L, Guccione S, Nielsen TK, Ficner R, Schwienhorst A. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124(1):258–270. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 84.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci USA. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci USA. 1996;93(23):13143–13147. doi: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and Gelsolin. Cancer Res. 2000;60(21):6068–6074. [PubMed] [Google Scholar]
- 87.Kwon SH, Ahn SH, Kim YK, Bae G-U, Yoon JW, Hong S. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human-acute promyelocytic leukemia cells. J Biol Chem. 2002;277:2073–2080. doi: 10.1074/jbc.M106699200. [DOI] [PubMed] [Google Scholar]
- 88.Singh SB, Zink DL, Liesch JM, Mosley RT, Dombrowski AW, Bills GF. Structure and chemistry of apicidins, a class of novel cyclic tetrapeptides without a terminal α-keto epoxide as inhibitors of histone deacetylase with potent antiprotozoal activities. J Org Chem. 2002;67(3):815–825. doi: 10.1021/jo016088w. [DOI] [PubMed] [Google Scholar]
- 89.Singh SB, Zink DL, Polishook JD, Dombrowski AW, Darkin-Rattray SJ, Schmatz DM, Goetz MA. Apicidins: Novel cyclic tetrapeptides as coccidiostats and antimalarial agents from Fusarium pallidoroseum. Tetrahedron Lett. 1996;37(45):8077–8080. [Google Scholar]
- 90.Colletti SL, Myers RW, Darkin-Rattray SJ, Gurnett AM, Dulski PM, Galuska S, Allocco JJ, Ayer MB, Li C, Lim J, Crumley TM, Cannova C, Schmatz DM, Wyvratt MJ, Fisher MH, Meinke PT. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: structure-activity relationships of apicidin. Part 1. Bioorg Med Chem Lett. 2001;11(2):107–111. doi: 10.1016/s0960-894x(00)00604-1. [DOI] [PubMed] [Google Scholar]
- 91.Komatsu Y, Tomizaki K, Tsukamoto M, Kato T. Cyclic Hydroxamic-acid-containing Peptide 31, a potent synthetic histone deacetylase inhibitor with antitumor activity. Cancer Res. 2001;61:4459–4466. [PubMed] [Google Scholar]
- 92.Horne WS, Olsen CA, Beierle JM, Montero A, Ghadiri MR. Probing the bioactive conformation of an archetypal natural product HDAC inhibitor with conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew Chem Int Ed. 2009;48(26):4718–4724. doi: 10.1002/anie.200805900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakao Y, Yoshida S, Matsunaga S, Shindoh N, Terada Y, Nagai K, Yamashita JK, Ganesan A, van Soest RWM, Fusetani N. Azumamides A-E: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge. mycale izuensis Angew Chem Int Ed. 2006;45(45):7553–7557. doi: 10.1002/anie.200602047. [DOI] [PubMed] [Google Scholar]
- 94.Maulucci N, Chini MG, Di Micco S, Izzo I, Cafaro E, Russo A, Gallinari P, Paolini C, Nardi MC, Casapullo A, Riccio R, Bifulco G, De Riccardis F. Molecular insights into azumamide e histone deacetylases inhibitory activity. J Am Chem Soc. 2007;129(10):3007–3012. doi: 10.1021/ja0686256. [DOI] [PubMed] [Google Scholar]
- 95.Chandrasekhar S, Rao CL, Seenaiah M, Naresh P, Jagadeesh B, Manjeera D, Sarkar A, Bhadra MP. Total synthesis of azumamide E and sugar amino acid-containing analogue. J Org Chem. 2009;74(1):401–404. doi: 10.1021/jo8020264. [DOI] [PubMed] [Google Scholar]
- 96.Wen S, Carey KL, Nakao Y, Fusetani N, Packham G, Ganesan A. Total synthesis of azumamide a and azumamide e, evaluation as histone deacetylase inhibitors, and design of a more potent analogue. Org Lett. 2007;9(6):1105–1108. doi: 10.1021/ol070046y. [DOI] [PubMed] [Google Scholar]
- 97.Nishino N, Jose B, Shinta R, Kato T, Komatsu Y, Yoshida M. Chlamydocin-hydroxamic acid analogues as histone deacetylase inhibitors. Bioorg Med Chem. 2004;12:5777–5784. doi: 10.1016/j.bmc.2004.08.041. [DOI] [PubMed] [Google Scholar]
- 98.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968. J Antibiot. 1994;47:301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 99.Shigematsu N, Ueda H, Takase S, Tanaka H. FR9011228: a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum no. 968. J Antibiot. 1994;47:311–314. doi: 10.7164/antibiotics.47.311. [DOI] [PubMed] [Google Scholar]
- 100.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8:718–728. [PubMed] [Google Scholar]
- 101.Marshall JL, Rizvi N, Kauh J, Dahut W, Figuera M. A phase I trial of depsipeptide (FR901228) in patients with advanced cancer. J Exp Ther Oncol. 2002;2:325–332. doi: 10.1046/j.1359-4117.2002.01039.x. [DOI] [PubMed] [Google Scholar]
- 102.Masuoka Y, Nagai A, Shinya K, Furihata K, Nagai K, Suzuki K-i, Hayakawa Y, Seto H. Spiruchostatins A and B, novel gene expression-enhancing substances produced by pseudomonas sp. Tetrahedron Lett. 2001;42(1):41–44. [Google Scholar]
- 103.Lijima Y, Munakata A, Shinya K, Ganesan A, Doi T, Takahashi T. A solid-phase total synthesis of the cyclic depsipeptide HDAC inhibitor spiruchostatin A. Tetrahedron Lett. 2009;50(24):2970–2972. [Google Scholar]
- 104.Calandra NA, Cheng YL, Kocak KA, Miller JS. Total synthesis of spiruchostatin A via chemoselective macrocyclization using an accessible enantiomerically pure latent thioester. Org Lett. 2009;11(9):1971–1974. doi: 10.1021/ol900436f. [DOI] [PubMed] [Google Scholar]
- 105.Takizawa T, Watanabe K, Narita K, Kudo K, Oguchi T, Abe H, Katoh T. Total synthesis of spiruchostatin A, a potent histone deacetylase inhibitor. Heterocycles. 2008;76(1):275–290. doi: 10.1039/b718310k. [DOI] [PubMed] [Google Scholar]
- 106.Takizawa T, Watanabe K, Narita K, Oguchi T, Abe H, Katoh T. Total synthesis of spiruchostatin B, a potent histone deacetylase inhibitor, from a microorganism. Chem Commun. 2008;14:1677–1679. doi: 10.1039/b718310k. [DOI] [PubMed] [Google Scholar]
- 107.Yurek-George A, Habens F, Brimmell M, Packham G, Ganesan A. Total synthesis of spiruchostatin A, a potent histone deacetylase inhibitor. J Am Chem Soc. 2004;126(4):1030–1031. doi: 10.1021/ja039258q. [DOI] [PubMed] [Google Scholar]
- 108.Narita K, Kikuchi T, Watanabe K, Takizawa T, Oguchi T, Kudo K, Matsuhara K, Abe H, Yamori T, Yoshida M, Katoh T. Total synthesis of the bicyclic depsipeptide HDAC inhibitors spiruchostatins A and B, 5″-epi-spiruchostatin B, FK228 (FR901228) and preliminary evaluation of their biological activity. Chem Eur J. 2009;15:11174–11186. doi: 10.1002/chem.200901552. [DOI] [PubMed] [Google Scholar]
- 109.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J Am Chem Soc. 2008;130(33):11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J Am Chem Soc. 2008;130(26):8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 111.Ghosh AK, Kulkarni S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org Lett. 2008;10(17):3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. Synthesis and conformation–activity relationships of the peptide isosteres of FK228 and largazole. J Am Chem Soc. 2009;131(8):2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seiser T, Kamena F, Cramer N. Synthesis and biological activity of largazole and derivatives. Angew Chem Int Ed. 2008;47:6483–6485. doi: 10.1002/anie.200802043. [DOI] [PubMed] [Google Scholar]
- 114.Bowers A, West N, Newkirk T, Troutman-Youngman A, Schreiber SL, Wlest O, Bradner J, Williams R. Synthesis and histone deacetylase inhibitory activity of largazole analogs: alteration of the zinc-binding domain and macrocyclic scaffold. Org Lett. 2009;11(16):1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen F, Gao AH, Li J, Nan FJ. Synthesis and biological evaluation of C7-demethyl largazole analogues. Chem Med Chem. 2009;4:1269–1272. doi: 10.1002/cmdc.200900125. [DOI] [PubMed] [Google Scholar]
- 116.Ying Y, Liu Y, Byeon SR, Kim H, Luesch H, Hong J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org Lett. 2008;10(18):4021–4024. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- 117.Curtin ML, Garland RB, Heyman HR, Frey RR, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Davidsen SK. Succinimide hydroxamic acids as potent inhibitors of histone deacetylase (HDAC) Bioorg Med Chem. 2002;12(20):2919–2923. doi: 10.1016/s0960-894x(02)00622-4. [DOI] [PubMed] [Google Scholar]
- 118.Liu T, Kapustin G, Etzkorn FA. Design and synthesis of a potent histone deacetylase inhibitor. J Med Chem. 2007;50(9):2003–2006. doi: 10.1021/jm061082q. [DOI] [PubMed] [Google Scholar]
- 119.Pirali T, Faccio V, Mossetti R, Grolla AA, Micco SD, Bifulco G, Genazzani AA, Tron GC. Synthesis, molecular docking and biological evaluation as HDAC inhibitors of cyclopeptide mimetics by a tandem three-component reaction and intramolecular [3+2] cycloaddition. Mol Divers. 2010;14:109–121. doi: 10.1007/s11030-009-9153-9. [DOI] [PubMed] [Google Scholar]
- 120.Randolph JT, Waid P, Nichols C, Sauer D, Haviv F, Diaz G, Bammert G, Besecke LM, Segreti JA, Mohning KM, Bush EN, Wegner CD, Greer J. Nonpeptide luteinizing hormone-releasing hormone antagonists derived from erythromycin a: design, synthesis, and biological activity of cladinose replacement analogues. J Med Chem. 2004;47(5):1085–1097. doi: 10.1021/jm030418i. [DOI] [PubMed] [Google Scholar]
- 121.Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg Med Chem. 2008;16(9):4839–4853. doi: 10.1016/j.bmc.2008.03.050. [DOI] [PubMed] [Google Scholar]
- 122.Mwakwari SC, Guerrant W, Patil V, Khan SI, Tekwani BL, Oyelere AK. Non-peptide macrocyclic histone deacetylase inhibitors derived from tricyclic ketolide skeleton. 2010 doi: 10.1021/jm100507q. Manuscript under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oku N, Nagai K, Shindoh N, Terada Y, Soest RWMv, Matsunaga S, Fusetani N. Three new cyclostellettamines, which inhibit histone deacetylase, from a marine sponge of the genus Xestospongia. Bioorg Med Chem Lett. 2004;14(10):2617–2620. doi: 10.1016/j.bmcl.2004.02.062. [DOI] [PubMed] [Google Scholar]
- 124.Fusetani N, Asai N, Matsunaga S, Honda K, Yasumuro K. Cyclostellettamines A-F, pyridine alkaloids which inhibit binding of methyl quinuclidinyl benzilate (QNB) to muscarinic acetylcholine receptors, from the marine sponge. Stelletta maxima Tetrahedron Lett. 1994;35(23):3967–3970. [Google Scholar]
- 125.Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98(9):2865–2868. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]
- 126.Harrison SJ, Quach H, Yuen K. High response rates with the combination of bortezomib, dexamethasone and the pan-histone deacetylase inhibitor romidepsin in patients with relapsed or refractory multiple myeloma in a phase I/II clinical trial. ASH Ann Meet Abstr. 2008;112:3698. [Google Scholar]
- 127.Doss HH, Jones SF, nfante JR, Spigel DR, Willcutt N, Lamar R, Barton J, Keegan M, Burris HA., III A phase I trial of romidepsin in combination with gemcitabine in patients with pancreatic and other advanced solid tumors. J Clin Oncol. 2008;26(15S):2567. [Google Scholar]
- 128.Whitehead RP, Rankin C, Hoff PM, Gold PJ, Billingsley KG, Chapman RA, Wong L, Ward JH, Abbruzzese JL, Blanke CD. Phase II Trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: a southwest oncology group study (S0336) Invest New Drugs. 2008;27:469–475. doi: 10.1007/s10637-008-9190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, Chan KK, Grever MR. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 130.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Kruchin E, Wright JJ, Rosing DR, Sparreboom A, Figg WD, MSS Clinical and molecular responses in lung cancer patients receiving romidepsin. Clin Cancer Res. 2008;14:188–198. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 131.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, Panageas K, Wright JJ, Pandolfi PP, Nimer SD. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826–832. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 132.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]
- 133.Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, Patterson S, Riggs CE, Jr, Higano C, Stadler WM, McCulloch W, Dearnaley D, Parker C, de Bono JS. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Ann Oncol. 2009;21:109–113. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 134.Shah MH, Villalona-Calero MA, Marcucci G, Byrd JC, Grever MR. HDAC inhibitors and cardiac safety. Clin Cancer Res. 2007;13:1068. doi: 10.1158/1078-0432.CCR-06-1715. [DOI] [PubMed] [Google Scholar]
- 135.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15:3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 136.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm189629.htm, 2009.