

Abstract
Background
Intracerebral hemorrhage (ICH) is a devastating stroke subtype affecting 120,000 Americans annually. Of those affected, 40-50% will die within the first thirty days, while the survivors are left with a lifetime of neurobehavioral disabilities. Recently, it has been shown that volatile anesthetics such as isoflurane can reduce brain injury after an ischemic stroke. As a result, in the present study, we investigated the effects of isoflurane as a posttreatment therapeutic modality in ICH-injured mice. Specifically investigating whether isoflurane posttreatment can preserve the structural integrity of the brain by reducing apoptotic damage and in turn, improve functional outcome by amelioration of brain edema and neurobehavioral deficits.
Methods
Male CD1 mice (n=53) were divided into the following groups: sham (n=14), ICH (n=14), ICH treated with 1.5% isoflurane posttreatment for 1 hour (n=15), and ICH treated with 1.5% isoflurane posttreatment for 2 hours (n=10). The blood injection ICH model was adapted; this involved extracting autologous blood from the mouse tail and injecting it directly into the right basal ganglia. One hour after surgery, treated mice were placed in a glass chamber maintained at 37°C and infused with 1.5% isoflurane for one or two hours. At 24hrs postinjury, mice were assessed for neurobehavioral deficits using the modified Garcia score and then sacrificed and assessed for brain water content. Double immunofluorescent staining was performed using neuronal marker MAP-2 and TUNEL under a fluorescent microscope to assess for apoptosis.
Results
Our results indicated that after one hour 1.5% isoflurane posttreatment, there was a significant reduction in brain edema, a decrease in apoptotic cell death, and a significant improvement in neurobehavioral deficits.
Conclusions
Our results suggest that isoflurane may be an effective posttreatment therapeutic option for ICH because of its ability to reduce structural damage and subsequently preserve functional integrity.
Introduction
Intracerebral hemorrhage (ICH) is a devastating stroke subtype affecting 120,000 individuals annually in the United States alone.1 Of those affected, 40-50% will die within the first thirty days, while the survivors are left with a lifetime of neurobehavioral disabilities.2 Because of the devastating impact on human health and the growing financial burden on society, an exorbitant amount of research has been put forth in an effort to determine ways in which to attenuate the devastating consequences of ICH injury. However, currently, there are no clinically viable therapeutic options for ICH patients.
One of the challenges faced with treating ICH victims has been due to the complexity of edema formation. After injury, close to 40% of victims experience a 1-2% percent increase in brain edema at 24hrs.This translates into a 4-8% increase in brain volume, leading to a severe increase in intracranial pressure and brain herniation.3 Previous studies attempting to combat the formation of edema have faced difficulties likely because of the multiple sources from which edema can develop, partly attributed to blood-brain-barrier (BBB) disruption, i.e., vasogenic edema, and partly attributed to cell death, i.e., cytotoxic edema.4
Within the last decade, one of the major advances in anesthesiology has been the recognition of volatile anesthetics as neuroprotective in adults.5 Specifically, isoflurane has been acknowledged for its role in reducing apoptosis and preventing the death of key neuronal and non-neuronal cells. In a rat focal cerebral ischemic injury model, isoflurane posttreatment significantly reduced apoptosis as measured by brain infarction volume, and improved neurobehavioral deficits.6 Yet to date, no study has cross-examined these potential neuroprotective effects against ICH-induced brain injury.
As a result, in the present study we investigated the potential therapeutic effects of isoflurane treatment after ICH injury. Specifically observing the potential of isoflurane to reduce apoptotic damage and ameliorate brain edema accumulation and neurobehavioral deficits, we hypothesized that 1.5% isoflurane will allow for structural preservation and in turn, improve functional outcome after ICH injury in mice.
Materials and Methods
This study was in accordance with the guidelines of the National Institute of Health for the treatment of animals and was approved by the Institutional Animal Care and Use Committee at Loma Linda University. Male CD1 mice (n=53; weight 35-45 grams, Charles River, MA, USA) were housed in a 12-hour light/dark cycle at a controlled temperature and humidity with free access to food and water. Mice were divided into the following groups: sham (n=14), ICH (n=14), ICH treated with 1.5% isoflurane posttreatment for 1 hour (n=15), and ICH treated with 1.5% isoflurane posttreatment for 2 hours (n=10).
Operative Procedure. ICH was induced using the autologous blood injection model which was modified from previous descriptions.7,8,9 Briefly, mice were anesthetized with a ketamine (100 mg/kg) and xylazine (10 mg/kg) cocktail (2:1 v/v, intraperitoneal injection) and positioned prone in a stereotactic head frame (Kopf Instruments, Tujunga, CA). A scalp incision was made along the midline and a burr hole (1 mm) was drilled on the right side of the skull (0.2 mm anterior and 2.0 mm lateral of the bregma). The mouse tail was warmed with hot water for 2 min and then cleaned with 70% ethanol before cutting off 1.0 cm of the tail tip with sterilized surgical scissors. Next, 30 ul of autologous tail blood was collected in a capillary tube without heparin and blown into a 1cc insulin syringe. The syringe was fixed onto the microinjection pump while the needle was stereotaxically inserted into the brain through the burr hole. At first the needle was stopped at 0.7 mm above the target position and 5 ul of blood was delivered at a rate 2 μl/min. This was done to allow for a clot to form just above the target site, that way, the chance of reflux up the needle tract is minimized. The needle was then advanced to the target position. After 7 min, the remaining 25 ul blood was injected at a rate of 2 μl/min. The needle was left in place for an additional 10 min after injection to prevent possible leakage and withdrawn slowly over 7 min. After the removal of the needle, the burr hole was sealed with bone wax, the incision was closed with sutures, and the mice were allowed to recover. To avoid postsurgical dehydration, 0.5 ml of normal saline was given to each mouse by subcutaneous injection after surgery.
Treatment Method. Mice were allowed to rest for one hour on a warm blanket before initiating therapy. Afterwards, mice designated for treatment were placed into a cylindrical glass chamber containing 1.5% isoflurane carried by 30% oxygen-70% nitrogen for either 1 or 2 hours duration. The temperature was continuously monitored and maintained at 37°C. The chamber was then opened to room air where the mice were returned to their appropriate cages.
Brain water content measurement. Brain water content was measured at 24hrs post-ICH injury as previously described.10,11,12 Briefly, mice were decapitated under deep anesthesia. Brains were immediately removed and cut into 4 mm sections. Each section was divided into four parts: ipsilateral and contralateral basal ganglia, ipsilateral and contralateral cortex. The cerebellum was collected as an internal control. Tissue samples were weighed on an electronic analytical balance (APX-60, Denver Instrument) to the nearest 0.1 mg to obtain the wet weight (WW). The tissue was then dried at 100°C for 24 h to determine the dry weight (DW). Brain water content (%) was calculated as [(WW - DW)/WW] × 100.
Assessment of Neurobehavioral Deficits. Neurological outcomes were assessed by a blinded observer at 24hrs post-ICH using the Modified Garcia Score.13 The Modified Garcia Score is a 21-point sensorimotor assessment system consisting of seven tests with scores of 0–3 for each test (max score = 21). These seven tests included: (i) spontaneous activity, (ii) side stroking, (iii) vibris touch, (iv) limb symmetry, (v) climbing, (vi) lateral turning, and (vii) forelimb walking.
Immunohistochemistry. Mice (n=12) were transcardially perfused under deep anesthesia with phosphate buffered saline followed by 4% paraformaldehyde. The brains were then removed and postfixed in formalin. Optimum cutting temperature-embedded brains were sectioned into 10-μm-thick slices by cryostat (CM3050S; Leica Microsystems). Double immunofluorescent staining was performed using neuronal marker MAP-2 (Cambridge, MA) and TUNEL (Roche, Indianapolis, IN). Once stained, specimens were examined under a fluorescent microscope (OLYMPUS BX51). TUNEL positive cells were counted at the center of the hemorrhagic lesion. Three perihematomal regions of the ipsilateral cerebral hemisphere were used for cell counting. Total cell counts were converted into TUNEL positive cell densities. Four mice per group were used for intergroup comparisons.
Statistical Analysis. All data were expressed as mean±SEM. Statistical differences between two groups were analyzed using the two sided t-test with unequal variances. Multiple comparisons were statistically analyzed with one-way analysis of variance (ANOVA) followed by Tukey multiple comparison post hoc analysis or Student-Newman-Keuls test on ranks. A p-value of less than 0.05 was considered statistically significant.
Results
Isoflurane posttreatment reduces ICH-induced brain edema accumulation. Brain edema is evident in both human and experimental studies as a result of ICH.14 Accordingly, we found a significant accumulation of brain edema in the ipsilateral cortex and basal ganglia, corresponding directly to the site of injury, as compared to our non-operated sham mice (Figure 1). This increase in brain edema was reversed by 1hr isoflurane posttreatment in both the ipsilateral cortex and basal ganglia (p=0.002).
Figure 1. Isoflurane Posttreatment Reduces Brain Edema.
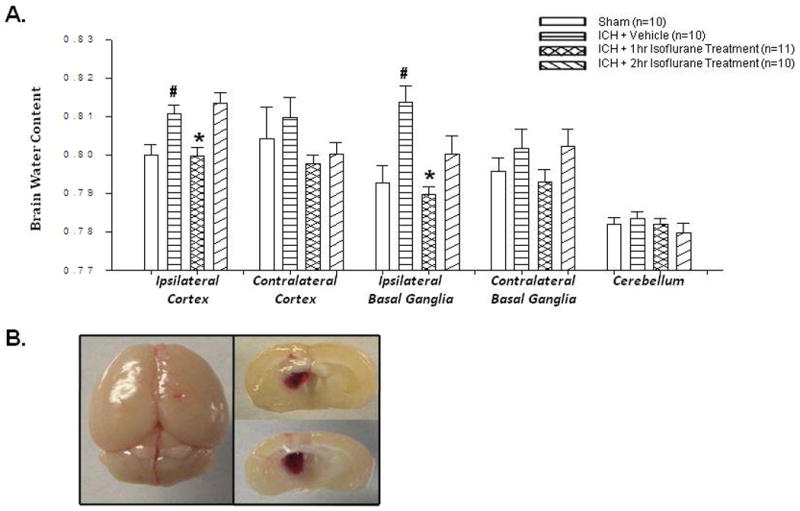
(A) Brain edema 24hrs after operation in sham, vehicle, and isoflurane-treated mice (p-value vehicle vs. treatment group = 0.002). # Significant Difference vs. Sham (p<0.05); * Significant Difference vs. Vehicle (p<0.05); (B) Gross pictures of the mouse brain displaying the needle site with corresponding hematoma in the right basal ganglia
Isoflurane posttreatment improves neurobehavioral deficits as assessed by the Modified Garcia Score. Surviving ICH patients are left with severe chronic neurobehavioral disabilities. The Modified Garcia Score is an accurate means of assessing both motor and sensory function.13 In our study, after ICH injury, there was a significant increase in neurobehavioral deficits compared to the sham group, with a subsequent reduction after 1hr isoflurane posttreatment (p=0.003; Figure 2). Although 2hr isoflurane posttreatment showed a trend towards improvement, it was not statistically significant (p=0.17).
Figure 2. Isoflurane Posttreatment Reduces Neurobehavioral Deficits.
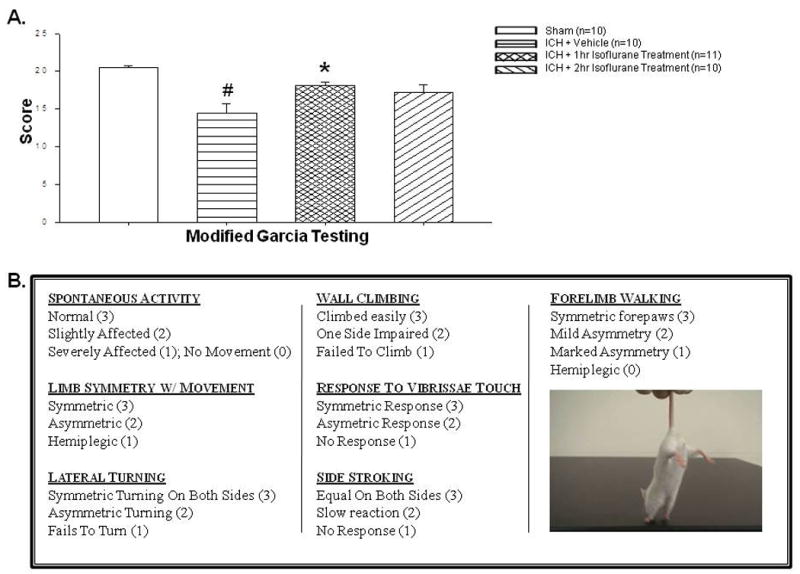
(A) Modified Garcia test 24hrs after operation in sham, vehicle, and isoflurane-treated mice (p-value vehicle vs. 1hr treatment group =0.003; vehicle vs 2hr treatment group 0.17). # Significant Difference vs. Sham (p<0.05); * Significant Difference vs. Vehicle (p<0.05); (B) Neurological outcomes were assessed by a blinded observer at 24hrs post-intracerebral hemorrhage (ICH) using the Modified Garcia Score, a 21-point sensorimotor seven –test assessment system with scores of 0–3 for each test (max score = 21).
Immunohistochemistry reveals a reduction in apoptosis after isoflurane posttreatment. In the perihematomal brain regions of the untreated mice, TUNEL was localized in circular layers of cells corresponding in size to microvascular profiles. This finding, which strongly suggests endothelial cell death, was much less abundant in the1hr isoflurane-treated mice (Figure 3a). In order to determine the neuron-specific antiapoptotic effects of isoflurane after ICH, an immunohistochemical study was undertaken using TUNEL and MAP-2 staining. Double staining revealed a marked increase in neuronal cell death after ICH injury. After 1hr isoflurane posttreatment, TUNEL up-take in neurons was largely diminished, translating into a reduction in cell death. These results were also quantified to further support what was found qualitatively (Figure 3b).
Figure 3. Isoflurane Posttreatment Reduces Apoptotic Cell Death.
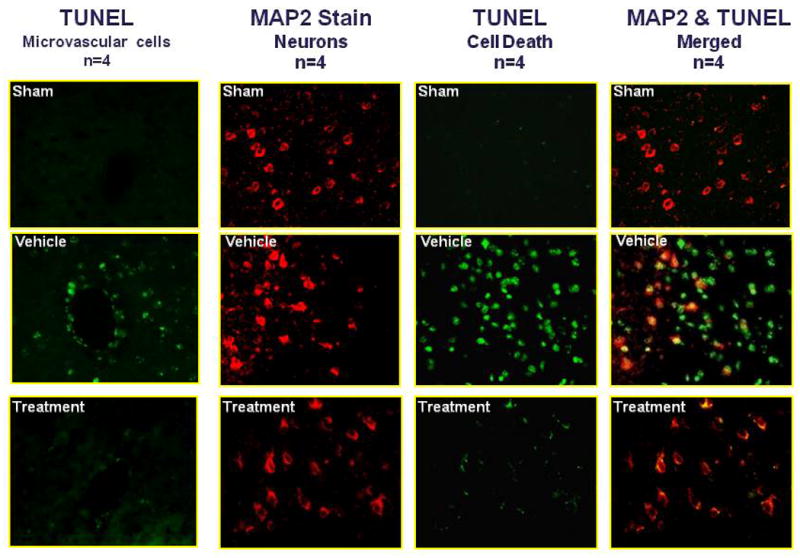
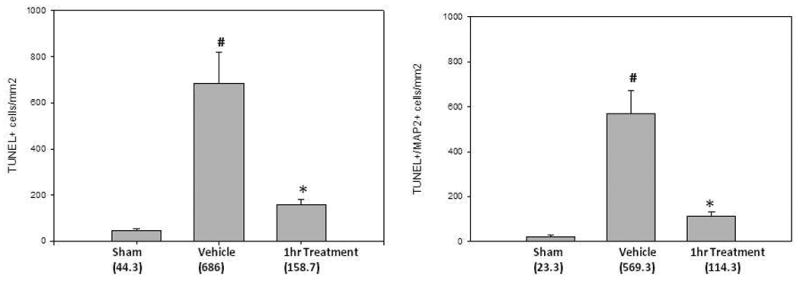
(A) Representative photographs of immunofluorescence staining using TUNEL and MAP-2 staining; (B) Corresponding cell death quantification (p-values for vehicle vs treatment group = 0.002 and 0.003); # Significant Difference vs. Sham (p<0.05); * Significant Difference vs. Vehicle (p<0.05);
Reductions in cell death correlate with a reduction in neurobehavioral deficits. In order to better observe a correlation between cell death and neurobehavioral deficits, a separate figure plotting these two results against one another was performed (Figure 4). The results showed that the two end points, i.e., cell death and neurobehavioral deficits, are interchangeable when considering differences among groups, but seemingly independent within groups.
Figure 4. Reductions in Cell Death Correlate with Reduced Neurobehavioral Deficits.
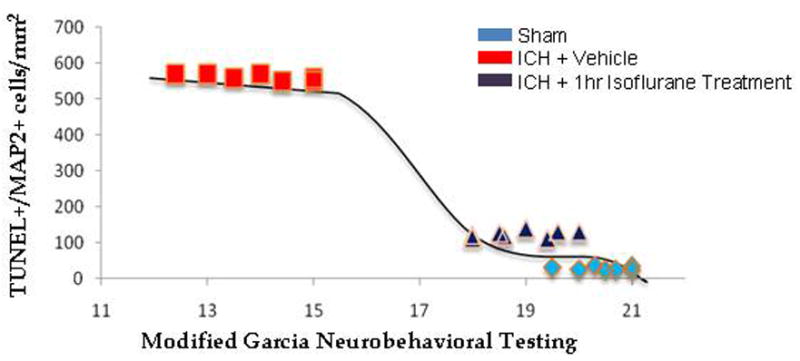
TUNEL+/MAP2+ cells were plotted against neurobehavioral scores. A Lowess regression line (tension parameter of 0.5) was added to assess the shape of the relationship.
Discussion
ICH is a fatal stroke subtype with no effective treatment options. Even if patients survive the initial attack, the growing hematoma triggers a series of life-threatening events leading to the accumulation of cerebral edema, progression of neurobehavioral deficits, and in some cases even death.15 In the present study we sought to determine the efficacy of isoflurane as a posttreatment therapeutic modality for ICH brain injury. We found that one hour 1.5% isoflurane posttreatment resulted in a significant reduction in brain edema, a qualitative decrease in apoptotic cell death (including both neuronal an non-neuronal cells), and a significant improvement in neurobehavioral deficits. To the best of our knowledge this is the first study demonstrating the effectiveness of isoflurane posttreatment after ICH injury by reducing structural damage and subsequently preserving functional integrity.
The movement of water from the vasculature to the brain is highly regulated by the BBB, which maintains both a physiologic and physical barrier, thereby preventing the accumulation of fluid in the injured brain. During an ICH injury, the BBB is disrupted by a variety of products including inflammatory mediators (i.e., infiltrating immune cells), thrombin, hemoglobin breakdown products, oxidative stress, and matrix metalloproteinases signals.14 A disruption in BBB patency leads to dysregulation of water homeostasis between the vasculature and brain parenchyma. This translates into severe increases in intracranial pressure, a reduction in cerebral blood flow, subsequent increase in ischemic damage, and further increase in cerebral edema accumulation secondary to apoptotic cell death.16 Clinically, this means an increased risk for herniation from the ensuing increase in intracranial pressure as well as severe neurobehavioral deficits from neuronal and non-neuronal cell death.
Similar to what we would expect physiologically in the ICH patient, we found a significant increase in brain edema accumulation between our non-operated sham group and ICH injured group. Two sources likely contributed to the accumulation of edema in our ICH population. First, from the disruption of the BBB facilitating increases in vascular permeability from endothelial cell death; and second, from direct neuronal and non-neuronal cell deaths observed on TUNEL and MAP-2 staining. These effects were subsequently ameliorated after one hour, 1.5% isoflurane posttreatment, suggesting that isoflurane plays a key role in reducing apoptosis after cerebrovascular injury. Additionally, the preservation of structural integrity translated into a significant improvement in neurobehavioral deficits. These results are in line with previous works which have also suggested an antiapoptotic role for isoflurane. 20
Previous studies have implied that through modulation of intracellular calcium levels, isoflurane has been implicated in indirect activation of antiapoptotic factors including Ca/calmodulin-dependent protein kinase II, the phosphatidylinositol-3-kinase (PI3K)/AKT cascade, and mitogen-activated protein kinase.17,18 However it is our belief that isoflurane works to block the release of pro-apoptotic factors intracellularly through another mechanism. Activation of the PI3K signaling pathway promotes cell survival. One way it does so is through phosphorylation-inhibition of glycogen synthase kinase 3β (GSK3β), a key serine/threonine protein kinase that regulates the opening of the mitochondrial permeability transition pores (MPTP). We hypothesize that by inhibiting the activation of GSK3β indirectly by isoflurane's activation of PI3K, the MPTP is unable to release pro-apoptotic factors that normally occur with neurnonal cell environment disruption. Although we understand this is simple speculation until further mechanistic studies are conducted and furthermore a limitation in our study, we do acknowledge the works that have been previously published on myocardial ischemia models and isoflurane that support this mechanistic theory.21
An additional question that would need to be addressed through further studies would be the optimal duration time for posttreatment with isoflurane. In our study we found that only one hour isoflurane posttreatment significantly improved our measured outcomes while two hour treatment failed to do so. This could be attributed to isoflurane's ability to increase cerebral blood flow, where one hour therapy is just enough to balance an increase in blood flow with a reduction in penumbral ischemic damage, whereas longer durations can in fact enhance or exacerbate damage. Because this is only speculative, further studies will be needed to determine exactly how isoflurane provides neuroprotection and determine optimal therapy durations. An additional question that would need to be addressed in further studies would be the effects of isoflurane posttreatment on long-term outcomes. Currently, our study only evaluated the short-term benefits of isoflurane; future investigations would need to determine if the neuroprotection afforded by isoflurane at 24hrs can be sustained at longer time points.
Isoflurane has come under intense scrutiny from studies suggesting that it only protects the brain acutely and postpones or delays the onset of injury.19 However, the evidence-at-large remains contradictory and a host of studies both affirm and refute the notion of injury postponement.6,19 As demonstrated in this study, low-dose isoflurane posttreatment may be effective at protecting the injured hemorrhagic brain and may be a promising therapeutic option as an acute treatment after ICH injury.
Acknowledgments
Funding: This study was partially supported by NIH NS06093 and NS053407.
Footnotes
The authors declare no conflicts of interest.
Reprints will not be available from the authors.
Disclosures:
Name: Nikan H. Khatibi, MD, MBA
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: Nikan H. Khatibi has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files.
Name: Qingyi Ma, BS
Contribution: This author helped conduct the study.
Attestation: Qingyi Ma has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: William Rolland, BS
Contribution: This author helped conduct the study.
Attestation: William Rolland has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Robert Ostrowski, MD, PhD
Contribution: This author helped conduct the study.
Attestation: Robert Ostrowski has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Nancy Fathali, PhD
Contribution: This author helped conduct the study and write the manuscript.
Attestation: Nancy Fathali has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Robert Martin, MD
Contribution: This author helped design the study and write the manuscript.
Attestation: Robert Martin has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Richard Applegate, MD
Contribution: This author helped design the study, analyze the data, and write the manuscript.
Attestation: Richard Applegate has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Gary Stier, MD
Contribution: This author helped design the study, analyze the data, and write the manuscript.
Attestation: Gary Stier has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Jiping Tang, MD
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: Jiping Tang has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: John H. Zhang, MD, PhD
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: John H. Zhang has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Contributor Information
Nikan H. Khatibi, Dept of Anesthesiology, Loma Linda University Medical Center, Loma Linda, CA.
Qingyi Ma, Dept of Physiology and Pharmacology, Loma Linda University Medical Center, Loma Linda, CA.
William Rolland, Dept of Physiology and Pharmacology, Loma Linda University Medical Center, Loma Linda, CA.
Robert Ostrowski, Dept of Physiology and Pharmacology, Loma Linda University Medical Center, Loma Linda, CA.
Nancy Fathali, Dept of Physiology and Pharmacolog, Loma Linda University Medical Center, Loma Linda, CA.
Robert Martin, Dept of Anesthesiology, Loma Linda University Medical Center, Loma Linda, CA.
Richard Applegate, Dept of Anesthesiology, Loma Linda University Medical Center, Loma Linda, CA.
Gary Stier, Dept of Anesthesiology, Loma Linda University Medical Center, Loma Linda, CA.
Jiping Tang, Dept of Physiology and Pharmacology, Loma Linda University Medical Center, Loma Linda, CA.
John H. Zhang, Dept of Neurosurgery, Anesthesiology, Physiology and Pharmacology, Loma Linda University Medical Center, Loma Linda, CA.
References
- 1.Ribo M, Grotta JC. Latest advances in intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2006;6:17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- 2.Dennis MS, Burn JP, Sandercock PA, Bamford JM, Wade DT, Warlow CP. Long-term survival after first-ever stroke: the Oxfordshire Community Stroke Project. Stroke. 1993;24:796–800. doi: 10.1161/01.str.24.6.796. [DOI] [PubMed] [Google Scholar]
- 3.Davalos A, Toni D, Iweins F, Lesaffre E, Bastianello S, Castillo J. Neurological deterioration in acute ischemic stroke: potential predictors and associated factors in the European cooperative acute stroke study (ECASS) I. Stroke. 1999;30(12):2631–2636. doi: 10.1161/01.str.30.12.2631. [DOI] [PubMed] [Google Scholar]
- 4.Klatzo I. Pathophysiological aspects of brain edema. Acta Neuropathol. 1987;72(3):236–9. doi: 10.1007/BF00691095. [DOI] [PubMed] [Google Scholar]
- 5.Matchett GA, Allard MW, Martin RD, Zhang JH. Neuroprotective effect of volatile anesthetic agents: molecular mechanisms. Neurol Res. 2009;31(2):128–34. doi: 10.1179/174313209X393546. [DOI] [PubMed] [Google Scholar]
- 6.Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–1062. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belayev L, Saul I, Curbelo K, Busto R, Belayev A, Zhang Y, Riyamongkol P, Zhao W, Ginsberg MD. Experimental intracerebral hemorrhage in the mouse: histological, behavioral, and hemodynamic characterization of a double-injection model. Stroke. 2003;34:2221–7. doi: 10.1161/01.STR.0000088061.06656.1E. [DOI] [PubMed] [Google Scholar]
- 8.Rynkowski MA, Kim GH, Komotar RJ, Otten ML, Ducruet AF, Zacharia BE, Kellner CP, Hahn DK, Merkow MB, Garrett MC, Starke RM, Cho BM, Sosunov SA, Connolly ES. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–8. doi: 10.1038/nprot.2007.513. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Fields J, Dore S. The development of an improved preclinical mouse model of intracerebral hemorrhage using double infusion of autologous whole blood. Brain Res. 2008;1222:214–21. doi: 10.1016/j.brainres.2008.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–45. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil-Granger D, Zhang JH. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–50. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- 12.Tejima E, Zhao BQ, Tsuji K, Rosell A, van Leyen K, Gonzalez RG, Montaner J, Wang X, Lo EH. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27:460–8. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- 13.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–34. doi: 10.1161/01.str.26.4.627. discussion 35. [DOI] [PubMed] [Google Scholar]
- 14.Keep RF. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–7. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- 15.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27(3):268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 16.Diringer MN. Intracerebral hemorrhage: pathophysiology and management. Crit Care Med. 1993;21(10):1591–603. doi: 10.1097/00003246-199310000-00032. [DOI] [PubMed] [Google Scholar]
- 17.Cheng A, Wang S, Yang D, Xiao R, Mattson MP. Calmodulin mediates brain-derived neurotrophic factor cell survival signaling upstream of Akt kinase in embryonic neocortical neurons. J Biol Chem. 2003;278(9):7591–7599. doi: 10.1074/jbc.M207232200. [DOI] [PubMed] [Google Scholar]
- 18.Fahlman CS, Bickler PE, Sullivan B, Gregory GA. Activation of the neuroprotective ERK signaling pathway by fructose-1,6-bisphosphate during hypoxia involves intracellular Ca2+ and phospholipase C. Brain Res. 2002;958(1):43–51. doi: 10.1016/s0006-8993(02)03433-9. [DOI] [PubMed] [Google Scholar]
- 19.Kawaguchi M, Drummond JC, Cole DJ, Kelly PJ, Spurlock MP, Patel PM. Effect of Isoflurane on neuronal apoptosis in rats subjected to focal cerebral ischemia. Anesth Analg. 2004;98(3):798–805. doi: 10.1213/01.ane.0000105872.76747.f6. table of contents. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Zuo Z. Isoflurane preconditioning improves short-term and long-term neurological outcome after focal brain ischemia in adult rats. Neuroscience. 2009 Dec 1;164(2):497–506. doi: 10.1016/j.neuroscience.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J, Rebecchi M, Tan M, Glass P, Brink P, Liu L. Age-associated differences in activation of Akt/GSK-3 [Google Scholar]