

Abstract
Pancreatic Neuroendocrine Tumors (PanNETs) are a rare but clinically important form of pancreatic neoplasia. To explore the genetic basis of PanNETs, we determined the exomic sequences of ten non-familial PanNETs and then screened the most commonly mutated genes in 58 additional PanNETs. Remarkably, the most frequently mutated genes specify proteins implicated in chromatin remodeling: 44% of the tumors had somatic inactivating mutations in MEN-1, which encodes menin, a component of a histone methyltransferase complex; and 43% had mutations in genes encoding either of the two subunits of a transcription/chromatin remodeling complex consisting of DAXX (death-domain associated protein) and ATRX (alpha thalassemia/mental retardation syndrome X-linked). Clinically, mutations in the MEN1 and DAXX/ATRX genes were associated with better prognosis. We also found mutations in genes in the mTOR (mammalian target of rapamycin) pathway in 14% of the tumors, a finding that could potentially be used to stratify patients for treatment with mTOR inhibitors.
PanNETs are the second most common malignancy of the pancreas. The ten-year survival rate of patients with PanNETs is only 40% (1-3). They are usually sporadic, but they can arise in multiple endocrine neoplasia type 1 and more rarely in other syndromes, including von Hippel-Lindau (VHL) syndrome and tuberous sclerosis (4). “Functional” PanNETs secrete hormones that cause systemic effects, while “Nonfunctional” PanNETs do not and therefore cannot always be readily distinguished from other neoplasms of the pancreas. Nonfunctional PanNETs grow silently and patients often present with either an asymptomatic abdominal mass or symptoms of abdominal pain secondary to compression by a large tumor. Surgical resection is the treatment of choice, but many patients present with unresectable tumors or extensive metastatic disease, and medical therapies are relatively ineffective in these cases.
There is currently insufficient information about this tumor to either predict prognosis of patients diagnosed with PanNETs or to develop companion diagnostics and personalized treatments to improve disease management. Biallelic inactivation of the MEN1 gene, usually through a mutation in one allele coupled with loss of the remaining wild-type allele, occurs in 25-30% of PanNETs (5, 6). MEN1 is a tumor suppressor gene which, when mutated in the germline, predisposes to multiple endocrine neoplasia type 1 syndrome. Chromosomal gains and losses and expression analyses have revealed candidate loci for genes involved in the development of PanNETs, but these have not been substantiated by genetic or functional analyses (7-9).
To gain insights into the genetic basis of this tumor type, we determined the exomic sequence of ~18,000 protein-coding genes in a Discovery set of ten well-characterized sporadic PanNETs. A clinically homogeneous set of tumors of high neoplastic cellularity is essential for the successful identification of genes and pathways involved in any tumor type. Thus, we excluded small cell and large cell neuroendocrine carcinomas and studied only samples that were not part of a familial syndrome associated with PanNETs (table S1) (1). We microdisected tumor samples to achieve a neoplastic cellularity of >80%. DNA from the enriched neoplastic samples and from matched non-neoplastic tissue from ten patients was used to prepare fragment libraries suitable for massively parallel sequencing. The coding sequences were enriched by capture with the SureSelect Enrichment System and sequenced using an Illumina GAIIx platform (10). The average coverage of each base in the targeted regions was 101-fold and 94.8 % of the bases were represented by at least 10 reads (table S2).
We identified 157 somatic mutations in 149 genes among the ten tumors used in the Discovery set. The mutations per tumor ranged from 8 to 23, with a mean of 16 (table S3). Of these mutations, 91 % were validated by Sanger sequencing. There were some obvious differences between the genetic landscapes of PanNETs and those of pancreatic ductal adenocarcinomas (PDAC, ref. 11). First, there were 60% fewer genes mutated per tumor in PanNETs than in PDACs. Second, the genes most commonly affected by mutation in PDACs (KRAS, TGF-β pathway, CDKN2A, TP53) were rarely altered in PanNETs and vice versa (Table 1). Third, the spectrum of mutations in PDAC and PanNET were different, with C to T transitions more common in PDACs than in PanNETs, and C to G transversions more common in PanNETs than in PDACs (table S4). This suggests that mutations in PanNETs and PDAC arise through different mechanisms, perhaps due to exposure to different environmental carcinogens or through the action of different DNA repair pathways.
Table 1. Comparison of commonly mutated genes in PanNETs and PDACc.
| Genesa | PanNET | PDACb |
|---|---|---|
| MEN1 | 44% | 0% |
| DAXX, ATRX | 43% | 0% |
| Genes in mTOR pathway | 15% | 0.80% |
| TP53 | 3% | 85% |
| KRAS | 0% | 100% |
| CDKN2A | 0% | 25% |
| TGFBR1, SMAD3, SMAD4 | 0% | 38% |
Includes point mutations and indels.
Data from Jones et al ., Science 321, 1801 (2008).
Based on 68 PanNETs and 114 PDACs.
We next selected genes for further analysis that were well-documented components of a pathway that was genetically altered in more than one tumor, because alterations in these genes are most likely to be clinically relevant. Four genes were mutated in at least two tumors in the Discovery set: MEN1 in five, DAXX in three, PTEN in two, and TSC2 in two. ATRX was mutated in only one sample in the Discovery set, but its product forms a heterodimer with DAXX and therefore is part of the same pathway, so it was also evaluated in the Validation set. Similarly, PIK3CA was included because its product is part of the mTOR pathway that includes PTEN and TSC2 (12-14). The sequences of these genes were then determined by Sanger sequencing in a Validation set consisting of 58 additional PanNETs and their corresponding normal tissues (Fig. 1, A and B). In total, somatic mutations in MEN1, DAXX, ATRX, PTEN, TSC2, and PIK3CA were identified in 44.1%, 25%, 17.6%, 7.3%, 8.8%, and 1.4% PanNETs, respectively (Table 2).
Figure 1.
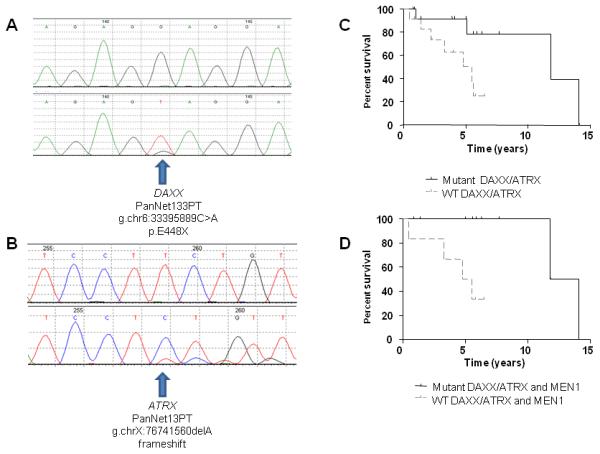
Table 2.
Mutations in MEN1, DAXX, ATRX, PTEN, TSC2, PIK3CA, and TP53 in Human Pancreatic Neuroendocrine Tumors.
| Sample # | Gene | Transcript Accession | Nucleotide (genomic)* | Nucleotide (cDNA) | Amino acid (protein)$ | Mutation type |
|---|---|---|---|---|---|---|
| PanNET3PT | ATRX | CCDS14434.1 | g.chrX:76716462G>A (hom) | c.6235C>T(hom) | p.R2079X | Nonsense |
| PanNET5PT | ATRX | CCDS14434.1 | g.chrX:76742636G>A | c.5620C>T | p.Q1874X | Nonsense |
| PanNET13PT | ATRX | CCDS14434.1 | g.chrX:76741560delA | c.5932delT | fs | Indel |
| PanNET27PT | ATRX | CCDS14434.1 | g.chrX:76700959_76700962delATAA | c.6338_6341delTTAT | fs | Indel |
| PanNET35PT | ATRX | CCDS14434.1 | g.chrX:76806893_76806909delAATTTCTTCTAAAAGCA | c.3824_3840delTGCTTTTAGAAGAAATT | fs | Indel |
| PanNET52PT | ATRX | CCDS14434.1 | g.chrX:76796337_76796340delCTTT | c.4221_4224delAAAG | fs | Indel |
| PanNET59PT | ATRX | CCDS14434.1 | g.chrX:76761014C>A | c.5364G>T | p.Q1788H | Missense |
| PanNET78PT | ATRX | CCDS14434.1 | g.chrX:76665406C>T | c.6829G>A | p.E2277K | Missense |
| PanNET85PT | ATRX | CCDS14434.1 | g.chrX:76794404dupC | c.4414dupG | fs | Indel |
| PanNET98PT | ATRX | CCDS14434.1 | g.chrX:76700832T>A(hom) | c.6468A>T(hom) | p.Q2156H | Missense |
| PanNET100PT | ATRX | CCDS14434.1 | g.chrX:76762518_76762521delCACT(hom) | c.5270_5272delAGTG(hom) | fs | Indel |
| PanNET112PT | ATRX | CCDS14434.1 | g.chrX:76826041T>A(hom) | c.1363A>T(hom) | p.K455X | Nonsense |
| PanNET25PT | DAXX | CCDS4776.1 | g.chr6:33394939delT | c.1976delA | fs | Indel |
| PanNET31PT | DAXX | CCDS4776.1 | g.chr6:33394935delC(hom) | c.1980delG(hom) | fs | Indel |
| PanNET44PT | DAXX | CCDS4776.1 | g.chr6:33394795delG | c.2120delC | fs | Indel |
| PanNET56PT | DAXX | CCDS4776.1 | g.chr6:33397319delG | c.211delC | fs | Indel |
| PanNET77PT | DAXX | CCDS4776.1 | g.chr6:33396614G>A | c.916C>T | p.R306X | Nonsense |
| PanNET84PT | DAXX | CCDS4776.1 | g.chr6:33395309delG | c.1766delC | fs | Indel |
| PanNET87PT | DAXX | CCDS4776.1 | g.chr6:33397141A>C | c.389T>G | p.L130R | Missense |
| PanNET93PT | DAXX | CCDS4776.1 | g.chr6:33396641C>G | c.889G>C | p.A297P | Missense |
| PanNET94PT | DAXX | CCDS4776.1 | g.chr6:33394872_33394873insA | c.2042_2043insT | fs | Indel |
| PanNET95PT | DAXX | CCDS4776.1 | g.chr6:33397221_33397224delCGCC | c.306_309delGGCG | fs | Indel |
| PanNET96PT | DAXX | CCDS4776.1 | g.chr6:33396167delC | c.1219delG | fs | Indel |
| PanNET97PT | DAXX | CCDS4776.1 | g.chr6:33395838C>A(hom) | c.1393G>T(hom) | p.E465X | Nonsense |
| PanNET102PT | DAXX | CCDS4776.1 | g.chr6:33397515T>A | c.166A>T | p.K56X | Nonsense |
| PanNET103PT | DAXX | CCDS4776.1 | g.chr6:33397579delA | c.102delT | fs | Indel |
| PanNET104PT | DAXX | CCDS4776.1 | g.chr6:33396604_33396605insACT(hom) | c.925_926insAGT(hom) | p.L309QF | Missense |
| PanNET108PT | DAXX | CCDS4776.1 | g.chr6:33395828delT(hom) | c.1403delA(hom) | fs | Indel |
| PanNET133PT | DAXX | CCDS4776.1 | g.chr6:33395889C>A(hom) | c.1342G>T(hom) | p.E448X | Nonsense |
| PanNET3PT | MEN1 | CCDS8083.1 | g.chr11:64331709C>A(hom) | c.689G>T(hom) | p.G230V | Missense |
| PanNET5PT | MEN1 | CCDS8083.1 | g.chr11:64332046A>G(hom) | c.562T>C(hom) | p.W188R | Missense |
| PanNET6PT | MEN1 | CCDS8083.1 | g.chr11:64333812_64333828delCACGGCTGGAGACACCC | c.329_345delGGGTGTCTCCAGCCGTG | fs | Indel |
| PanNET10PT | MEN1 | CCDS8083.1 | g.chr11:64334105_64334108delTCGT(hom) | c.50_53delACGA(hom) | fs | Indel |
| PanNET23PT | MEN1 | CCDS8083.1 | g.chr11:64331233_64331234delAG(hom) | c.832_833delCT(hom) | fs | Indel |
| PanNET29PT | MEN1 | CCDS8083.1 | g.chr11:64334070C>A(hom) | c.88G>T(hom) | p.E30X | Nonsense |
| PanNET31PT | MEN1 | CCDS8083.1 | g.chr11:64328587G>T | c.1643C>A | p.S548X | Nonsense |
| PanNET39PT | MEN1 | CCDS8083.1 | g.chr11:64333993_64333999delAGGGATG(hom) | c.159_165delCATCCCT(hom) | fs | Indel |
| PanNET44PT | MEN1 | CCDS8083.1 | g.chr11:64333955delG | c.203delC | fs | Indel |
| PanNET45PT | MEN1 | CCDS8083.1 | g.chr11:64330370G>C | c.974C>G | p.P325R | Missense |
| PanNET52PT | MEN1 | CCDS8083.1 | g.chr11:64333876delG | c.282delC | fs | Indel |
| PanNET57PT | MEN1 | CCDS8083.1 | g.chr11:64334002delG(hom) | c.156delC(hom) | fs | Indel |
| PanNET59PT | MEN1 | CCDS8083.1 | g.chr11:64329234G>A | c.1213C>T | p.Q405X | Nonsense |
| PanNET61PT | MEN1 | CCDS8083.1 | g.chr11:64334049_64334201delGGAGCACCAGGTCCGGCTCCTCT CGGCCCAGCTCGGCAGCAAACAGGCGCACCACGTCGTCGATGGAGC GCAGCGGGAACAGCGTCTTCTGGGCGGCCTTCAGCCCCATGGCGGC GGGCGGTGGGCGGCGGCCTGCAAGGCAAGCCGGGGGAG(hom) |
c.1_109delATGGGGCTGAAGGCCGCCCAGAA GACGCTGTTCCCGCTGCGCTCCATCGACGACG TGGTGCGCCTGTTTGCTGCCGAGCTGGGCCGA GAGGAGCCGGACCTGGTGCTCC(hom) |
fs | Indel |
| PanNET64PT | MEN1 | CCDS8083.1 | g.chr11:64333781delC | c.377delG | fs | Indel |
| PanNET69PT | MEN1 | CCDS8083.1 | g.chr11:64330291delA | c.1053delT | fs | Indel |
| PanNET77PT | MEN1 | CCDS8083.1 | g.chr11:64334063_64334079delGGCTCCTCTCGGCCCAG | c.79_95delCTGGGCCGAGAGGAGCC | fs | Indel |
| PanNET78PT | MEN1 | CCDS8083.1 | g.chr11:64332045C>T | c.563G>A | p.W188X | Nonsense |
| PanNET83PT | MEN1 | CCDS8083.1 | g.chr11:64330369delG | c.975delC | fs | Indel |
| PanNET84PT | MEN1 | CCDS8083.1 | g.chr11:64334139G>A | c.19C>T | p.Q7X | Nonsense |
| PanNET85PT | MEN1 | CCDS8083.1 | g.chr11:64332011_64332012insCTGT | c.596_597insACAG | fs | Indel |
| PanNET93PT | MEN1 | CCDS8083.1 | g.chr11:64333906_64333909delAGAC | c.249_252delGTCT | fs | Indel |
| PanNET94PT | MEN1 | CCDS8083.1 | g.chr11:64333906_64333909delAGAC | c.249_252delGTCT | fs | Indel |
| PanNET95PT | MEN1 | CCDS8083.1 | g.chr11:64332032delC(hom) | c.576delG(hom) | fs | Indel |
| PanNET96PT | MEN1 | CCDS8083.1 | g.chr11:64331269T>C | c.IVS799-2A>G | c.IVS799-2A>G | SpliceSite |
| PanNET99PT | MEN1 | CCDS8083.1 | g.chr11:64331938C>A | c.IVS669+1G>T | c.IVS669+1G>T | SpliceSite |
| PanNET100PT | MEN1 | CCDS8083.1 | g.chr11:64331940_64331941delCG(hom) | c.667_668delCG(hom) | fs | Indel |
| PanNET102PT | MEN1 | CCDS8083.1 | g.chr11:64332102G>A | c.506C>T | p.A169V | Missense |
| PanNET108PT | MEN1 | CCDS8083.1 | g.chr11:64331200_64331251delGCAGCCTGGCCACTTCCCTCTACT GACCTTTCCAGATGTCCCAGGTCATAGA(hom) |
c.815_837delTCTATGACCTGGGACATCTGGA A(hom) |
del exon and intron | Indel |
| PanNET109PT | MEN1 | CCDS8083.1 | g.chr11:64334093A>C | c.65T>G | p.L22R | Missense |
| PanNET10PT | PIK3CA | CCDS43171.1 | g.chr3:180418785G>A | c.1633G>A | p.E545K | Missense |
| PanNET10PT | PTEN | CCDS31238.1 | g.chr10:89707693delG | c.738delG | fs | Indel |
| PanNET31PT | PTEN | CCDS31238.1 | g.chr10:89682819T>G | c.323T>G | p.L108R | Missense |
| PanNET29PT | PTEN | CCDS31238.1 | g.chr10:89710790_89710791insTGACAAGGAATATCTAGTACTTAC TTTAA |
c.961_c.962insTGACAAGGAATATCTAGTACTT ACTTTAA |
fs | Indel |
| PanNET96PT | PTEN | CCDS31238.1 | g.chr10:89675287T>C(hom) | c.202T>C(hom) | p.Y68H | Missense |
| PanNET104PT | PTEN | CCDS31238.1 | g.chr10:89701856G>A(hom) | c.494G>A(hom) | p.G165E | Missense |
| PanNET24PT | TP53 | CCDS11118.1 | g.chr17:7518284G>A | c.722C>T | p.S241F | Missense |
| PanNET91PT | TP53 | CCDS11118.1 | g.chr17:7519210delA(hom) | c.445delT(hom) | fs | Indel |
| PanNET100PT | TP53 | CCDS11118.1 | g.chr17:7520101delT(hom) | c.311delA(hom) | fs | Indel |
| PanNET2PT | TSC2 | CCDS10458.1 | g.chr16:2070191C>T | c.3422C>T | p.A1141V | Missense |
| PanNET31PT | TSC2 | CCDS10458.1 | g.chr16:2074957G>A | c.4498G>A | p.V1500M | Missense |
| PanNET44PT | TSC2 | CCDS10458.1 | g.chr16:2074337_2074338delTG | c.4113_c.4114delTG | fs | Indel |
| PanNET70PT | TSC2 | CCDS10458.1 | g.chr16:2078571C>T | c.5383C>T | p.R1795C | Missense |
| PanNET93PT | TSC2 | CCDS10458.1 | g.chr16:2038643C>A | c.26C>A | p.S9X | Nonsense |
| PanNET112PT | TSC2 | CCDS10458.1 | g.chr16:2076836A>G | c.4952A>G | p.N1651S | Missense |
Coordinates refer to human reference genome hg18 release (NCBI 36.1, March 2006).
Samples PanNET3, PanNET7, PanNET10, PanNET21, PanNET23, PanNET24, PanNET25, PanNET31, PanNET36, and PanNET93 were used for the initial (Discovery Set) screen.
Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S,Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
(hom): these mutations appear homozygous
Of the 30 mutations in MEN1, 25 were inactivating mutations (18 insertions or deletions (indels), 5 nonsense and 2 splice-site mutations), while five were missense. At least 11 were homozygous; in the others, the presence of “contaminating” DNA from normal cells made it difficult to reliably distinguish heterozygous from homozygous changes. MEN1 encodes menin, a nuclear protein that acts as a scaffold to regulate gene transcription by coordinating chromatin remodeling. It is an essential component of the MLL SET1-like histone methyltransferase (HMT) complex (15-19). Overall, MEN1 was mutated in 30 of the 68 PanNETs used in the Discovery and Validation sets combined.
DAXX and ATRX were mutated in 17 and 12 PanNETs, respectively. No tumor with a mutation in DAXX had a mutation in ATRX, consistent with their presumptive function within the same pathway. Overall 29 of 68 PanNETs (42.6%) had a mutation in this pathway. There were 11 insertions or deletions (indels) and 4 nonsense mutations in DAXX, and six indels and 3 nonsense mutations in ATRX. The three ATRX missense mutations were within the conserved helicase domain while the DAXX missense mutations were non-conserved changes. Five DAXX and four ATRX mutations were homozygous, indicating loss of the other allele. The high ratio of inactivating to missense mutations in both genes establishes them as PanNET tumor suppressor genes. Loss of immunolabelling for DAXX and ATRX correlated with mutation of the respective gene (fig. S1, A and B, and table S5). From these data, we hypothesize that both copies of DAXX are generally inactivated, one by mutation and the other either by loss of the non-mutated allele or by epigenetic silencing. We also hypothesize that both copies of ATRX are inactivated, one by mutation and the other by chromosome X inactivation. Recently, it has been shown that DAXX is an H3.3-specific histone chaperone (20). ATRX encodes for a protein that at the amino-terminus has an ADD (ATRX-DNMTT3-DNMT3L) domain and a carboxy-terminal helicase domain. Almost all missense disease causing mutations are within these two domains (21). DAXX and ATRX interact and both are required for H3.3 incorporation at the telomeres and ATRX is also required for suppression of telomeric repeat-containing RNA expression (22-24). ATRX was recently shown to target CpG islands and G-rich tandem repeats (25), which exist close to telomeric regions.
We identified five PTEN mutations, two indels and three missense; six TSC2 mutations, one indel, one nonsense and four missense; and one PIK3CA missense mutation. Previously published expression analyses have indicated that the expression of genes in the mTOR pathway is altered in most PanNETs (26,27). Our data suggest that, at least at the genetic level, only a subset of PanNETs have alterations of this pathway. This finding may have direct clinical application through prioritization of patients for therapy with mTOR pathway inhibitors. Everolimus (Afinitor, RAD-001, 40-O-(hydroxyethyl)-rapamycin) has been shown to increase progression free survival in a subset of PanNET patients with advanced disease (28). If the mutational status of genes coding for proteins in the mTOR pathway predicts clinical response to mTOR inhibitors, it should be possible to select patients who would benefit most from an mTOR inhibitor through analysis of these genes in patients’ tumors (29, 30).
All 68 tumors evaluated in this study were from patients undergoing aggressive intervention (table S6) and included patients undergoing curative resection as well as those with metastatic disease. Interestingly, mutations in MEN1, DAXX/ATRX or the combination of both MEN1 and DAXX/ATRX were associated with prolonged survival relative to those patients whose tumors lacked these mutations (Fig. 1, C and D and table S7). This was particularly evident in patients with metastatic disease and with mutations in both MEN1 and DAXX/ATRX: 100% of patients with PanNETs that had these mutations survived at least ten years while over 60% of the patients without these mutations died within five years of diagnosis (Fig. 1D). One possible explanation for the difference in survival is that mutations in MEN1 and DAXX/ATRX identify a biologically specific subgroup of PanNETs.
In summary, whole exome sequencing of pancreatic neuroendocrine tumors has led to the identification of novel tumor suppressor genes and illuminated the genetic differences between the two major neoplasms of the pancreas. The mutations may serve to aid prognosis and provide a way to prioritize patients for therapy with mTOR inhibitors.
Supplementary Material
Acknowledgments
We thank M. Whalen for expert technical assistance. Supported by a research grant from the Caring for Carcinoid Foundation, the Lustgarten Foundation for Pancreatic Cancer Research, the Sol Goldman Pancreatic Cancer Research Center, The Joseph L. Rabinowitz Fund for Pancreatic Cancer Research, The Virginia and D. K. Ludwig Fund for Cancer Research, the Raymond and Beverly Sackler Research Foundation , the AACR Stand Up To Cancer-Dream Team Translational Cancer Research Grant, and National Institutes of Health grants CA57345, CA121113, P50CA062924, P01CA134292, and R01CA113669. N.P., B.V., L.D., V.E.V., and K.W.K. are members of the Scientific Advisory Board of Inostics, a company that is developing technologies for the molecular diagnosis of cancer. N.P., B.V., L.D., V.E.V., and K.W.K. are co-founders of Inostics and Personal Genome Diagnostics and are members of their Scientific Advisory Boards. The authors are entitled to a share of the royalties received by the University on sales of products related to genes described in this manuscript. N.P., B.V., K.W.K., L.D., and V.E.V own Inostics and Personal Genome Diagnostics stock, which is subject to certain restrictions under University policy.
Footnotes
The terms of these arrangements are managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
References
- 1.Hruban RH, Pitman MB, Klimstra DS. Tumors of the Pancreas. Atlas of Tumor Pathology. American Registry of Pathology and Armed Forces Institute of Pathology; Washington, DC: 2007. p. 6. Fourth Series, Fascicle. [Google Scholar]
- 2.Fredrich M, Reisch A, Illing RB. Exp Brain Res. 2009;195:241. doi: 10.1007/s00221-009-1776-7. [DOI] [PubMed] [Google Scholar]
- 3.Ekeblad S, Skogseid B, Dunder K, Oberg K, Eriksson B. Clin Cancer Res. 2008;14:7798. doi: 10.1158/1078-0432.CCR-08-0734. [DOI] [PubMed] [Google Scholar]
- 4.Francalanci P, et al. Am J Surg Pathol. 2003;27:1386. doi: 10.1097/00000478-200310000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Corbo V, et al. Endocr Relat Cancer. 2010;17:771. doi: 10.1677/ERC-10-0028. [DOI] [PubMed] [Google Scholar]
- 6.Capelli P, et al. Arch Pathol Lab Med. 2009;133:350. doi: 10.5858/133.3.350. [DOI] [PubMed] [Google Scholar]
- 7.Chung DC, et al. Cancer Res. 1998;58:3706. [PubMed] [Google Scholar]
- 8.Floridia G, et al. Cancer Genet. Cytogenet. 2005;156:23. doi: 10.1016/j.cancergencyto.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Hu W, et al. Genes Cancer. 2010;1:360. doi: 10.1177/1947601910371979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.See supporting material on Science Online.
- 11.Jones S, et al. Science. 2008;321:1801. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons DW, et al. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 13.Guertin DA, Sabatini DM. Cancer Cell. 2007;12:9. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Shaw RJ, Cantley LC. Nature. 2006;441:424. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 15.Hughes CM, et al. Mol Cell. 2004;13:587. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama A, et al. Mol Cell Biol. 2004;24:5639. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grembecka J, Belcher AM, Hartley T, Cierpicki T. J Biol Chem. 2010 Oct 20; doi: 10.1074/jbc.M110.172783. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, et al. Cancer Res. 2003;63:6135. [PubMed] [Google Scholar]
- 19.Agarwal SK, et al. Cell. 1999;96:143. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 20.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Proc Natl Acad Sci U S A. 2010;107:14075. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbons RJ, et al. Human Mutation. 2008;29:796. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- 22.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. Genes Dev. 2010;24:1253. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg AD, et al. Cell. 2010;140:678. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong LH, et al. Genome Res. 2010;20:351. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law MJ, et al. Cell. 2010;143:367. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 26.Missiaglia E, et al. J Clin Oncol. 2010;28:245. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perren A, et al. Am. J. Pathol. 2000;157:1097. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao J. A randomized, double-blind, placebo-controlled, multicenter pahseIII trial of everolimus in patients with advanced pancreatic neuroendocrine tumors (PNET) (RADIANT-3) Annals of Oncology. 21(Supplemment 8):viii4. ESMO, Milan 8-12 October 2010. Published in. Abstract LBA9. [Google Scholar]
- 29.Liu P, Cheng H, Roberts TM, Zhao JJ. Nat Rev Drug Discov. 2009;8:627. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krueger DA, et al. N Engl J Med. 2010;363:1801. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.