

Abstract
BACKGROUND AND PURPOSE
Neonatal pulmonary hypertension (PPHN) is characterized by pulmonary vasoconstriction, due in part to dysregulation of the thromboxane prostanoid (TP) receptor. Hypoxia induces TP receptor–mediated hyperresponsiveness, whereas serine phosphorylation mediates desensitization of TP receptors. We hypothesized that prostacyclin (IP) receptor activity induces TP receptor phosphorylation and decreases ligand affinity; that TP receptor sensitization in hypoxic myocytes is due to IP receptor inactivation; and that this would be reversible by the cAMP-specific phosphodiesterase inhibitor milrinone.
EXPERIMENTAL APPROACH
We examined functional regulation of TP receptors by serine phosphorylation and effects of IP receptor stimulation and protein kinase A (PKA) activity on TP receptor sensitivity in myocytes from neonatal porcine resistance pulmonary arteries after 72 h hypoxia in vitro. Ca2+ response curves to U46619 (TP receptor agonist) were determined in hypoxic and normoxic myocytes incubated with or without iloprost (IP receptor agonist), forskolin (adenylyl cyclase activator), H8 (PKA inhibitor) or milrinone. TP and IP receptor saturation binding kinetics were measured in presence of iloprost or 8-bromo-cAMP.
KEY RESULTS
Ligand affinity for TP receptors was normalized in vitro by IP receptor signalling intermediates. However, IP receptor affinity was compromised in hypoxic myocytes, decreasing cAMP production. Milrinone normalized TP receptor sensitivity in hypoxic myocytes by restoring PKA-mediated regulatory TP receptor phosphorylation.
CONCLUSIONS AND IMPLICATIONS
TP receptor sensitivity and EC50 for TP receptor agonists was regulated by PKA, as TP receptor serine phosphorylation by PKA down-regulated Ca2+ mobilization. Hypoxia decreased IP receptor activity and cAMP generation, inducing TP receptor hyperresponsiveness, which was reversed by milrinone.
Keywords: persistent pulmonary hypertension of the newborn, hypoxia, prostanoid, prostacyclin, phosphodiesterase inhibitor
Introduction
Persistent pulmonary hypertension of the newborn (PPHN) is a multifactorial disorder of postnatal pulmonary vasoconstriction and remodelling, resulting in respiratory failure, hypoxemia and 20% mortality. Common PPHN aetiologies include meconium aspiration (nearly 40% of cases) and sepsis (25%) (Clark et al., 2000; Konduri et al., 2004; Finer and Barrington, 2006). Infants with PPHN who have sepsis or inflammation are among the poorest responders to vasodilators.
The crucial endogenous pathways that regulate perinatal pulmonary vascular tone include the nitric oxide–endothelin and prostacyclin–thromboxane systems (Weinberger et al., 2001). Thromboxane A2 (TxA2), an inflammatory prostanoid with vasoconstrictive and mitogenic properties, contracts pulmonary arterial myocytes via the Gαq-coupled thromboxane receptor (TP) (nomenclature follows Alexander et al., 2009), leading to increased intracellular [Ca2+], force generation and sensitization of the contractile apparatus to Ca2+ (Cogolludo et al., 2003). TxA2 is abundant in the neonatal circulation at birth, participating in the closure of the umbilical vessels and the ductus arteriosus (Reyes, 1993). Hypoxic PPHN increases the serum TxA2 to prostacylin (PGI2) ratio (Fike et al., 2003). Hypoxia increases pulmonary vasoconstriction to the TP receptor agonist U46619 by over fivefold, relative to its effect under normoxic conditions, and underlies hypersensitization to agonist ligands (Snow et al., 2008). Hypoxia has a priming effect on pulmonary arterial myocytes, increasing inositol 1,4,5-trisphosphate (IP3) generation in response to vasoconstrictors (Peacock et al., 1998).
Postnatal prostacyclin (PGI2) production is normally high, but hypoxia inverts the TxA2 : PGI2 ratio by decreasing prostacyclin synthesis (Fike et al., 2003). PGI2 mediates vasodilatory and anti-inflammatory actions via the prostacyclin (IP) receptor, which is coupled via Gαs to adenylyl cyclase (Vane and Botting, 1995). Clinical use of prostacyclin in pulmonary hypertension is limited by rapid agonist-induced down-regulation of the IP vasodilator response (Sobolewski et al., 2004).
Phosphorylation is a primary regulatory mechanism for vascular prostanoid receptors, in response to ligand binding, or due to signalling interactions with other prostanoid receptors (Breyer et al., 2001). TP receptor phosphorylation is induced by pulmonary vasodilators, including PGI2 (Reid and Kinsella, 2003). IP receptor–mediated phosphorylation of TP receptors has been ascribed to protein kinase C (PKC) (Kelley-Hickie and Kinsella, 2004) and protein kinase A (PKA) (Walsh et al., 2000; Reid and Kinsella, 2003) and leads to TP receptor desensitization (Reid and Kinsella, 2003; O'Meara and Kinsella, 2004) and internalization (Parent et al., 1999). The ligand affinity of human TP receptor isoforms is regulated by PKA-mediated phosphorylation at serine 329 (Habib et al., 1997; Foley et al., 2001; Reid and Kinsella, 2003).
We reported that in normal neonatal pulmonary artery myocytes, TP receptors are maintained in a state of low affinity by tonic phosphorylation. Hypoxia decreases inhibitory TP receptor phosphorylation, causing hypersensitivity of these receptors. Calcium mobilization in pulmonary arterial myocytes is increased after hypoxia both in vivo and in vitro, with sustained effects even after subsequent cell culture in normoxia (Hinton et al., 2006). Hypoxic sensitization of TP receptors occurs due to a decreased ligand Kd shown as a shift of the agonist competitive binding curve to the left (Hinton et al., 2007), indicating an increased TP receptor affinity for agonists (Gong et al., 2010).
Milrinone, a phosphodiesterase (PDE)-3 inhibitor, is an inotrope and vasodilator, increasing cellular cAMP and improving hypoxic pulmonary haemodynamics (Joynt et al., 2008; Lakshminrusimha et al., 2009). Studies on the functional effects of milrinone have recently focused on prostanoid receptors, as milrinone attenuates inflammatory lung injury (Bueltmann et al., 2009), potentiates the anti-remodelling effect of PGI2 on hypoxic artery (Phillips et al., 2005) and enhances relaxation to PGI2 in PPHN (Lakshminrusimha et al., 2009).
In this study, our objective was to study whether the loss of TP receptor regulation after hypoxia in vitro may be due to diminished activity of PKA. We hypothesized that hypoxia inhibits IP receptor signalling and depresses myocyte PKA activity, thereby inhibiting TP receptor phosphorylation and enhancing TP receptor affinity. Since milrinone increases PKA activity, we further hypothesized that sensitization of TP receptors induced by hypoxia would be reversed by milrinone treatment, reactivating PKA-mediated TP receptor regulation.
Methods
All animal care and experimental protocols were approved by the University of Manitoba Central Animal Care committee, in accordance with Canadian Council on Animal Care and US National Institutes of Health guidelines. Cultured myocytes from resistance pulmonary arteries of newborn (<24 h age) swine were examined in first passage, following 72 h normoxic or hypoxic exposure.
Cell culture
Pulmonary artery smooth muscle cells were obtained from newborn swine using a dispersed cell culture method selective for myocytes (Shimoda et al., 2000). Fourth- to sixth-generation pulmonary arteries were obtained by microdissection into Ca2+-free Krebs–Henseleit physiological buffer and were allowed to recover in cold HEPES-buffered saline solution (HBS; in mM: 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, 10 glucose, pH 7.4) supplemented with antibiotic/antimycotic mixture and gentamicin. Arteries were washed in Ca2+-reduced HBS (20 µM CaCl2), finely minced, then the tissue transferred to a digestion solution containing Ca2+-reduced HBS, type I collagenase (1750 U·mL−1), dithiothreitol (1 mM), BSA (2 mg·mL−1) and papain (9.5 U·mL−1) for 15 min at 37°C with gentle agitation. Dispersed myocytes were collected by centrifugation at 800×g for 5 min, washed in Ca2+-free HBS to remove digestion solution and then resuspended in culture medium. Cells were plated at a density of 4.4 × 104 cells cm−2 in Ham's F-12 medium with L-glutamine supplemented with 10% fetal calf serum, 1% penicillin and 1% streptomycin. Once confluent, myocytes were serum-deprived for 2 days (Ham's F-12 with L-glutamine/penicillin/streptomycin and 1% insulin–transferrin–selenium) to synchronize cells in a contractile phenotype, then split into two groups for the final 3 days of culture: (i) control normoxic myocytes, maintained serum-free in 21% O2, 5% CO2; and (ii) hypoxic myocytes, maintained serum-free in 10% O2, 5% CO2 for 3 days.
Live cell calcium mobilization
Myocytes were rinsed free of media in Hanks balanced salt solution (HBSS; in mM: 1.26 CaCl2, 0.493 MgCl2·6 H2O, 0.407 MgSO4·7 H2O, 5.33 KCl, 0.441 KH2PO4, 4.17 NaHCO3, 137.93 NaCl, and 0.338 NaHPO2) with 0.1% BSA. Myocytes were loaded with the Ca2+-sensitive fluorescent dye fura 2-acetoxymethyl ester (fura-2AM) dissolved in dimethyl sulphoxide, as 5 µM in an HBSS/0.1% BSA solution, with 1.0 µg·mL−1 pluronic acid (for AM ester solubilization), for 1 h at 37°C. Extracellular fura-2AM was washed off with HBSS/0.1% BSA. Cells were allowed to recover for 30 min at room temperature, for complete cleavage of intracellular AM esters. Coverglass plates were secured on an inverted microscope (Olympus, Markham, Ontario, Canada) in 21% O2, and studied at 20× magnification. Cells were challenged with serial concentrations of the TP receptor agonist U46619 after pre-incubation with or without 10 µM forskolin (adenylyl cyclase activator), 1 µM H8 (PKA inhibitor), 1 µM iloprost (IP receptor agonist) for 1 h; or 1 µM deoxy-prostaglandin-E1 (stable, partially selective EP2 receptor agonist), 0.1 µM adenosine-5′-N-ethylcarboxamide (NECA, selective adenosine A2B receptor agonist) or 5 nM milrinone for 15 min. Ratiometric imaging of intracellular Ca2+ concentration utilized excitation wavelengths of 340 and 380 nm and emission wavelength of 510 nm; data were captured by a charge-coupled device camera and Perkin Elmer software (Montreal, Quebec, Canada). Each recording consisted of a stable baseline and a return to same. Following subtraction of baseline fluorescence, Ca2+ mobilization was analyzed from at least four equally sized regions from each microscope field, tracing three to five cells with minimal cell-free areas. Emission ratios from 340/380 λ excitations were quantified against a calcium standard (Grynkiewicz et al., 1985).
PKA assay
PKA activity was measured in a 96-well ProFluor assay kit (Promega, Nepean, Ontario, Canada), using PKA substrate peptide bisamide rhodamine 110 (PKA R110, 25 µL) pre-incubated with 25 µL ATP reagent in reaction buffer. Following 10 min incubation with 1 µM 8-bromo-cAMP, 1 µM H8 (Maggi et al., 1996), 1 µM iloprost, 10 nM milrinone or diluent, lysates from hypoxic and normoxic pulmonary artery myocytes were added to the reaction mixture and incubated for 3 min. Reaction was terminated with 25 µL protease reagent in termination buffer. After a 30 min incubation, and addition of 25 µL stabilizing reagent, fluorescence was measured at 485 nm excitation and 530 nm emission λ, against reagent buffer blanks, using a FLUOstar Optima microplate reader (BMG Labtec GMBH, Hanns-Martin-schleyer-strio, Offenburg, Germany). Phosphorylated PKA R110 substrate is resistant to digestion by the protease reagent and remains non-fluorescent; fluorescence thus inversely correlates with PKA activity. Measured fluorescence was inverted to calculate PKA activity (arbitrary units) and expressed as fold-change compared to (untreated) normoxic lysates.
Sample preparation for receptor kinetics
Myocytes were pre-treated for 24 h with 1 µM U46619, 1 µM iloprost, 1 µM 8-bromo cAMP or diluent. Cells were twice rinsed in PBS, then lysed in binding buffer (25 mM Tris, 10 mM CaCl2, 0.01 mM indomethacin and 75 µg·mL−1 PMSF, pH 7.4). Unlysed cells and particulate matter were removed by brief centrifugation at 2000×g. The supernatant was then centrifuged at 100 000×g for 60 min at 4°C, and membrane fractions were resuspended in binding buffer. Aliquots (30–60 µg protein) of this membrane preparation were used for radioligand experiments.
Saturation binding kinetics
Saturation binding kinetics was quantified in membrane fractions of hypoxic and normoxic myocytes pre-treated for 24 h with 1 µM U46619, 1 µM iloprost or 1 µM 8-bromo-cAMP. Samples were incubated with 3H-SQ-29548 (0.1–50 nM; diluted in binding buffer) with or without an excess of unlabelled U46619 (10 µM), in a total reaction volume of 100 µL for 1 h at room temperature. Reactions were terminated by vacuum filtration, and membranes were washed twice with ice-cold binding buffer. Filters were agitated in 500 µL distilled water to release adsorbed radioisotope and then allowed to equilibrate in 5 mL CytoScint (ICN) for 5 h before counting. Unbound radioisotope was also collected. Counts per minute were analyzed for 3 min per sample.
PGI2 receptor (IP) abundance
Hypoxic and normoxic myocytes were scraped and sonicated in 300 µL ice-cold radioimmunoprecipitation assay buffer (PBS containing 0.1%SDS, 1% Triton X100, 1 mM EDTA, 3.3 mM PMSF, 10 mM sodium orthovanadate and protease inhibitor [4-(2-aminoethyl) benzenesulfonyl fluoride] 0.1 mM, aprotinin 0.08 µM, bestatin 4 µM, N-(trans-epoxysuccinyl)-L-leucine 4-guanidinobutylamide 1.4 µM, leupeptin 2.2 µM, pepstatin A 1.5 µM). IP receptor protein abundance was measured in 20 µg protein by Western blot following SDS-PAGE on polyvinylidene fluoride membrane, using polyclonal rabbit antibody to human IP receptors (Cayman Chemical, Ann Arbor, MI, USA) at 1:250 dilution overnight and visualized after incubation with 1:5000 goat anti-rabbit IgG peroxidase conjugate.
cAMP assay
Cells were treated with 1 µM iloprost for serial time intervals. Media was removed, and cytosolic protein was precipitated with acidic ethanol. Samples were separated by brief centrifugation at 5000×g, then supernatant was neutralized with KOH prior to cAMP assay. cAMP was measured using an assay kit (TRK 432, Amersham/GE, Baie d'urfe, Quebec, Canada), based on competitive binding between unlabelled cAMP and a fixed quantity of the supplied 3H-labelled binding protein with high specificity and affinity for cAMP. Results were normalized to extract protein content.
PDE assay
cAMP-specific PDE assay used reagents provided in QuantiZyme Cyclic Nucleotide PDE colorimetric assay kit (Biomol, Burlington, Ontario, Canada), with a modified protocol to assess cAMP degradation due to lysate enzyme activity. Normoxic and hypoxic myocytes were incubated for 5 min with selective PDE inhibitors: 1 µM vinpocetine (PDE-1-selective; Enzo Life Sciences, Burlington, Ontario, Canada), Bay-60-755 (PDE-2; Cayman Chemical), milrinone (PDE-3), rolipram (PDE-4; Sigma, Oakville, Ontario, Canada) or diluent. Plates were then washed twice with PBS, and cells were scraped into lysis buffer containing protease and phosphatase inhibitors. Cytosolic fractions were isolated by fractional centrifugation. The assay mixture, containing 20 µL of 0.5 mM cAMP substrate, 75 µg cytosolic protein and assay buffer, was incubated at 30°C for 30 min. Reactions were terminated by the addition of Biomol Green reagent. AMP resulting from the action of cytosolic PDE was degraded by 5′-nucleotidase to release free phosphate, generating a colorimetric reaction quantified at 620 nm in a microplate reader (against a blank containing cytosolic protein without substrate). Phosphate release per unit time was calculated from a linear standard curve generated using 5′-AMP concentrations of 0.05–3.0 nM and 5′-nucleotidase, normalized to lysate protein content (Kolosionek et al., 2009).
IP3 measurement
Confluent serum-starved normoxic and hypoxic pulmonary arterial myocytes were pre-treated for 30 min with 1 µM iloprost, 10 nM 8Br-cAMP or diluent, then challenged with 1 µM U46619 for 1 min. Unstimulated controls were used to determine basal levels of IP3. Cells were lysed, and intracellular IP3 was extracted by ice-cold 20% trichloracetic acid for 20 min. Precipitated proteins were sedimented by centrifugation; supernatants were collected and neutralized with 10 M KOH. IP3 was quantified using a TRK1000 radio-competition binding assay employing [3H]-D-myo-IP3 as the radioactive standard (GE Life Sciences, Baie d'urfe, Quebec, Canada) and expressed as pmol IP3 mg−1 lysate protein (measured in the pelleted cellular extract).
Statistical analyses
Quantitative data were analysed by anova. Ca2+ mobilization was calculated from measured 340/380 nm emission ratios, and log EC50 determined by sigmoidal curve fit of transformed data. Data are expressed as mean ± SD or mean ± SEM; P < 0.05 was taken to be significant.
Materials
3H SQ29548 (0.1 mCi·mL−1) was obtained from Perkin Elmer and 3H iloprost (0.1 mCI·mL−1) from GE Life Sciences. Forskolin, 8-Br-cAMP, U46619, milrinone, NECA, rolipram and indomethacin were from Sigma Aldrich (Oakville, Ontario, Canada); fura-2AM from Molecular Probes (Burlington, Ontario, Canada); iloprost, Bay-60 and 11-deoxy-PGE1 from Cayman Chemicals; H8 and vinpocetine from Biomol Enzo Life Sciences.
Results
Hypoxia causes thromboxane receptor hypersensitivity and hyperreactivity
Neonatal porcine pulmonary arterial myocytes in the first passage were loaded with the calciometric dye fura-2AM and challenged with increasing concentrations of TP receptor agonist U46619, following 72 h exposure to hypoxia, or normoxia in vitro. The dose–response relation for U46619 in hypoxic myocytes was shifted to the left (hypersensitive) and upwards (hyperreactive), compared with that for normoxic myocytes (Figure 1A), resulting in sensitization of the normalized dose–response curve and a lower concentration required for 50% maximal response (Figure 1B; EC50 in hypoxic myocytes 0.53 µM and in normoxic myocytes 1.2 µM; P < 0.0005). Both hypersensitivity and hyperreactivity are elements of pharmacological hyperresponsiveness.
Figure 1.
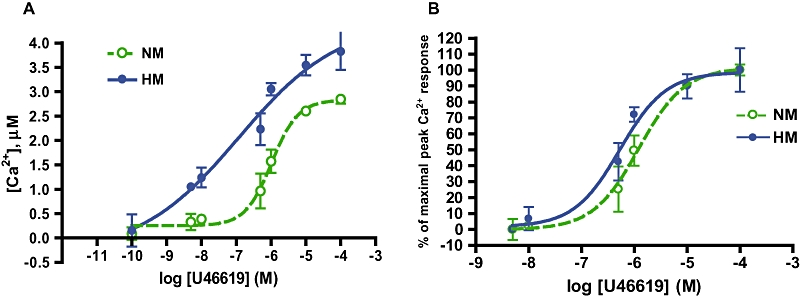
Hypoxia causes TP receptor hypersensitivity and hyperreactivity. (A) Hypoxic (HM) and normoxic (NM) pulmonary arterial myocytes were loaded with fura-2AM and challenged with TP receptor agonist U46619. Peak calcium mobilization calculated from 340/380 nm emission ratio. HM calcium response is left-shifted (hypersensitive) and up-shifted (hyperreactive) relative to NM curve. (B) Concentration response curve for U46619, standardized to maximal response. NM EC50, 1.2 µM; HM EC50, 0.53 µM (data in curves presented as mean ± SD; P < 0.0005, n = 15).
Effect of hypoxia on PKA activity
Hypoxia decreases PKA activity in myocyte lysates by nearly 40% (Figure 2). Treatment with the cAMP analogue, 8-bromo-cAMP (1 µM) or with the PDE-3 inhibitor milrinone (1 µM), increased PKA activity in hypoxic myocytes to normoxic levels, while treatment with the non-selective PKA inhibitor H8 (1 µM) decreased normoxic PKA activity, to the level observed in untreated hypoxic myocytes.
Figure 2.
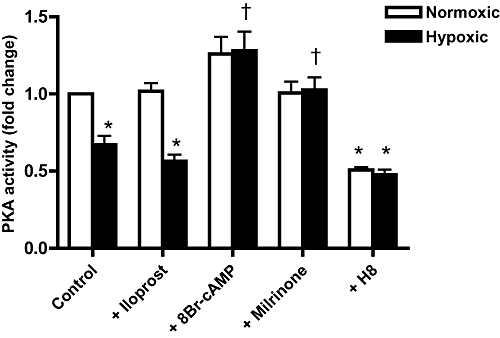
Hypoxia decreases PKA activity. PKA activity determined in myocyte lysates obtained following pre-incubations with 1 µM iloprost, 8-bromo-cAMP or H8, 10nM milrinone or diluent. Mean ± SEM; n = 6–8 separate experiments; *P < 0.05 compared with normoxic untreated myocytes; †P < 0.05 compared with hypoxic untreated myocytes.
Effect of PKA activation state on TP receptor sensitivity
We examined regulation of TP receptorsensitivity by adenylyl cyclase, or its downstream effector PKA. Calcium mobilization was quantified to serial concentrations of U46619 in hypoxic and normoxic myocytes pre-incubated for 15 min with forskolin, H8 or buffer. Forskolin had no effect on responses of normoxic myocytes (Figures 3A and 4A) but shifted the response of hypoxic myocytes to U46619 to the right, resulting in normalization of the dose–response curve for hypoxic myocytes (Figures 3C and 4C). H8 induced a leftward displacement of the U46619 response curve in normoxic myocytes, towards the position of the curve for hypoxic myocytes (Figures 3B and 4B) while causing no further change in response of hypoxic myocytes (Figures 3D and 4D). EC50s for each condition were calculated from percent (normalized) dose–response curves. All dose–response relationships in conditions of phosphorylated TP receptors showed an approximately threefold increase in EC50 (Table 1).
Figure 3.
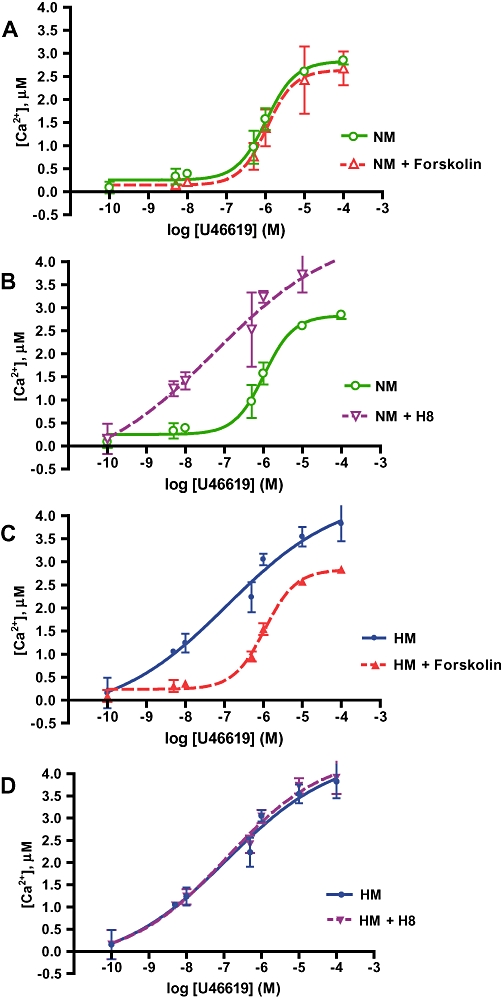
PKA activation state determines TP receptor reactivity and maximal calcium mobilization. Concentration response curves for TP receptor agonist U46619, showing peak Ca2+ response to U46619 challenge quantified in fura-2AM-loaded pulmonary arterial myocytes pre-incubated for 15 min with 1 µM forskolin (activating PKA) or H8 (inactivating PKA), or buffer. Baseline Ca2+ subtracted from all peak measurements. All curves presented as mean ± SD; n = 15–20 per point. (A) Forskolin has no effect on the dose–response curve to U46619 in normoxic myocytes (NM). (B) H8 shifted the curve in NM to the left and upwards to resemble the curve in hypoxic myocytes (HM). (C) Forskolin exposure normalizes TP receptor hyperresponsiveness in HM. (D) H8 has no effect on the dose–response to the TP receptor agonist in HM.
Figure 4.
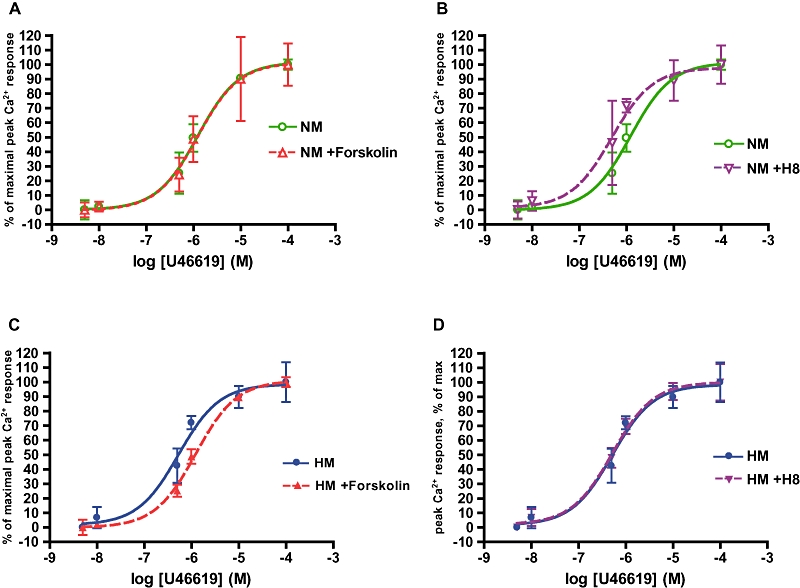
PKA activation state determines TP receptor sensitivity and EC50 of Ca2+ mobilization. Concentration response curves for the TP receptor agonist U46619, in pulmonary arterial myocytes pre-incubated with 1 µM forskolin (activating PKA) or H8 (inactivating PKA), or buffer, by fura-2AM. All Ca2+ values normalized as % of maximal concentration. Curves presented as mean ± SD. (A) Forskolin had no effect on the dose–response curve to U46619 in normoxic myocytes (NM). (B) H8 shifts the curve in NM to the left to resemble that of hypoxic myocytes (HM). (C) In HM, forskolin shifts the dose–response curve to the right to resemble that in NM. (D) H8 has no effect on the dose–response curve to the TP receptor agonist in HM.
Table 1.
Log EC50 and EC50 values for PKA-activated and -inhibited myocytes from normoxic (NM) or hypoxic (HM) cultures
| NM | NM + Forskolin | NM + H8 | HM | HM + Forskolin | HM + H8 | |
|---|---|---|---|---|---|---|
| Log EC50± SEM | −5.916 ± 0.037 | −5.901 ± 0.066 | −6.313 ± 0.253 | −6.272 ± 0.163 | −5.910 ± 0.025 | −6.294 ± 0.118 |
| EC50 (µM) | 1.2 | 1.3 | 0.49 | 0.53 | 1.2 | 0.51 |
The values of log EC50 and EC50 for each group show differences between PKA-active (with forskolin) and PKA-inhibited (with H8) groups (P < 0.0005, n = 15). PKA activation decreased EC50 values approximately 2.5-fold.
TP receptor kinetics
TP receptor saturation binding kinetics were determined in cell membrane fractions from hypoxic and normoxic myocytes, using 3H-SQ29548, a TP receptor–specific antagonist (Figure 5A). The dissociation constant (slope or Kd, in nM) and the maximal concentration of available binding sites (Bmax, expressed as fmoL·mg−1) were significantly decreased in hypoxic, compared with normoxic myocytes (Table 2). Incubation of cell extracts with the IP receptor agonist iloprost (1 µM) markedly desensitized TP receptors, increasing Kd in hypoxic and normoxic myocytes (Figure 5B, P < 0.01), and eliminating the difference between these groups (Table 2).
Figure 5.
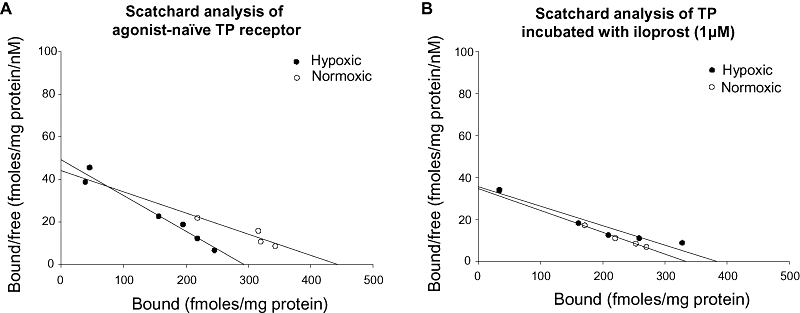
Ligand affinity of TP receptors in hypoxic myocytes can be normalized by incubation with iloprost. (A) Representative Scatchard plot for TP receptors in cell membrane fractions from hypoxic myocytes (HM) and normoxic myocytes (NM), using the radiolabelled TP receptor antagonist 3H-SQ29548. Dissociation constant (Kd) is lower in HM than NM (P < 0.01; data represent three separate experiments). (B) Representative Scatchard plot for TP receptors. Pre-incubation of membrane fractions with iloprost (1 µM) increased Kd in both NM and HM membranes, eliminating the difference between these groups (P = ns; represents three separate experiments).
Table 2.
TP receptor ligand association kinetics
| Bmax | Kd | |
|---|---|---|
| TP | ||
| Normoxic | 412 ± 7.2 | 8.8 ± 0.21 |
| Hypoxic | 315 ± 7.6 | 6.55 ± 0.05** |
| TP + Iloprost | ||
| Normoxic | 342 + 6.0 | 11.43 + 0.28 |
| Hypoxic | 361 + 9.4 | 10.29 + 0.28 |
Mean kinetics for TP receptor ligand association in hypoxic and normoxic myocyte cell membrane fractions, using the radiolabelled TP receptor antagonist 3H-SQ29548. Dissociation constant (Kd) was lower in hypoxic than in normoxic myocytes (**P < 0.01; data represent three separate experiments). Pre-incubation of membrane fractions with 1 µM iloprost increased Kd of both hypoxic and normoxic cells, eliminating the difference between the groups (P = ns; data from three separate experiments).
IP receptor abundance, activity and cAMP generation
The effect of the IP agonist iloprost (1 µM) on intracellular cAMP was examined in a time-dependent study, under hypoxic and normoxic conditions. Challenge of normoxic myocytes with iloprost stimulated significant cAMP generation at 1 min after challenge (P < 0.01), with return to baseline by 5 min. Hypoxic myocytes had a lower basal cAMP level (P < 0.01) and exhibited no measurable rise in cAMP after iloprost stimulation (Figure 6A). IP receptor protein expression was comparable in hypoxic and normoxic myocytes (Figure 6B). Decreased cAMP in hypoxic myocytes was not explained by increased breakdown, as cAMP-specific PDE activity in HM was diminished at all time points measured (Figure 6C, P < 0.05). Fractionation of measured cAMP-specific PDE activities indicated that the primary PDE isoforms in pulmonary arterial myocytes are PDE-3 and PDE-4, inhibited by rolipram and milrinone, respectively (Figure 6D). Scatchard analysis of saturation binding of 3H-iloprost shows the Kd for this IP receptor agonist was elevated in hypoxic myocytes, suggesting that it is impaired IP receptor ligand affinity in hypoxia that induced the decreased cAMP signal (Figure 6E, F). The Kd of IP receptors was further increased (desensitized) by pre-treatment with iloprost, a known effect of this agonist. Stimulation of TP receptors with U46619 had no effect on the Kd value for the IP receptor ligand in hypoxic or normoxic myocytes, indicating the absence of receptor cross-regulation.
Figure 6.
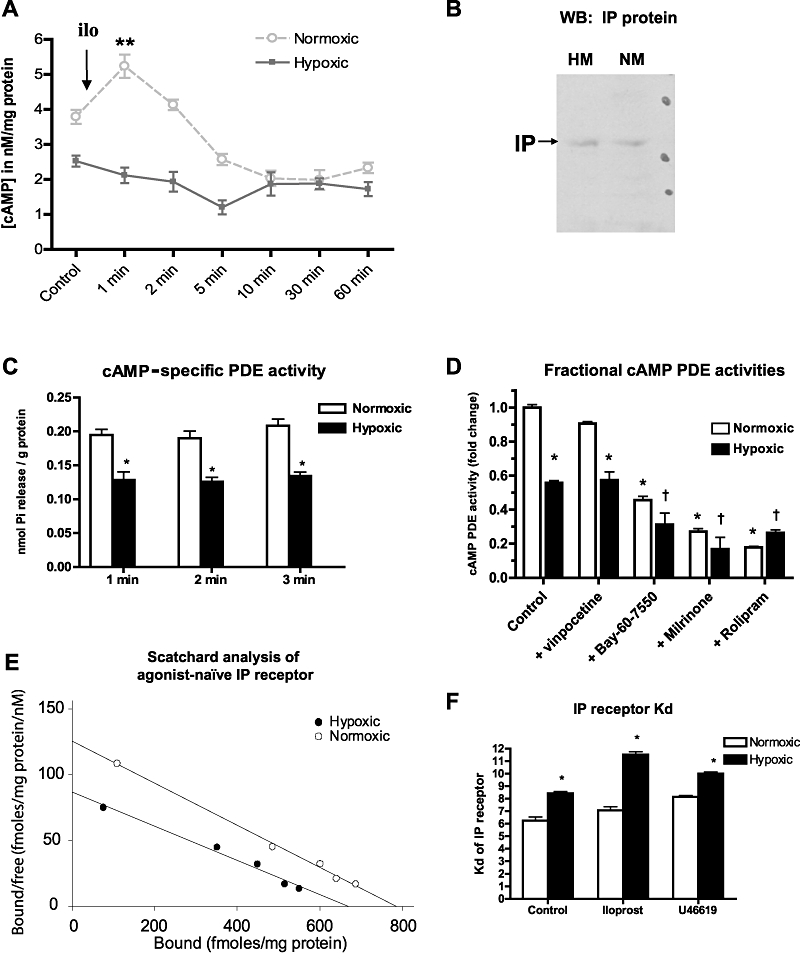
Attenuated IP receptor signalling in hypoxic myocytes is associated with decreased receptor affinity. (A) Stimulation of hypoxic (HM) and normoxic myocytes (NM) with the IP receptor agonist iloprost (ilo) results in cAMP release from NM (mean ± SEM; P < 0.01) but no measurable cAMP release from HM (n = 4). (B) Western blot of NM and HM lysates probed with polyclonal antibody to IP receptors (IP). (C) cAMP-specific PDE activity (as phosphate release from PDE substrate, mean ± SEM) is lower in HM (P < 0.05) compared with NM at all time points. (D) cAMP-specific PDE activity in NM and HM, fractionated by selective inhibition of PDE-1 (vinpocetine, 1 µM), PDE-2 (Bay-60–7550, 1 µM), PDE-3 (milrinone, 1 µM) or PDE-4 (rolipram, 1 µM). Mean ± SEM; n = 6; *P < 0.05 compared with normoxic control; †P < 0.05 compared with hypoxic control. (E) Representative Scatchard plot for IP receptors from agonist-naïve HM and NM cell membrane fractions, using 3H-iloprost. (F) Dissociation constant (Kd in nM) of IP receptors is higher in HM, indicating impaired ligand affinity of IP receptors induced by hypoxia (mean ± SEM; P < 0.05; n = 3 separate experiments). Addition of iloprost further increases Kd (decreases affinity) in HM. Challenge with the TP receptor agonist U46619 has no effect on Kd of IP receptors.
Iloprost cannot normalize IP3 or Ca2+ responses to TP receptor agonist in hypoxic myocytes
Generation of second messenger IP3 was higher in basal and U46619-stimulated hypoxic myocytes than in normoxic myocytes. Pre-treatment with 1 µM iloprost did not attenuate agonist-mediated IP3 release in hypoxic myocytes; however, the cAMP analogue, 8-bromo-cAMP did reduce IP3 release in these cells (Figure 7A). Calcium responses to serial concentrations of U46619 were elevated in hypoxic myocytes compared with those in normoxic myocytes (Figure 7B, C) and were not normalized by iloprost pre-treatment.
Figure 7.
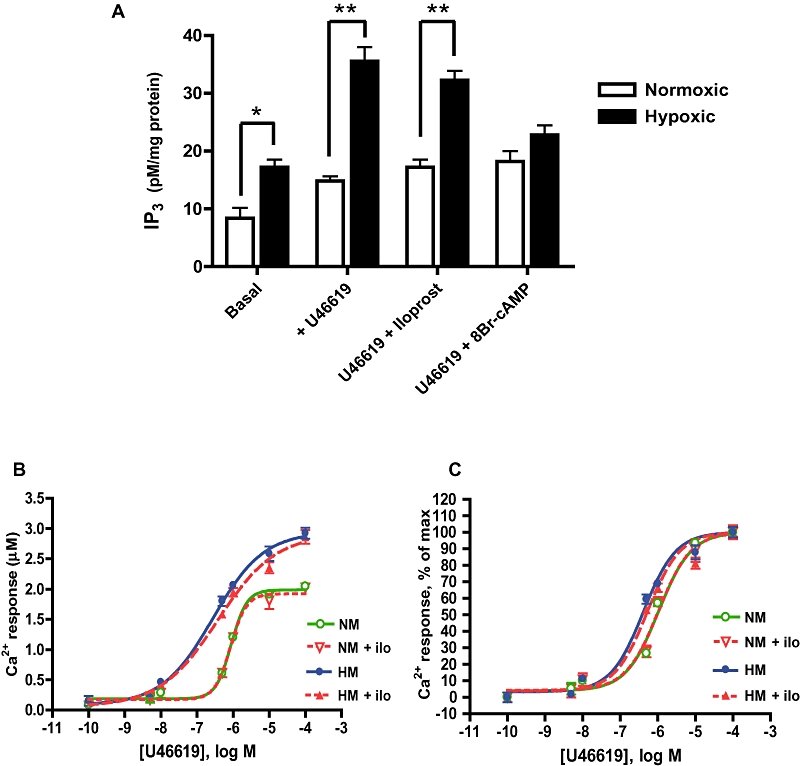
IP receptor stimulation cannot normalize IP3 or Ca2+ responses to TP receptor stimulation in hypoxic myocytes. Hypoxic (HM) and normoxic (NM) myocytes were pre-incubated with 1 µM iloprost or 1 µM 8Br-cAMP prior to U46619 challenge. (A) Second messenger IP3 was higher in basal and U46619-stimulated HM compared with NM (mean ± SEM; *P < 0.01, **P < 0.001). Pre-treatment with the cAMP analogue 8Br-cAMP, but not with iloprost, attenuated IP3 production in HM (P < 0.001, n = 3). (B) In fura-2AM loaded myocytes, iloprost (ilo) pre-treatment had no effect on U46619-Ca2+ dose–response curves in NM or HM and (C) did not alter normalized dose–response curves (data in curves presented as mean ± SD; n = 16).
Effect of milrinone on TP receptor–mediated Ca2+ mobilization
Hypoxic and normoxic myocytes loaded with fura-2AM were pre-incubated with 50 nM milrinone or vehicle, then stimulated with serial concentrations of the thromboxane analogue U46619. Peak calcium mobilization responses were quantified to derive normalized dose–response curves for each experimental condition. Milrinone had no effect on the TP receptor calcium dose–response curve in normoxic myocytes (Figure 8A, B), but it did shift the dose–response curve in hypoxic myocytes markedly to the right. The EC50 for U46619 in hypoxic myocytes treated with milrinone was completely normalized and comparable with previously stated EC50 values for TP receptors in PKA-active myocytes (Figure 8C, D; P < 0.005).
Figure 8.
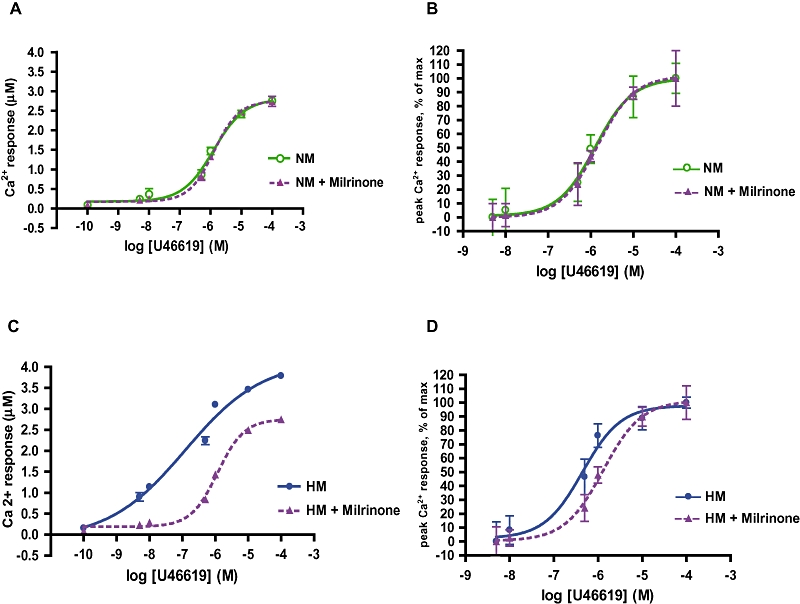
Both hypersensitivity and hyperreactivity of the TP receptors in hypoxic myocytes are normalized by exposure to milrinone. Hypoxic (HM) and normoxic (NM) myocytes loaded with fura-2AM were pre-incubated with 50 nM milrinone or diluent, then stimulated with serial concentrations of U46619 to derive concentration response curves. All curves presented as mean ± SD. (A) Milrinone had no effect on the maximal U46619 response in NM and (B) did not alter receptor sensitivity (NM EC50 1.3 µM, NM + milrinone EC50 1.4 µM, P = ns). (C) Milrinone attenuated the maximal Ca2+ response of HM to U46619 stimulation, and (D) decreased the sensitivity of the U46619 response curve (HM EC50 0.45 µM; HM + milrinone EC50 1.3 µM, P < 0.005; n = 6).
Effects of EP receptor or adenosine A2B receptor stimulation on TP receptor–mediated calcium mobilization
To determine whether the attenuation of IP receptor–mediated regulation of TP receptors represented a more generalized derangement of cAMP generation, or a defect peculiar to the IP receptor, we examined the effects of two other pulmonary arterial Gαs-coupled vasodilator receptors: prostaglandin EP receptors and adenosine A2B receptors. Stimulation of EP receptors, with 11-deoxy prostaglandin E1, normalized the dose–response curve to U46619 in hypoxic myocytes (Figure 9B; EC50 1.2 µM, compared to 0.45 µM in untreated hypoxic myocytes). Stimulation of adenosine A2B receptors with NECA, shifted the dose–response curves to U46619 further to the right in both normoxic myocytes (EC50 18.3 µM) and hypoxic myocytes (EC50, 13 µM); (Figure 9C).
Figure 9.
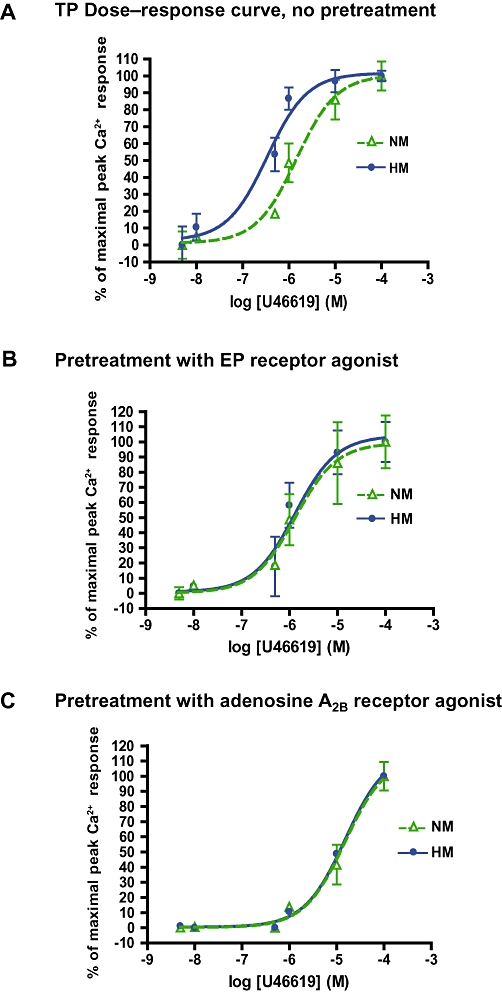
Hypersensitivity of TP receptors in hypoxic myocytes was normalized by activation of non- PGI2 Gαs-coupled receptors. (A) Dose–response curve (percent normalized, curves presented as mean ± SD) for agonist challenge of TP receptors, by fura-2 calciometry; n = 18. NM EC50, 1.5 µM; HM EC50, 0.34 µM. (B) TP receptor dose–response curve following 15 min pre-incubation of NM and HM myocytes with 1 µM 11-deoxy-prostaglandin E1; HM EC50, 1.3 µM. (C) TP dose–response curve following pre-incubation for 15 min with 1 µM NECA (NM EC50, 18.3 µM, HM EC50, 13 µM; n = 16–18).
Discussion
We previously reported sensitization of the response to U46619 in pulmonary arterial myocytes exposed to hypoxia and in myocytes derived from swine with hypoxic PPHN (Hinton et al., 2006); this sensitization was associated with an increase in TP receptor phosphorylation (Hinton et al., 2007). From the present study, we conclude that: (i) TP receptor ligand affinity and TP receptor–mediated signalling were increased by hypoxic exposure, resulting in displacement of the agonist dose–response curve to the left and upwards; (ii) sensitivity of TP receptors was regulated by the state of PKA activation; (iii) TP receptor activity could be normalized by raising cAMP, but not by direct stimulation of hypoxic myocytes with an IP receptor agonist; (iv) loss of regulation of TP receptors is due in part to IP receptor desensitization, which leads to diminished IP receptor signalling capability in hypoxic myocytes; and finally; and (v) TP receptors in hypoxic myocytes can be effectively desensitized by the PDE inhibitor milrinone (Figure 10).
Figure 10.
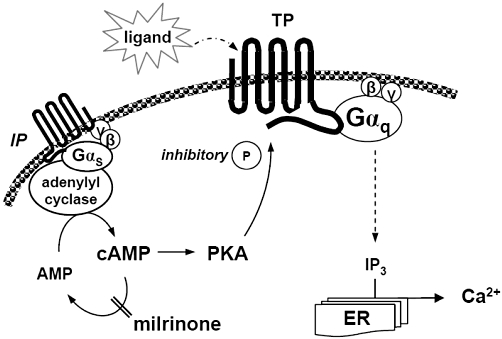
Proposed pathway of TP receptor regulation. We propose that milrinone down-regulates TP receptor affinity by restoring PKA activity in hypoxic myocytes, where IP receptor activity is impaired. ER, endoplasmic reticulum; IP, prostacyclin receptor; TP, thromboxane receptor; IP3, inositol trisphosphate; P, phosphate group; PKA, protein kinase A.
An imbalance between production of TxA2 and PGI2 is implied in the pathogenesis of pulmonary hypertension accompanying hypoxic respiratory failure (Christman et al., 1992) and septic pulmonary hypertension (Hammerman et al., 1988; Ermert et al., 2003). Infants with PPHN have markedly higher serum ratios of TxA2 metabolite to PGE2, indicating a predominance of vasoconstrictor eicosanoid activity (Sood et al., 2007). Pulmonary arterial pressure correlates strongly with plasma TxA2 (Bui et al., 1991a; Bui et al., 1992); increases in TxA2 correspond with worsening post-ductal arterial hypoxemia (Nakayama et al., 1992), while successful treatment of severe PPHN with extracorporeal oxygenation normalizes elevated plasma TxA2 (Bui et al., 1991b). TxA2 underlies the early development of pulmonary arterial constriction in chronic hypoxia (Fike et al., 2002). Both TxA2 inhibition (Fike et al., 2005) and TP receptor blockade (Cathcart et al., 2008) can decrease hypoxic pulmonary hypertension, suggesting that prostanoids are crucial in the pulmonary vascular response to hypoxia.
Our data indicate that hypoxia decreases myocyte sensitivity to PGI2, resulting in a loss of regulatory crosstalk and attenuated intrinsic inhibition of TP receptors. This increases the ability of TP receptors to bind agonist, leading to increased pro-contractile signalling. In hypoxic myocytes, both TP receptor sensitivity (EC50 for U46619) and reactivity (maximal Ca2+ response at receptor saturation) are enhanced. Both aberrations are corrected by forskolin and milrinone (which increase PKA activity). Thromboxane A2, of which U46619 is a mimetic, has an exceedingly short half-life, and its serum concentration varies over a large range. The observed degree of sensitization would significantly increase TP receptor occupancy and, in context of fluctuating blood levels of agonist, result in greater time above the response threshold. In studies of the actions of milrinone in samples of radial artery, a 3.8-fold increase in the EC50 of the α-adrenoceptor agonist phenylephrine was considered clinically very significant (He and Yang, 2000).
TP receptors are phosphorylated at C-terminal serine residues through the action of the pulmonary vasodilator prostanoid PGI2, influencing TP receptor sensitivity (Reid and Kinsella, 2003; O'Meara and Kinsella, 2004). PGI2 activates the IP receptor coupled to adenylyl cyclase (Vane and Botting, 1995). IP–TP receptor interactions, through heterodimerization and regulatory crosstalk, generate both positive and negative feedback balancing their respective signals (Gleim et al., 2009); TP receptor desensitization induced by IP receptor activation is primarily attributed to PKA (Walsh and Kinsella, 2000). We now report that TP receptor regulation via PKA, activated by the IP receptor, tightly controlled the dose–response relationship of TP receptor ligands and that the IP receptor was desensitized (decreased ligand Kd) in hypoxic myocytes. Myocyte PKA activity is decreased during hypoxia. Loss of protective IP receptor signalling in hypoxic myocytes induced TP receptor dysregulation. We conclude that PKA activity was both necessary and sufficient for desensitization of TP receptors. PKA activation in hypoxic myocytes via adenylyl cyclase (but not via IP receptor stimulation) normalized the TP receptor dose–response curve, and inhibition of PKA in normoxic myocytes shifted the dose–-response curve to the left and upwards to resemble that in hypoxic myocytes.
Superimposition of TP receptor agonist response curves from all treatment groups reveals only two curve morphologies and two EC50 value ranges for this receptor, as determined by PKA activation state (i.e. characteristic phospho-TP or non-phospho-TP receptor curves). This finding implies single-point regulation of TP receptor affinity. Robust activation of PKA with 8-bromo-cAMP raises PKA activity in hypoxic and normoxic myocytes by just 25% above the untreated normoxic control. However, this level of activation corresponds to a rise in PKA-dependent phosphorylation sufficient to fully desensitize the TP receptor. PDE-3 inhibition had no additional activating effect on PKA nor any inhibitory effects on Ca2+ mobilization after TP receptor stimulation in normoxic myocytes, suggesting this may reach a phosphorylation maximum or default basal state, below which TP receptor activity cannot be further reduced by phosphorylation. Conversely, inhibition of PKA with H8 reduces the measured PKA activity to half its control level, falling below a threshold level of PKA activity which allows full activation of the TP receptor. In discussing the effects of PKA activation and inhibition, we note that the protein kinase inhibitor H8 used in these studies may be incompletely selective, with some inhibitory activity against PKG and PKC (at higher concentrations) as well as PKA (Chen et al., 2000). However, while we have previously reported phosphorylation of TP receptors by more than one protein kinase, only PKA-mediated phosphorylation measurably altered TP receptor function (Hinton et al., 2007). We have not quantified TP receptor phosphorylation in this study; however, we did fully describe TP receptor dephosphorylation after hypoxic exposure in an earlier study (Hinton et al., 2007). We have thus limited the current study to analysis of upstream regulatory events.
Loss of pulmonary arterial IP receptor activity after prolonged agonist exposure is well described (Schermuly et al., 2007). The rapid onset of IP receptor dysfunction in hypoxic myocytes is a new finding. Agonist exposure of the IP receptor is diminished under hypoxic conditions, as lowered PGI2 and cAMP, as well as increased thromboxane levels, are reported in plasma and cerebrospinal fluid from hypoxic neonates (Liu et al., 2003). While prolonged arterial remodelling in idiopathic pulmonary hypertension may decrease IP receptor expression (Lai et al., 2008), such expression appears unaltered in hypoxic models of pulmonary hypertension (Abe et al., 2001; Hoshikawa et al., 2001). We also report unchanged IP receptor abundance following in vitro hypoxic exposure but a notable decrease in the activity of this receptor.
We describe two important effects of hypoxia on adenylyl cyclase–dependent signalling: (i) lower basal cAMP and PKA activity; and (ii) an attenuated cAMP response to IP receptor stimulation, due to decreased IP receptor–ligand interaction and/or receptor uncoupling. Hypoxia-induced defects in IP receptor–Gαs association and signalling, particularly attenuated receptor–ligand affinity, may cause the loss of IP receptor signal. Our data indicate that activation of the cAMP pathway downstream to the IP receptor (via direct adenylyl cyclase activation by forskolin, or via a stable cAMP analogue) can effectively regulate the TP receptor in hypoxic myocytes, while IP stimulation is ineffective. The decreased cAMP elicited in hypoxic myocytes results from loss of Gαs-linked adenylyl cyclase activation rather than accelerated cAMP degradation, as both resting and stimulated cAMP-specific PDE activity were lower in hypoxic cells. This may appropriately reflect lower basal and/or stimulated cAMP abundance (Tilley and Maurice, 2002). Inhibition of cAMP-specific PDEs increases cAMP, leading to relaxation of arterial and venous smooth muscle via decreased intracellular calcium. In models of pulmonary hypertension, PDE-3 expression is up-regulated and pulmonary artery relaxation enhanced by milrinone (Wagner et al., 1997), while the inhibition of PDE-4 reverses hypertensive pulmonary arterial remodelling (Izikki et al., 2009). In our studies, replacement of PKA activity, either using a cAMP analogue or by PDE-3 inhibition, down-regulates the sensitivity of hypoxic pulmonary arterial myocytes to a TP receptor agonist and restores their normal agonist dose–response curve. Additionally, we report normalization of the TP receptor dose–response relationship following pre-treatment with 11-deoxy-PGE1, a prostanoid EP receptor agonist; and with adenosine-NECA, an adenosine A2B receptor agonist. The ready ability of these unrelated Gαs-coupled receptors to influence hypoxic TP receptor sensitivity suggests that cAMP generation by receptors other than IP receptors is preserved in hypoxic myocytes. The diminished IP receptor affinity observed in hypoxia may at least partly explain loss of stimulated cAMP response. Selective desensitization of other prostanoid receptors has been reported in hypoxic pulmonary arterial myocytes (Millen et al., 2006). Further study of prostanoid receptor coupling and hypoxic regulation of adenylyl cyclase is warranted.
The cAMP pathway has been explored for therapeutic use in nitric oxide-refractory pulmonary hypertension (Rimensberger et al., 2001). Milrinone is a PDE-3 inhibitor and pulmonary artery relaxant, which improves hypoxic pulmonary haemodynamics (Joynt et al., 2008). Its mechanism of action is distinct from that of the well-described PDE-5 inhibitor sildenafil (Zhao et al., 2001; Kirsch et al., 2008). Pulmonary hypertension induced by TP receptor stimulation was alleviated by milrinone, with concurrent improvement of systemic haemodynamics (Gelvez et al., 2004; Lobato et al., 2006). The combination of milrinone and nitric oxide produced greater pulmonary relaxation than either drug alone (Holzmann et al., 2001; Khazin et al., 2004). There is considerable clinical interest in the pulmonary vasodilator effect of milrinone in neonates, as successful use of milrinone has been reported in small cohorts of infants with severe PPHN, refractory to nitric oxide (Bassler et al., 2006; McNamara et al., 2006). The milrinone concentration used in this study to attenuate TP receptor activity was 50 nM; higher doses completely ablated TP receptor responses (data not shown). This concentration elicits near maximal biochemical effect in cardiomyocytes in vitro (Mylotte et al., 1985). Milrinone has direct vasodilator effects at concentrations above 10 nM (Liu et al., 1997), with an EC50 for relaxation of pulmonary arterioles of 100 nM (Jhaveri et al., 2004); its IC50 for selective PDE-3 inhibition is reported to be over a range from 490 nM (Zhao et al., 2007) to as low as 56 nM (Tang et al., 1994). We therefore consider that the observed effect of milrinone on TP receptor responsiveness in pulmonary arterial myocytes may occur in vivo at pharmacological concentrations of milrinone.
In view of the known effects of milrinone in attenuating inflammatory lung injury (Bueltmann et al., 2009) and enhancing relaxation PGI2 (Lakshminrusimha et al., 2009), it is possible that the functional target of milrinone in pulmonary arterial myocytes may involve its interaction with prostanoid receptors. Our data provide the first clear evidence that milrinone may exert its biological effect, at least in part, by ameliorating TP receptor sensitization in hypoxic myocytes (Figure 10).
We conclude that hypoxia influences the balance of signalling in pulmonary arterial prostanoid receptors, favouring vasoconstriction mediated by TP receptors. PDE inhibition may attenuate these effects. In meconium aspiration and sepsis, both PPHN aetiologies marked by thromboxane signalling, treatment strategies that include milrinone may deserve further investigation.
Acknowledgments
This work was funded by Sick Kids Foundation (XG07-031 to SD), Winnipeg Rh Institute Foundation (to SD) and Ikaria Advancing Newborn Medicine fellowship grant (to OE/SD). KTS was supported by a Manitoba Institute of Child Health post-doctoral fellowship. NECA was the kind gift of Dr F. Parkinson (University of Manitoba).
Glossary
Abbreviations
- EP
prostaglandin E receptor
- IP
prostacyclin receptor
- IP3
inositol-1,4,5-trisphosphate
- NECA
adenosine-5′-N-ethylcarboxamide
- PDE
phosphodiesterase
- PKA
protein kinase A
- TP
thromboxane receptor
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–10 as PowerPoint slide.
References
- Abe Y, Tatsumi K, Sugito K, Ikeda Y, Kimura H, Kuriyama T. Effects of inhaled prostacyclin analogue on chronic hypoxic pulmonary hypertension. J Cardiovasc Pharmacol. 2001;37:239–251. doi: 10.1097/00005344-200103000-00002. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler D, Choong K, McNamara P, Kirpalani H. Neonatal persistent pulmonary hypertension treated with milrinone: four case reports. Biol Neonate. 2006;89:1–5. doi: 10.1159/000088192. [DOI] [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Bueltmann M, Kong X, Mertens M, Yin N, Yin J, Liu Z, et al. Inhaled milrinone attenuates experimental acute lung injury. Intensive Care Med. 2009;35:171–178. doi: 10.1007/s00134-008-1344-9. [DOI] [PubMed] [Google Scholar]
- Bui KC, Hammerman C, Hirschl RB, Hill V, Snedecor SM, Schumacher R, et al. Plasma prostanoids in neonates with pulmonary hypertension treated with conventional therapy and with extracorporeal membrane oxygenation. J Thorac Cardiovasc Surg. 1991a;101:973–983. [PubMed] [Google Scholar]
- Bui KC, Hammerman C, Hirschl RB, Hill V, Snedecor SM, Schumacher R, et al. Plasma prostanoids in neonates with pulmonary hypertension treated with conventional therapy and with extracorporeal membrane oxygenation. J Thorac Cardiovasc Surg. 1991b;101:973–983. [PubMed] [Google Scholar]
- Bui KC, Martin G, Kammerman LA, Hammerman C, Hill V, Short BL. Plasma thromboxane and pulmonary artery pressure in neonates treated with extracorporeal membrane oxygenation. J Thorac Cardiovasc Surg. 1992;104:124–129. [PubMed] [Google Scholar]
- Cathcart MC, Tamosiuniene R, Chen G, Neilan TG, Bradford A, O'Byrne KJ, et al. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J Pharmacol Exp Ther. 2008;326:51–58. doi: 10.1124/jpet.107.134221. [DOI] [PubMed] [Google Scholar]
- Chen Y, McCarron RM, Ohara Y, Bembry J, Azzam N, Lenz FA, et al. Human brain capillary endothelium: 2-arachidonoglycerol (endocannabinoid) interacts with endothelin-1. Circ Res. 2000;87:323–327. doi: 10.1161/01.res.87.4.323. [DOI] [PubMed] [Google Scholar]
- Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med. 2000;342:469–474. doi: 10.1056/NEJM200002173420704. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–663. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- Ermert M, Kuttner D, Eisenhardt N, Dierkes C, Seeger W, Ermert L. Cyclooxygenase-2-dependent and thromboxane-dependent vascular and bronchial responses are regulated via p38 mitogen-activated protein kinase in control and endotoxin-primed rat lungs. Lab Invest. 2003;83:333–347. doi: 10.1097/01.lab.0000059924.47118.88. [DOI] [PubMed] [Google Scholar]
- Fike CD, Pfister SL, Kaplowitz MR, Madden JA. Cyclooxygenase contracting factors and altered pulmonary vascular responses in chronically hypoxic newborn pigs. J Appl Physiol. 2002;92:67–74. doi: 10.1152/jappl.2002.92.1.67. [DOI] [PubMed] [Google Scholar]
- Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol. 2003;284:L316–L323. doi: 10.1152/ajplung.00228.2002. [DOI] [PubMed] [Google Scholar]
- Fike CD, Zhang Y, Kaplowitz MR. Thromboxane inhibition reduces an early stage of chronic hypoxia-induced pulmonary hypertension in piglets. J Appl Physiol. 2005;99:670–676. doi: 10.1152/japplphysiol.01337.2004. [DOI] [PubMed] [Google Scholar]
- Finer NN, Barrington KJ. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev. 2006;18:CD000399. doi: 10.1002/14651858.CD000399.pub2. [DOI] [PubMed] [Google Scholar]
- Foley JF, Kelley LP, Kinsella BT. Prostaglandin D(2) receptor-mediated desensitization of the alpha isoform of the human thromboxane A(2) receptor. Biochem Pharmacol. 2001;62:229–239. doi: 10.1016/s0006-2952(01)00661-x. [DOI] [PubMed] [Google Scholar]
- Gelvez J, Fakioglu H, Olarte JL, Soliz A, Totapally BR, Torbati D. Effect of aerosolized milrinone during drug-induced pulmonary hypertension in lambs. Pharmacol Res. 2004;50:87–91. doi: 10.1016/j.phrs.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Gleim S, Kasza Z, Martin K, Hwa J. Prostacyclin receptor/thromboxane receptor interactions and cellular responses in human atherothrombotic disease. Curr Atheroscler Rep. 2009;11:227–235. doi: 10.1007/s11883-009-0035-5. [DOI] [PubMed] [Google Scholar]
- Gong Y, Yi M, Fediuk J, Lizotte PP, Dakshinamurti S. Hypoxic neonatal pulmonary arterial myocytes are sensitized to ROS-Generated 8-Isoprostane. Free Radic Biol Med. 2010;48:882–894. doi: 10.1016/j.freeradbiomed.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Habib A, Vezza R, Creminon C, Maclouf J, FitzGerald GA. Rapid, agonist-dependent phosphorylation in vivo of human thromboxane receptor isoforms. Minimal involvement of protein kinase C. J Biol Chem. 1997;272:7191–7200. doi: 10.1074/jbc.272.11.7191. [DOI] [PubMed] [Google Scholar]
- Hammerman C, Komar K, Abu-Khudair H. Hypoxic vs septic pulmonary hypertension. Selective role of thromboxane mediation. Am J Dis Child. 1988;142:319–325. doi: 10.1001/archpedi.1988.02150030093030. [DOI] [PubMed] [Google Scholar]
- He GW, Yang CQ. Vasorelaxant effect of phosphodiesterase-inhibitor milrinone in the human radial artery used as coronary bypass graft. J Thorac Cardiovasc Surg. 2000;119:1039–1045. doi: 10.1016/S0022-5223(00)70102-4. [DOI] [PubMed] [Google Scholar]
- Hinton M, Mellow L, Halayko AJ, Gutsol A, Dakshinamurti S. Hypoxia induces hypersensitivity and hyperreactivity to thromboxane receptor agonist in neonatal pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;290:L375–L384. doi: 10.1152/ajplung.00307.2005. [DOI] [PubMed] [Google Scholar]
- Hinton M, Gutsol A, Dakshinamurti S. Thromboxane hypersensitivity in hypoxic pulmonary artery myocytes: altered TP receptor localization and kinetics. Am J Physiol Lung Cell Mol Physiol. 2007;292:L654–L663. doi: 10.1152/ajplung.00229.2006. [DOI] [PubMed] [Google Scholar]
- Holzmann A, Manktelow C, Weimann J, Bloch KD, Zapol WM. Inhibition of lung phosphodiesterase improves responsiveness to inhaled nitric oxide in isolated-perfused lungs from rats challenged with endotoxin. Intensive Care Med. 2001;27:251–257. doi: 10.1007/s001340000774. [DOI] [PubMed] [Google Scholar]
- Hoshikawa Y, Voelkel NF, Gesell TL, Moore MD, Morris KG, Alger LA, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314–318. doi: 10.1164/ajrccm.164.2.2010150. [DOI] [PubMed] [Google Scholar]
- Izikki M, Raffestin B, Klar J, Hatzelmann A, Marx D, Tenor H, et al. Effects of Roflumilast, a phosphodiesterase-4 inhibitor on hypoxia- and monocrotaline-induced pulmonary hypertension in rats. J Pharmacol Exp Ther. 2009;330:54–62. doi: 10.1124/jpet.108.148742. [DOI] [PubMed] [Google Scholar]
- Jhaveri R, Kim S, White AR, Burke S, Berkowitz DE, Nyhan D. Enhanced vasodilatory responses to milrinone in catecholamine-precontracted small pulmonary arteries. Anesth Analg. 2004;98:1618–1622. doi: 10.1213/01.ANE.0000115781.69209.59. table of contents. [DOI] [PubMed] [Google Scholar]
- Joynt C, Bigam DL, Charrois G, Jewell LD, Korbutt G, Cheung PY. Dose-response effects of milrinone on hemodynamics of newborn pigs with hypoxia-reoxygenation. Intensive Care Med. 2008;34:1321–1329. doi: 10.1007/s00134-008-1060-5. [DOI] [PubMed] [Google Scholar]
- Kelley-Hickie LP, Kinsella BT. EP1- and FP-mediated cross-desensitization of the alpha (alpha) and beta (beta) isoforms of the human thromboxane A2 receptor. Br J Pharmacol. 2004;142:203–221. doi: 10.1038/sj.bjp.0705695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazin V, Kaufman Y, Zabeeda D, Medalion B, Sasson L, Schachner A, et al. Milrinone and nitric oxide: combined effect on pulmonary artery pressures after cardiopulmonary bypass in children. J Cardiothorac Vasc Anesth. 2004;18:156–159. doi: 10.1053/j.jvca.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Kirsch M, Kemp-Harper B, Weissmann N, Grimminger F, Schmidt HH. Sildenafil in hypoxic pulmonary hypertension potentiates a compensatory up regulation of NO-cGMP signaling. Faseb J. 2008;22:30–40. doi: 10.1096/fj.06-7526com. [DOI] [PubMed] [Google Scholar]
- Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, et al. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Mol Biol Cell. 2009;20:4751–4765. doi: 10.1091/mbc.E09-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, et al. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics. 2004;113:559–564. doi: 10.1542/peds.113.3.559. [DOI] [PubMed] [Google Scholar]
- Lai YJ, Pullamsetti SS, Dony E, Weissmann N, Butrous G, Banat GA, et al. Role of the prostanoid EP4 receptor in iloprost-mediated vasodilatation in pulmonary hypertension. Am J Respir Crit Care Med. 2008;178:188–196. doi: 10.1164/rccm.200710-1519OC. [DOI] [PubMed] [Google Scholar]
- Lakshminrusimha S, Porta NF, Farrow KN, Chen B, Gugino SF, Kumar VH, et al. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2009;10:106–112. doi: 10.1097/PCC.0b013e3181936aee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chang L, Chen Y, Xia S, Zhang X. Clinical implication of the changes of cAMP, TXA2 and PGI2 in CSF of asphyxiated newborns. J Huazhong Univ Sci Technolog Med Sci. 2003;23:195–197. doi: 10.1007/BF02859956. 200. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Doolan LA, Xie B, Chen JR, Buxton BF. Direct vasodilator effect of milrinone, an inotropic drug, on arterial coronary bypass grafts. FANZCA. J Thorac Cardiovasc Surg. 1997;113:108–113. doi: 10.1016/s0022-5223(97)70405-7. [DOI] [PubMed] [Google Scholar]
- Lobato EB, Beaver T, Muehlschlegel J, Kirby DS, Klodell C, Sidi A. Treatment with phosphodiesterase inhibitors type III and V: milrinone and sildenafil is an effective combination during thromboxane-induced acute pulmonary hypertension. Br J Anaesth. 2006;96:317–322. doi: 10.1093/bja/ael009. [DOI] [PubMed] [Google Scholar]
- McNamara PJ, Laique F, Muang-In S, Whyte HE. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care. 2006;21:217–222. doi: 10.1016/j.jcrc.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Santicioli P, Giuliani S. Protein kinase A inhibitors selectively inhibit the tonic contraction of the guinea pig ureter to high potassium. Gen Pharmacol. 1996;27:341–348. doi: 10.1016/0306-3623(95)00103-4. [DOI] [PubMed] [Google Scholar]
- Millen J, MacLean MR, Houslay MD. Hypoxia-induced remodelling of PDE4 isoform expression and cAMP handling in human pulmonary artery smooth muscle cells. Eur J Cell Biol. 2006;85:679–691. doi: 10.1016/j.ejcb.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Mylotte KM, Cody V, Davis PJ, Davis FB, Blas SD, Schoenl M. Milrinone and thyroid hormone stimulate myocardial membrane Ca2+-ATPase activity and share structural homologies. Proc Natl Acad Sci USA. 1985;82:7974–7978. doi: 10.1073/pnas.82.23.7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama DK, Motoyama EK, Evans R, Hannakan C. Relation between arterial hypoxemia and plasma eicosanoids in neonates with congenital diaphragmatic hernia. J Surg Res. 1992;53:615–620. doi: 10.1016/0022-4804(92)90263-y. [DOI] [PubMed] [Google Scholar]
- O'Meara SJ, Kinsella BT. Investigation of the effect of the farnesyl protein transferase inhibitor R115777 on isoprenylation and intracellular signalling by the prostacyclin receptor. Br J Pharmacol. 2004;143:318–330. doi: 10.1038/sj.bjp.0705956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JL, Labrecque P, Orsini MJ, Benovic JL. Internalization of the TXA2 receptor alpha and beta isoforms. Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J Biol Chem. 1999;274:8941–8948. doi: 10.1074/jbc.274.13.8941. [DOI] [PubMed] [Google Scholar]
- Peacock AJ, Scott P, Plevin R, Wadsworth R, Welsh D. Hypoxia enhances proliferation and generation of IP3 in pulmonary artery fibroblasts but not in those from the mesenteric circulation. Chest. 1998;114(Suppl 1):24S. doi: 10.1378/chest.114.1_supplement.24s. [DOI] [PubMed] [Google Scholar]
- Phillips PG, Long L, Wilkins MR, Morrell NW. cAMP phosphodiesterase inhibitors potentiate effects of prostacyclin analogs in hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2005;288:L103–L115. doi: 10.1152/ajplung.00095.2004. [DOI] [PubMed] [Google Scholar]
- Reid HM, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for nitric oxide-mediated desensitization. Independent modulation of Tp alpha signaling by nitric oxide and prostacyclin. J Biol Chem. 2003;278:51190–51202. doi: 10.1074/jbc.M309314200. [DOI] [PubMed] [Google Scholar]
- Reyes JL. Arachidonic acid metabolites and haemodynamics of the neonate. Pediatr Nephrol. 1993;7:841–844. doi: 10.1007/BF01213371. [DOI] [PubMed] [Google Scholar]
- Rimensberger PC, Spahr-Schopfer I, Berner M, Jaeggi E, Kalangos A, Friedli B, et al. Inhaled nitric oxide versus aerosolized iloprost in secondary pulmonary hypertension in children with congenital heart disease: vasodilator capacity and cellular mechanisms. Circulation. 2001;103:544–548. doi: 10.1161/01.cir.103.4.544. [DOI] [PubMed] [Google Scholar]
- Schermuly RT, Pullamsetti SS, Breitenbach SC, Weissmann N, Ghofrani HA, Grimminger F, et al. Iloprost-induced desensitization of the prostacyclin receptor in isolated rabbit lungs. Respir Res. 2007;8:4. doi: 10.1186/1465-9921-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;279:L884–L894. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, et al. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol. 2008;104:110–118. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- Sobolewski A, Jourdan KB, Upton PD, Long L, Morrell NW. Mechanism of cicaprost-induced desensitization in rat pulmonary artery smooth muscle cells involves a PKA-mediated inhibition of adenylyl cyclase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L352–L359. doi: 10.1152/ajplung.00270.2003. [DOI] [PubMed] [Google Scholar]
- Sood BG, Delaney-Black V, Glibetic M, Aranda JV, Chen X, Shankaran S. PGE2/TXB2 imbalance in neonatal hypoxemic respiratory failure. Acta Paediatr. 2007;96:669–673. doi: 10.1111/j.1651-2227.2007.00237.x. [DOI] [PubMed] [Google Scholar]
- Tang KM, Jang EK, Haslam RJ. Photoaffinity labelling of cyclic GMP-inhibited phosphodiesterase (PDE III) in human and rat platelets and rat tissues: effects of phosphodiesterase inhibitors. Eur J Pharmacol. 1994;268:105–114. doi: 10.1016/0922-4106(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Maurice DH. Vascular smooth muscle cell phosphodiesterase (PDE) 3 and PDE4 activities and levels are regulated by cyclic AMP in vivo. Mol Pharmacol. 2002;62:497–506. doi: 10.1124/mol.62.3.497. [DOI] [PubMed] [Google Scholar]
- Vane JR, Botting RM. Pharmacodynamic profile of prostacyclin. Am J Cardiol. 1995;75:3A–10A. doi: 10.1016/s0002-9149(99)80377-4. [DOI] [PubMed] [Google Scholar]
- Wagner RS, Smith CJ, Taylor AM, Rhoades RA. Phosphodiesterase inhibition improves agonist-induced relaxation of hypertensive pulmonary arteries. J Pharmacol Exp Ther. 1997;282:1650–1657. [PubMed] [Google Scholar]
- Walsh MT, Kinsella BT. Regulation of the human prostanoid TPalpha and TPbeta receptor isoforms mediated through activation of the EP(1) and IP receptors. Br J Pharmacol. 2000;131:601–609. doi: 10.1038/sj.bjp.0703624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MT, Foley JF, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for prostacyclin-mediated desensitization. J Biol Chem. 2000;275:20412–20423. doi: 10.1074/jbc.M907881199. [DOI] [PubMed] [Google Scholar]
- Weinberger B, Weiss K, Heck DE, Laskin DL, Laskin JD. Pharmacologic therapy of persistent pulmonary hypertension of the newborn. Pharmacol Ther. 2001;89:67–79. doi: 10.1016/s0163-7258(00)00104-2. [DOI] [PubMed] [Google Scholar]
- Zhao L, Mason NA, Morrell NW, Kojonazarov B, Sadykov A, Maripov A, et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104:424–428. doi: 10.1161/hc2901.093117. [DOI] [PubMed] [Google Scholar]
- Zhao H, Quilley J, Montrose DC, Rajagopalan S, Guan Q, Smith CJ. Differential effects of phosphodiesterase PDE-3/PDE-4-specific inhibitors on vasoconstriction and cAMP-dependent vasorelaxation following balloon angioplasty. Am J Physiol Heart Circ Physiol. 2007;292:H2973–H2981. doi: 10.1152/ajpheart.00419.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.