

Abstract
BACKGROUND AND PURPOSE
High plasma levels of fenretinide [N-(4-hydroxyphenyl)retinamide (4-HPR)] were associated with improved outcome in a phase II clinical trial. Low bioavailability of 4-HPR has been limiting its therapeutic applications. This study characterized metabolism of 4-HPR in humans and mice, and to explore the effects of ketoconazole, an inhibitor of CYP3A4, as a modulator to increase 4-HPR plasma levels in mice and to increase the low bioavailability of 4-HPR.
EXPERIMENTAL APPROACH
4-HPR metabolites were identified by mass spectrometric analysis and levels of 4-HPR and its metabolites [N-(4-methoxyphenyl)retinamide (4-MPR) and 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR)] were quantified by high-performance liquid chromatography (HPLC). Kinetic analysis of enzyme activities and the effects of enzyme inhibitors were performed in pooled human and pooled mouse liver microsomes, and in human cytochrome P450 (CYP) 3A4 isoenzyme microsomes. In vivo metabolism of 4-HPR was inhibited in mice.
KEY RESULTS
Six 4-HPR metabolites were identified in the plasma of patients and mice. 4-HPR was oxidized to 4-oxo-4-HPR, at least in part via human CYP3A4. The CYP3A4 inhibitor ketoconazole significantly reduced 4-oxo-4-HPR formation in both human and mouse liver microsomes. In two strains of mice, co-administration of ketoconazole with 4-HPR in vivo significantly increased 4-HPR plasma concentrations by > twofold over 4-HPR alone and also increased 4-oxo-4-HPR levels.
CONCLUSIONS AND IMPLICATIONS
Mice may serve as an in vivo model of human 4-HPR pharmacokinetics. In vivo data suggest that the co-administration of ketoconazole at normal clinical doses with 4-HPR may increase systemic exposure to 4-HPR in humans.
Keywords: fenretinide, metabolism, ketoconazole, paediatric cancers
Introduction
Fenretinide [N-(4-hydroxyphenyl)retinamide; 4-HPR] is a synthetic analogue of all-trans retinoic acid (ATRA) that exhibits cytotoxic activity against a variety of human cancer cell lines in vitro at concentrations of 1–10 µM (Delia et al., 1993; Mariotti et al., 1994; Kalemkerian et al., 1995; Oridate et al., 1996; O'Donnell et al., 2002). 4-HPR has also been studied clinically both as a chemopreventive agent in breast (Veronesi et al., 1999), bladder (Sabichi et al., 2008) and oral mucosal cancers (Chiesa et al., 2005), and more recently as a chemotherapeutic agent in paediatric (Garaventa et al., 2003; Villablanca et al., 2006) and adult cancers (Puduvalli et al., 2004; Vaishampayan et al., 2005; Reynolds et al., 2007). Despite substantial in vitro cytotoxicity, response rates in clinical trials with 4-HPR have been less than anticipated, probably due to the low bioavailability of the oral capsule formulation employed (Cheng et al., 2001; Puduvalli et al., 2004; Vaishampayan et al., 2005; Reynolds et al., 2007; William et al., 2009).
After a single 10 mg·kg−1 dose of the capsules, the oral bioavailability of 4-HPR was approximately 16% in beagle dogs (Liu et al., 2007). In chemoprevention studies utilizing chronic, low-dose schedules (100–400 mg daily), 4-HPR plasma concentrations were ≤3 µM (Formelli and Cleris, 1993). A paediatric phase I study in neuroblastoma patients achieved a mean 4-HPR plasma level of 9.9 µM at the maximum tolerated dose, 2450 mg·m−2 per day (Villablanca et al., 2006). In adults, high dose schedules (1800 mg·m−2 per day) have achieved 4-HPR plasma concentrations of 5 to 7 µM with wide interpatient variation (Jasti et al., 2001; Garcia et al., 2004). Pharmacokinetic data in both children and adults have suggested that gastrointestinal absorption was limited at higher doses during phase I studies. New oral and intravenous formulations of 4-HPR have been developed to improve bioavailability of the drug (Liu et al., 2007; Maurer et al., 2007), and are currently being tested in phase I trials in paediatric and adult malignancies (Mohrbacher et al., 2007; Marachelian et al., 2009). The interim results from these trials suggest that increased clinical activity accompanies increased systemic exposure. Complete responses were observed in 4 of 18 neuroblastoma patients at the highest five dosing levels tested with an improved oral powder formulation [4-HPR/Lym-X-Sorb (LXS)™ oral powder] (Marachelian et al., 2009), and a 50% response rate (partial + complete) in 10 patients with relapsed T-cell lymphomas was observed using a novel intravenous emulsion 4-HPR formulation (MH Kang et al., unpublished data).
An alternative approach to dose escalation to increase systemic 4-HPR levels is pharmacological modulation of 4-HPR metabolism. However, before mouse models can be used for pharmacokinetic studies of 4-HPR pharmacological modulation, differences in drug metabolism, if any, between humans and mice need to be defined. In rodents, 4-HPR is primarily metabolized to N-(4-methoxyphenyl)retinamide (4-MPR) (Hultin et al., 1986), whereas in humans, 4-HPR metabolism to 4-MPR and an additional metabolite, 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR), have been reported (Villani et al., 2004). 4-MPR is reported to be the most abundant metabolite of 4-HPR in both rodent and human plasma, with 4-oxo-4-HPR being present at lower concentrations in humans (Villani et al., 2004). 4-MPR is non-cytotoxic in numerous malignant cell lines, in vitro (Mehta et al., 1998; Appierto et al., 2001; Villani et al., 2006), while 4-oxo-4-HPR was two- to fourfold more cytotoxic than 4-HPR in solid tumor cell lines (Villani et al., 2006). While individual metabolites have been reported both in humans and rodent models, the metabolic pathways for 4-HPR in each species still remain poorly delineated.
Several cytochrome P450 (CYP) enzymes, including CYP3A4, 2C8, 2C9 and 26A1, are reported to metabolize ATRA into polar derivatives such as 4-oxo-RA (Thatcher, 2005), and these observations have been used to make predictions concerning metabolic pathways for 4-HPR. Villani et al. (2004) reported that CYP26A1 catalyzed the formation of 4-oxo-4-HPR from 4-HPR in a human ovarian carcinoma cell line, in vitro. Also, in mice pretreated with the CYP enzyme inducer phenobarbital prior to 4-HPR administration, hepatic CYP enzyme levels were increased by threefold and 4-HPR concentrations in plasma and tissues were decreased by 50% (Hultin et al., 1986). Triazole antifungal agents are known to inhibit members of the CYP family of enzymes and to both decrease the levels of polar metabolites of ATRA and prolong the biological half-life of ATRA (Van Wauwe et al., 1988; 1990;). These observations suggest that inhibition of the mixed function oxidase system using triazoles could increase systemic 4-HPR levels. A similar approach employing concurrent ketoconazole, a triazole antifungal agent, was initially successful in increasing ATRA plasma levels for 1 or 2 days but failed to prevent the decrease of ATRA plasma levels at the end of a 2 week treatment course, possibly because the ATRA-induced CYP enzyme was not completely inhibited by ketoconazole over the extended treatment period (Lee et al., 1995). However, unlike ATRA, where auto-induction of metabolism leads to a significant decrease in half-life and drug exposures during therapy (Muindi et al., 1994), 4-HPR exposures are relatively constant during treatment courses and do not show evidence of auto-induction of metabolism (Formelli and Cleris, 1993). Thus, in contrast to experiences with ATRA, we hypothesized that the use of triazoles to inhibit specific CYP enzymes that metabolize 4-HPR would increase systemic 4-HPR concentrations.
The purpose of this study was therefore to characterize and compare inter-species differences in the metabolism of 4-HPR in humans and mice, and to determine whether pharmacological modulation of the metabolism of 4-HPR would result in higher 4-HPR plasma concentrations in mouse models. Our pharmacokinetic analyses of plasma from 4-HPR-treated patients and mice identified six common metabolites of 4-HPR in humans and mice. We also report in vitro and in vivo metabolism data suggesting that the inhibition of CYP3A4 using ketoconazole could result in higher systemic 4-HPR concentrations clinically.
Methods
Patients
All patients involved in this study gave written informed consent and the study received Institutional Review Board approval. Patients whose plasma samples were analyzed for 4-HPR and metabolites were participating in a New Approaches to Neuroblastoma Therapy (NANT) Consortium (http://www.NANT.org), phase I trial (Study N2004-04) for the treatment of relapsed and refractory high-risk neuroblastoma using 4-HPR delivered in an oral lipid matrix powder (4-HPR/LXS oral powder, 4-HPR/LXS). Trial design and interim results have been reported (Marachelian et al., 2009). Briefly, the trial enrolled 30 evaluable patients to eight dose levels (range 352–2210 mg·m−2 per day 4-HPR). 4-HPR/LXS was administered twice daily for one week every 21 days (one cycle of treatment). The 4-HPR plasma levels were measured for three specific patients treated at the 595 mg·m−2 per day dose level. Venous blood samples (5 mL) were taken from the patients on day 6 of cycle 1 just before one daily dose (i.e. 12 h since the previous dose on day 5) and for the next 8 h at 2 h intervals. Patient blood samples were collected in foil-wrapped heparinized tubes, centrifuged immediately and plasma was retained and stored at −80○C until analysis. All procedures were carried out in amber tubes and/or in the dark to prevent light exposure.
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee. Five- to 6 week old non-obese diabetic/severe combined immune deficiency (NOD/SCID) and athymic nude (nu/nu) mice were obtained from Charles River Laboratories International (Wilmington, MA, USA). For each experiment, 4-HPR/LXS, 4-HPR/LXS + ketoconazole, or the LXS powder alone (negative control) was slurried in sterile water and administered via gavage. Exact dosing and number of doses before pharmacokinetic analysis are given in the legend to Figure 4. After mice were anesthetized with isoflurane, blood samples (approximately 500 µL) for pharmacokinetic studies were collected by cardiac puncture and treated as described above for human samples. Mice were then killed by carbon dioxide narcosis.
Figure 4.
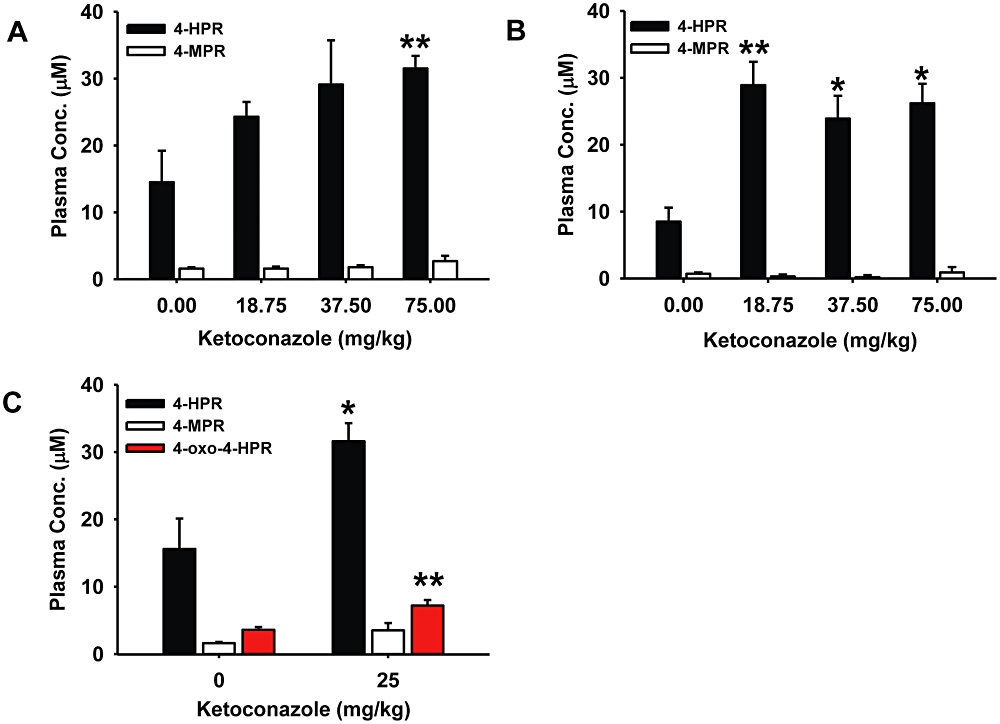
Co-administration of ketoconazole increased systemic N-(4-hydroxyphenyl)retinamide (4-HPR) levels in mice in vivo. (A) 4-HPR and N-(4-methoxyphenyl)retinamide (4-MPR) levels in plasma of nu/nu mice given 4-HPR with increasing doses of ketoconazole. The mice were killed 4 h after the ninth dose. (B) 4-HPR and 4-MPR levels in plasma of non-obese diabetic/severe combined immune deficiency (NOD/SCID) mice given 4-HPR with increasing doses of ketoconazole. Assay times were the same as described for Figure 4A. (C) 4-HPR, 4-MPR and 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR) levels in plasma of NOD/SCID mice given 4-HPR alone or with ketoconazole (25 mg·kg−1). Mice in both groups were killed 4 h after the fifth dose. In panels A–C, all mice received 4-HPR (180 mg·kg−1 per day) gavaged in two divided doses; control mice for all experiments received 4-HPR without ketoconazole. Cohorts of three mice were used for each condition and columns are the mean and error/vertical bars are standard deviation from experiments performed in triplicate. *P < 0.02, **P < 0.01.
High-performance liquid chromatography (HPLC) and tandem mass spectrometry (MS/MS) analysis
Levels of 4-HPR, 4-MPR and 4-oxo-4-HPR were quantified using an HPLC assay (Vratilova et al., 2004) with the following modifications: HPLC was an Agilent 1200 system (Agilent Technologies, Palo Alto, CA, USA); column was an Agilent Zorbax Eclipse reverse-phase C18 150 × 4.6 mm, 5 µm; gradient elution with 0.01 M ammonium acetate in water and methanol was used at 1 mL·min−1; 4-oxo-4-HPR standard was added; calibration curve concentration range from 0.019 to 40.0 µg·mL−1; injection volume into the HPLC was 30 µL; and 100 µL of each plasma sample was used for extraction and analysis. Data were acquired and integrated by ChemStation 3D software (Agilent).
Metabolites were identified using LC/MS/MS. An Agilent 1200 HPLC was used with an Applied Biosystems 4000 QTRAP MS/MS (Foster City, CA, USA) operated in electrospray positive ion mode. For sample preparation, the internal standard [N-(4-ethoxyphenyl)retinamide (4-EPR)] was added to 50 µL of plasma and 100 µL of acetonitrile was added, vortexed and centrifuged at 9300×g for 5 min. Supernatants were then collected and 130 µL of water with formic acid (0.1%) was added to adjust the acetonitrile concentration to approximately 30% by volume. Ten µL of supernatant with adjusted composition was then injected into the LC/MS/MS. A Waters Corporation reverse-phase Symmetry C18 column 150 × 4.6 mm, 3.5 µm (Milford, MA, USA) was used. The mobile phase consisted of acetonitrile and water with formic acid (0.1%) and was delivered via gradient at 1.0 mL·min−1 for 35 min. The initial mobile phase conditions of 30:70 (v/v, acetonitrile : aqueous formic acid) were held for 2 min. Concentrations were then changed to 95:5 over 23 min and were held for 3 min. Then at 28 min, concentrations adjusted back to initial conditions over 2 min and were held for 5 min. The system was operated in an information-dependent acquisition mode (threshold: 1000 cps), which was set to include an MS3 experiment following enhanced product ion spectrum for the parent ion in the transition. Ion source conditions were: curtain gas 25; collision gas Low; spray voltage 5000; temperature 700○C; source gas1 50; source gas2 50. Analyst (version 1.4.2) and LightSight Metabolite ID software (both from Applied Biosystems) were used for data acquisition and processing.
Human (HLMs) and mouse liver microsomes (MLMs)
4-HPR and 4-MPR metabolism assays were performed using 0.5 mg protein·mL−1 of pooled HLMs or pooled MLMs, or human CYP3A4 isoenzyme microsomes (BD Biosciences, Franklin Lakes, NJ, USA). The NADPH regenerating system consisted of solutions ‘A’ and ‘B’ (BD Gentest catalog no. 451220 and 451200). Inhibitors of CYP2C8 (gemfibrozil) (Scheen, 2007), CYP2C9 (fluconazole) (Nivoix et al., 2008) and CYP3A4 (ketoconazole) (Jia and Liu, 2007) were dissolved in methanol at stock concentrations up to 5 mM. Solutions ‘A’ and ‘B’, liver microsomes and inhibitors were combined in 0.1 M potassium phosphate buffer (pH 7.4), and incubated for 20 min at 37°C. Substrates were then added and the mixture was incubated while shaking in a 37°C water bath. When incubation times were completed, reactions were immediately terminated by addition of ice-cold acetonitrile fortified with internal standard (4-EPR), vortexed for 1 min and centrifuged at 2917×g for 10 min. Supernatants were collected and analyzed via HPLC as described earlier.
Cell lines
Human acute lymphoblastic leukemia (ALL) cell lines COG-LL-317, -332 and -329 (all T-cell ALL); COG-LL-319 and -355 (both Pre-B ALL); and COG-LL-356 (Pro-B ALL); and neuroblastoma lines CHLA-90 and -136 were obtained from the Children's Oncology Group Cell Culture Repository (http://www.COGcell.org). Cells were cultured in Iscove's Modified Dulbecco's Medium (Cambrex, Walkersville, MD, USA) supplemented with 3 mM L-glutamine, 5 µg·mL−1 insulin and 20% heat-inactivated fetal bovine serum (FBS). The human Pre-B ALL cell line NALM-6 (from Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany); and human cell lines CCRF-CEM, MOLT-3 and MOLT-4 (all T-cell ALL), RS4;11 (Pre-B ALL), GA-10 (B-cell Burkitt lymphoma), HuT-78 (cutaneous T-cell lymphoma) and Toledo (B-cell non-Hodgkin lymphoma), all from American Type Culture Collection, Manassas, VA, USA, were maintained in RPMI-1640 (Mediatech Inc., Herdon, VA, USA) supplemented with 10% heat-inactivated FBS. All lines tested mycoplasma-free. Lymphoid cell lines were maintained at 37○C in humidified incubators containing 5% O2, 5% CO2 and 90% N2, and neuroblastoma lines were maintained at 37○C in humidified incubators containing 95% room air and 5% CO2. Cell line identities were confirmed after each expansion but prior to freezing by short tandem repeat (STR) genotyping using Ampf/STRTM Identifier TM kit (Applied Biosystems) (Masters et al., 2001) and compared with the Children's Oncology Group STR database (http://www.COGcell.org). STR profiles were performed in all cell lines for cell line identification one month before the experiments.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR was performed using an Applied Biosystems Prism 7900HT system to quantify basal gene expression of CYP26A1 in each of fourteen malignant lymphoid cell lines and two neuroblastoma cell lines. The reagents, assay conditions and data normalization methods were as previously described (Kang et al., 2008). Primers and probes for CYP26A1 (TaqMan Gene Expression Assay proprietary sequences, P/N 4331102; Applied Biosystems) were added according to the manufacturer's instructions. Total RNA added per well was 300 ng for all cell lines except CCRF-CEM, for which 150 ng was added because of its apparently high level of CYP26A1 mRNA.
Statistical analysis
Statistical evaluation was performed using Student's t-test. P-values were two sided and tests were considered significant at P < 0.05. All in vitro experiments were performed in triplicate and were consistently repeatable; for simplicity, one representative experiment for each condition is shown.
Materials
4-HPR, 4-MPR and 4-EPR were supplied by the Developmental Therapeutics Program of the National Cancer Institute (NCI, Bethesda, MD, USA). 4-oxo-4-HPR was synthesized as previously described (Villani et al., 2004). 4-HPR/LXS™ oral powder and LXS™ oral powder was from Avanti Polar Lipids, Inc, Alabaster, AL, USA, manufactured under agreement with BioMolecular Products, Newburyport, MA, USA, and the formulation was supported by NCI Rapid Access to Intervention Development Program. Ketoconazole, gemfibrozil, and fluconazole were from Sigma-Aldrich (St Louis, MO, USA). Potassium phosphate monobasic, acetonitrile, methanol and formic acid were from Fisher Scientific (Pittsburgh, PA, USA).
Results
Identification of 4-HPR metabolites in the plasma of 4-HPR-treated patients and mice
To identify metabolites of 4-HPR, we used LC/MS/MS to analyze plasma samples from 4-HPR-treated patients and mice. HPLC chromatograms of patient plasma and mouse plasma are shown in Figure 1A left and right panels respectively. 4-HPR eluted at approximately 25.0 min and the detected metabolites had retention times ranging from 7.6 to 27.5 min. In both species, seven major peaks (peak area ratio > 0.04 relative to the parent drug in humans and >0.12 relative to the parent drug in mice) representing different 4-HPR metabolites were detected, M1-M7 in mouse plasma and H2-H8 in human plasma. Six of these seven peaks, M2–M7 in mouse plasma and H2–H7 in human plasma, were eluted at similar retention times (i.e. were metabolites, common to both species).
Figure 1.
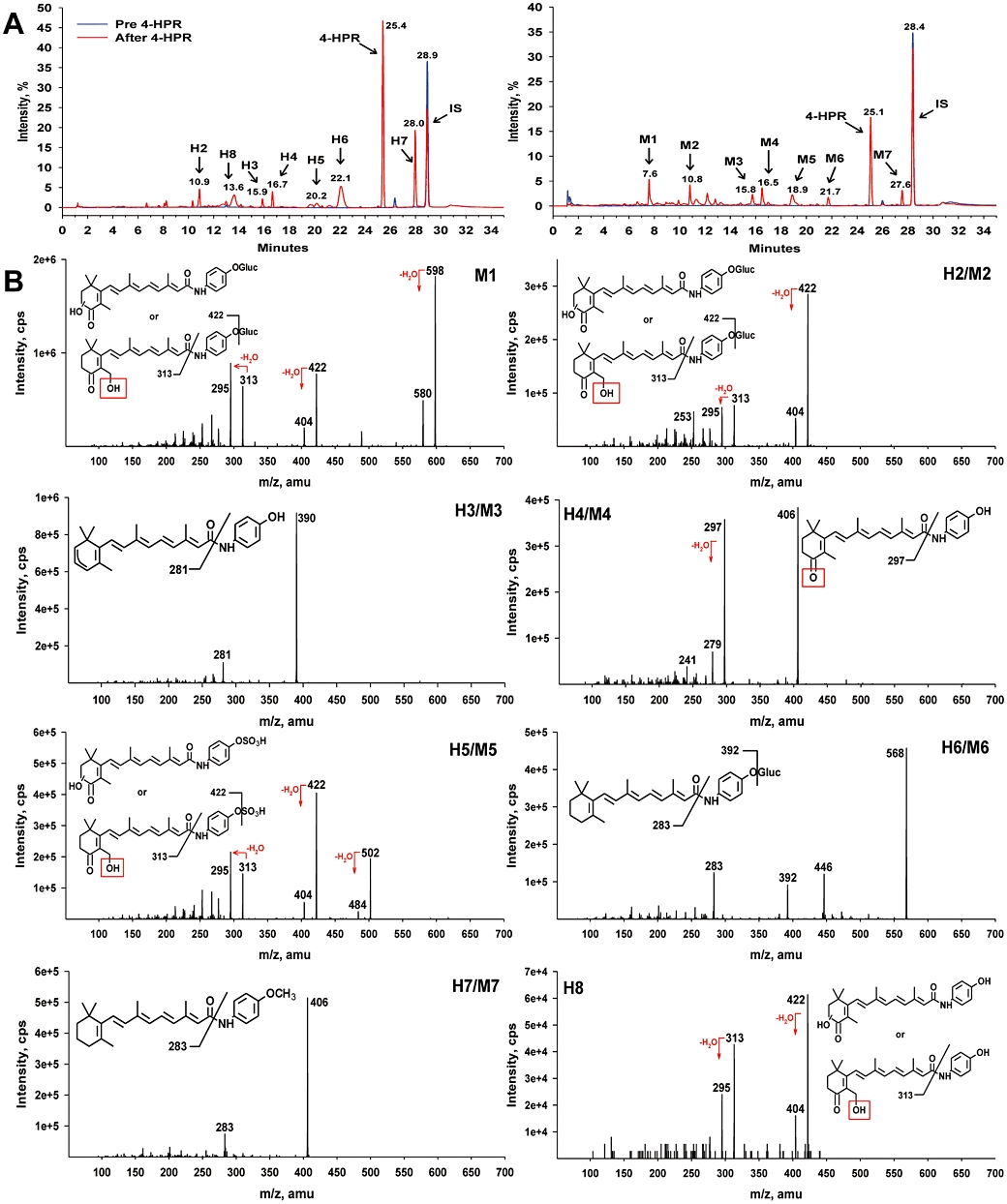
Identification of N-(4-hydroxyphenyl)retinamide (4-HPR) metabolites in plasma of 4-HPR-treated patients and mice. (A) HPLC chromatograms showing metabolite peaks in humans (H2–H8, left) and mice (M1–M7, right). Pre-4-HPR (blue), after 4-HPR (red). (B) Structural identification of H2–H8 and M1–M7 via tandem mass spectrometry (MS/MS). Dehydration that occurred during fragmentation of M1, H2/M2, H5/M5 and H8 is likely to involve the hydroxyl (-OH) group highlighted with red box.
The results of MS/MS analysis of patient and mouse plasma are shown in Figure 1B. Two of the metabolites in patients (H6 and H7) and two in mice (M6 and M7) were detected using a positive precursor ion scan at m/z 283, which is the fragment occurring after loss of the amino-phenyl-hydroxy group. The structure of H7 and M7 identified the two metabolites as 4-MPR, while H6 and M6 showed an [M + H]+ ion at m/z 568 and fragmentation spectra corresponding to the direct addition of glucuronide to 4-HPR. Metabolites H2–H4, H8 and M2–M4 were detected by a neutral loss scan of 109. The fragmentation spectra identified metabolites H4 and M4 as 4-oxo-4-HPR, and the spectra of H2, M2 and H8 each corresponded to a monohydroxy-4-oxo-4-HPR. The exact location of the hydroxyl group on the 4-oxo-substituted β-ionone ring could not be determined for H2, M2 or H8 as hydroxylation could occur at any of the C-2, C-3 or C-18 positions. The metabolites H3 and M3 were identified as dehydrogenated 4-HPR. Finally, metabolites M1, H5 and M5 were detected by a precursor ion scan at m/z 313, which is the fragment occurring after loss of the amino-phenyl-hydroxy group from monohydroxy-4-oxo-4-HPR. Fragmentation spectra suggested that M1 was a glucuronidated metabolite and that H5 and M5 were both sulfated metabolites.
In vitro metabolism of 4-HPR in liver microsomes and malignant mammalian cells
Identification of 4-oxo-4-HPR in the plasma of mice prompted us to measure and compare the Michaelis–Menten kinetic constants for the oxidation of 4-HPR to 4-oxo-4-HPR in pooled HLMs and pooled MLM using HPLC (Figure 2A and B). The Vmax was more than threefold greater for HLM compared with MLM (1.50 vs. 0.42 nmol·h−1 per mg protein), but the Km values were equivalent (23.9 vs. 25.8 µM), showing that 4-oxo-4-HPR generation in mice is slower but the total amount formed in mice is similar to humans. We also investigated whether 4-MPR, the major metabolite of 4-HPR, could be demethylated to form 4-HPR or 4-oxo-4-HPR in HLM (Figure 2C, D). Although the Vmax and Km values were low for 4-MPR, both 4-HPR and 4-oxo-4-HPR were detected as metabolites of 4-MPR in HLM. Neither 4-HPR nor 4-oxo-4-HPR were produced from 4-MPR at measurable concentrations in MLM (data not shown).
Figure 2.
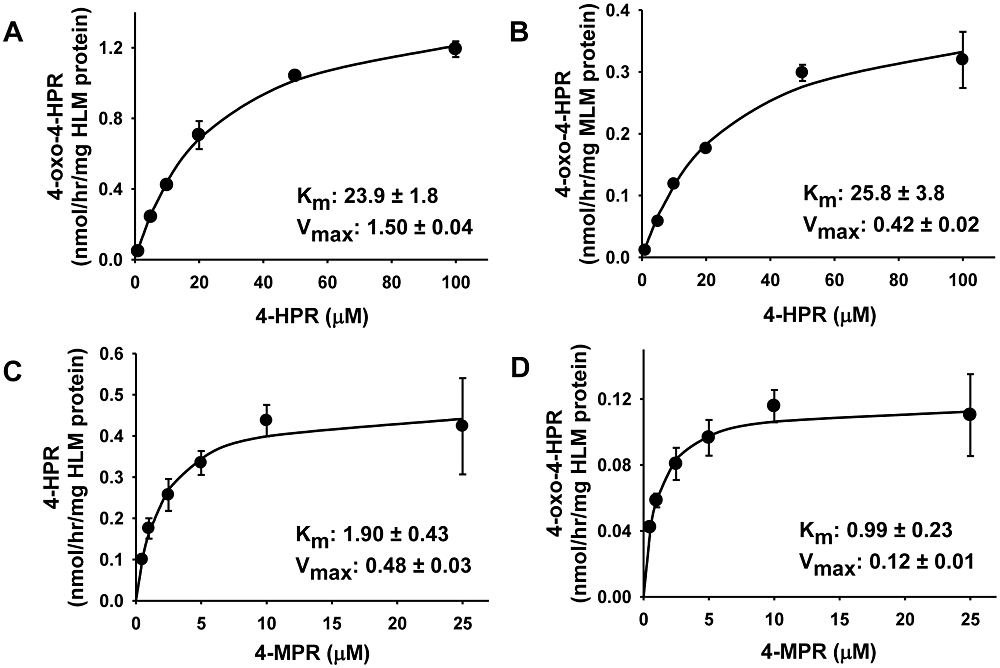
In vitro metabolism of N-(4-hydroxyphenyl)retinamide (4-HPR) and N-(4-methoxyphenyl)retinamide (4-MPR) in pooled human (HLM) and mouse liver microsomes (MLM). (A) Rate of 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR) formation from 4-HPR in HLM and (B) in MLM. (C) Rate of demethylation of 4-MPR to the parent drug in HLM. (D) Rate of 4-oxo-4-HPR formation from 4-MPR in HLM. (A–D) Substrates at indicated concentrations were incubated in HLM or MLM for 60 min. Points in each graph are the mean and error bars are standard deviation from experiments performed in triplicate. Amounts measured by HPLC were normalized to the amount of HLM or MLM protein. In each graph, the Km (µM) and Vmax (nmol·h−1 mg protein−1) are the mean ± SD.
As the CYP enzymes 3A4, 2C8, 2C9 and 26A1 have been shown to metabolize ATRA into polar metabolites including 4-oxo-ATRA (Thatcher, 2005), we investigated whether these enzymes contributed to the formation of 4-oxo-4-HPR from 4-HPR in humans and mice. Three of these enzymes (CYP2C8, 2C9 and 3A4) are inhibited by agents that are readily available and well tolerated in humans, making the inhibitors of the enzymes clinically applicable for co-administration with 4-HPR. To identify the CYP enzymes mediating the conversion of 4-HPR to 4-oxo-4-HPR in HLM and MLM, inhibitors to CYP2C8 (gemfibrozil), 2C9 (fluconazole) and 3A4 (ketoconazole) were added to MLM and HLM in combination with 4-HPR and the amount of 4-oxo-4-HPR formation was measured via HPLC (Figure 3). A 4-HPR concentration of 25 µM, an approximated Km from both MLM and HLM experiments, was used for the formation of 4-oxo-4-HPR from 4-HPR. In MLM (Figure 3A), the addition of fluconazole reduced 4-oxo-4-HPR formation by 40.9% ± 4.1 at 10 µM from 4-HPR alone (P < 0.01) and ketoconazole reduced 4-oxo-4-HPR formation by 96.5% ± 3.1 at 10 µM from 4-HPR alone (P < 0.001). In HLM (Figure 3B), only ketoconazole reduced 4-oxo-4-HPR formation (33.7% ± 8.8 from 4-HPR alone at 10 µM, P < 0.01). To confirm that human CYP3A4 catalyzed the formation of 4-oxo-4-HPR from 4-HPR, we incubated 4-HPR (at 10 µM anticipating that the CYP3A4 isoenzyme activity would be higher than in pooled HLMs) in human CYP3A4 isoenzyme microsomes with the NADPH generating system and analyzed amounts of 4-HPR and 4-oxo-4-HPR via HPLC. We found that 4-oxo-4-HPR concentrations in the CYP3A4 microsomes increased in a time-dependent manner (Figure 3C).
Figure 3.
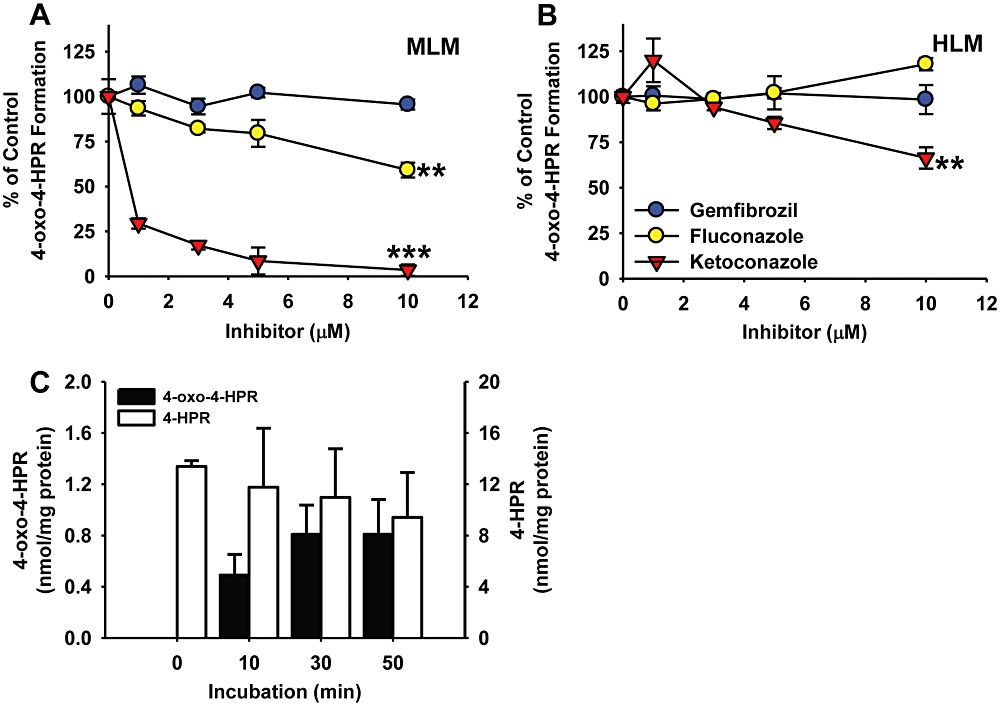
CYP enzymes catalyzing the formation of 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR) from N-(4-hydroxyphenyl)retinamide (4-HPR). (A, B) Analysis of the effects of CYP enzyme inhibitors on 4-oxo-4-HPR formation from 4-HPR in (A) pooled mouse (MLM) and (B) pooled human liver microsomes (HLM). 4-HPR (25 µM) was incubated with or without the indicated concentration of CYP enzyme inhibitor in MLM or HLM for 60 min before analysis via HPLC. (C) Formation of 4-oxo-4-HPR from 4-HPR via human CYP3A4 isoenzyme. In human CYP3A4 isoenzyme microsomes, 4-HPR (10 µM) was incubated for the times indicated. Amounts of 4-HPR and 4-oxo-4-HPR measured by HPLC were normalized to the amount of CYP3A4 protein. 4-HPR (10 µM) was used as a positive control in HLM to confirm the generation of 4-oxo-4-HPR. (A–C) The points/columns are the mean and error/vertical bars are standard deviation from experiments performed in triplicate. **P < 0.01, ***P < 0.001.
As ketoconazole at 10 µM partially inhibited 4-oxo-4-HPR production in HLM, and ketoconazole is reported to be specific for CYP3A4 at concentrations up to 1 µM but will inhibit other CYP isoforms at higher concentrations (Jia and Liu, 2007), we considered the possibility that other CYP enzymes might be contributing to 4-oxo-4-HPR formation in cancer patients. Expression of CYP26A1 was reported to be induced after exposure to 4-HPR in a human ovarian cancer cell line, and 4-oxo-4-HPR formation from 4-HPR was measured after overexpression of CYP26A1 (Villani et al., 2004), so we investigated whether CYP26A1 formed 4-oxo-4-HPR from 4-HPR in human malignant lymphoid cell lines and neuroblastoma cell lines. We first measured basal CYP26A1 mRNA levels via RT-PCR in fourteen malignant lymphoid cell lines grown at physiological bone marrow level hypoxia (5% O2) and two neuroblastoma cell lines grown at ‘normoxic’ tissue culture conditions (20% O2). mRNA for CYP26A1 was quantified in all cell lines and the levels were comparable between the malignant lymphoid cells (range 0.03–1.27) and the neuroblastoma cells (CHLA-90: 0.108 and CHLA-136: 0.076). However, when we used HPLC to examine cellular extracts from five of the malignant lymphoid cell lines (CCRF-CEM, COG-LL-317, HuT-78, MOLT-3 and RS4;11) and both neuroblastoma cell lines after treatment with 2–10 µM 4-HPR for 12 and 24 h. 4-oxo-4-HPR was not formed at measurable concentrations (data not shown).
Co-administration of ketoconazole with 4-HPR increased systemic 4-HPR levels in vivo
To examine whether the modulation of 4-HPR metabolism would alter systemic 4-HPR levels in vivo, we administered 4-HPR with or without ketoconazole at human treatment-equivalent dosing to nu/nu and NOD/SCID mice. In nu/nu mice (Figure 4A) the addition of 75 mg·kg−1 of ketoconazole (representing a human equivalent dose of 6 mg·kg−1, which is within the therapeutic dosing range for children and adults) increased 4-HPR plasma levels from 14.5 µM ± 4.7 to 31.5 µM ± 1.9 (a 2.2-fold increase over control, P < 0.01), and in NOD/SCID mice (Figure 4B) ketoconazole significantly increased 4-HPR plasma levels at all doses (at 18.75 mg·kg−1, an increase from 8.5 µM ± 4.4 to 28.9 µM ± 3.1, a 3.4-fold increase over control, P < 0.01). In a separate experiment, the addition of 25 mg·kg−1 ketoconazole to 4-HPR in NOD/SCID mice (Figure 4C) increased 4-HPR plasma levels from 15.6 µM ± 4.5 to 31.6 µM ± 2.7 (a twofold increase over control, P < 0.02) and also increased levels of 4-oxo-4-HPR from 3.6 µM ± 0.4 to 7.2 µM ± 0.8 (a twofold increase over control, P < 0.01).
Comparison of levels of 4-HPR and Its metabolites in the plasma of 4-HPR-treated patients and mice
Using HPLC, we analyzed the concentrations of 4-HPR, 4-MPR and 4-oxo-4-HPR in the plasma of three patients enrolled in a phase I clinical trial of 4-HPR/LXS (Table 1). Blood samples were taken from three patients treated at the 4-HPR dose-level of 595 mg·m−2 per day, just before the first dose of the day and then at 2 h intervals up to 8 h after treatment, on day 6 of cycle 1. In plasma prepared from a sample taken 4 h after treatment (the middle of the 8-h sampling period), 4-HPR, 4-MPR and 4-oxo-4-HPR levels were 12.82 µM ± 8.08, 5.72 µM ± 4.21 and 0.86 µM ± 0.66 respectively. By comparison, in the plasma of NOD/SCID mice that were administered 4-HPR/LXS at a 4-HPR dose approximately equivalent to the dose administered in the patients (patients: 595 mg·m−2 per day and mice: 180 mg·kg−1 per day) (Reagan-Shaw et al., 2008), average levels of 4-HPR, 4-MPR and 4-oxo-4-HPR were 15.6 µM ± 4.5, 1.6 µM ± 0.2 and 3.6 µM ± 0.4 respectively (control mice in Figure 4C).
Table 1.
N-(4-hydroxyphenyl)retinamide (4-HPR), N-(4-methoxyphenyl)retinamide (4-MPR) and 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR) levels in plasma of neuroblastoma patients
| Mean ± standard deviation (µM) | |||
|---|---|---|---|
| Hours after treatment (n = 3) | 4-HPR | 4-MPR | 4-oxo-4-HPRa |
| 0 | 7.04 ± 4.75 | 4.03 ± 2.98 | 0.91b |
| 2 | 9.69 ± 7.30 | 4.91 ± 3.69 | 1.27b |
| 4 | 12.82 ± 8.08 | 5.72 ± 4.21 | 0.86 ± 0.66c |
| 6 | 13.11 ± 8.50 | 6.61 ± 5.75 | 0.43b |
| 8 | 8.55 ± 6.42 | 4.69 ± 3.92 | 1.00 ± 0.72d |
Blood was taken from three patients treated at the 595 mg·m−2 per day dose level. All blood samples were collected on day 6 of cycle 1, just before and after the first dose of the day, and plasma was prepared as described. Doses of 4-HPR were given twice a day, 12 h apart and the zero time value represents blood taken just before the first dose of day 6, that is, 12 h after the last dose on day 5.
Not all patients were analyzed for 4-oxo-4-HPR at the indicated hour.
n = 1
n = 2
n = 3.
Discussions and conclusions
Low plasma drug levels and tumor response rates in clinical trials employing an oral corn-oil slurry based 4-HPR capsule have led investigators to suggest that higher systemic 4-HPR exposures may be required to improve clinical outcomes (Cheng et al., 2001; Puduvalli et al., 2004; Vaishampayan et al., 2005; Reynolds et al., 2007; William et al., 2009). As a result, new formulations that increase 4-HPR bioavailability have been developed (Liu et al., 2007; Maurer et al., 2007) and are currently being tested in phase I trials (Mohrbacher et al., 2007; Marachelian et al., 2009). However, the possibility that systemic 4-HPR exposures could be further improved by modulating 4-HPR metabolism in conjunction with oral delivery has not been investigated. Based on reports suggesting that both ATRA and 4-HPR metabolism occurs via CYP enzymes (Hultin et al., 1986; Van Wauwe et al., 1988; 1990; 1994; Thatcher, 2005; Villani et al., 2004), we hypothesized that pharmacological inhibition of CYP enzymes that metabolize 4-HPR using clinically-available agents might increase systemic 4-HPR concentrations.
Therefore, to explore this hypothesis, we first investigated how 4-HPR metabolism in humans compared with in vivo murine models as the metabolic pathways for 4-HPR are poorly defined in both species. In this study, we determined both similarities and differences in 4-HPR metabolism between humans and mice. Using human and MLMs we demonstrated that 4-oxo-4-HPR is formed from 4-HPR by CYP3A4 in mice and, in part, by CYP3A4 in human cells in vitro; we further demonstrated that the co-administration of the CYP3A4-inhibitor, ketoconazole, with 4-HPR in vivo increased 4-HPR plasma concentrations in mice.
The demonstrated similarities and differences in 4-HPR metabolism between humans and mice suggests that mouse models may be useful in vivo models of 4-HPR pharmacokinetics and studies of pharmacological manipulation in humans within certain limitations. Specifically, differences in the kinetic constants for oxidation of 4-HPR to 4-oxo-4-HPR in HLM and MLM were observed, suggesting that mice proportionately oxidize a greater portion of 4-HPR to 4-oxo-4-HPR than do humans. Also, the observation that ketoconazole inhibited 4-HPR conversion to 4-oxo-4-HPR at lower concentrations in MLM than in HLM, suggesting that mice may rely more heavily on CYP3A4 for 4-oxo-4-HPR formation than humans. In terms of similarities, the low kinetic constants for conversion of 4-MPR to 4-HPR or 4-oxo-4-HPR in HLM (Figure 2C and D) and the absence of conversion in MLM suggest that neither conversion occurs at high rates or amounts in humans or mice. Thus, while 4-MPR is reported to accumulate to high levels in plasma and tissues and to have a longer half-life than 4-HPR (Formelli and Cleris, 1993; Sabichi et al., 2003; Formelli et al., 2008), it is unlikely that back conversion (i.e. ‘recycling’) of the 4-MPR pool in plasma significantly contributes to systemic 4-HPR levels or tumor cell 4-HPR exposure. A proposed biotransformation pathway of 4-HPR is shown in Figure 5 based on the data from human and mouse plasma analyses and the in vitro enzyme kinetic studies.
Figure 5.
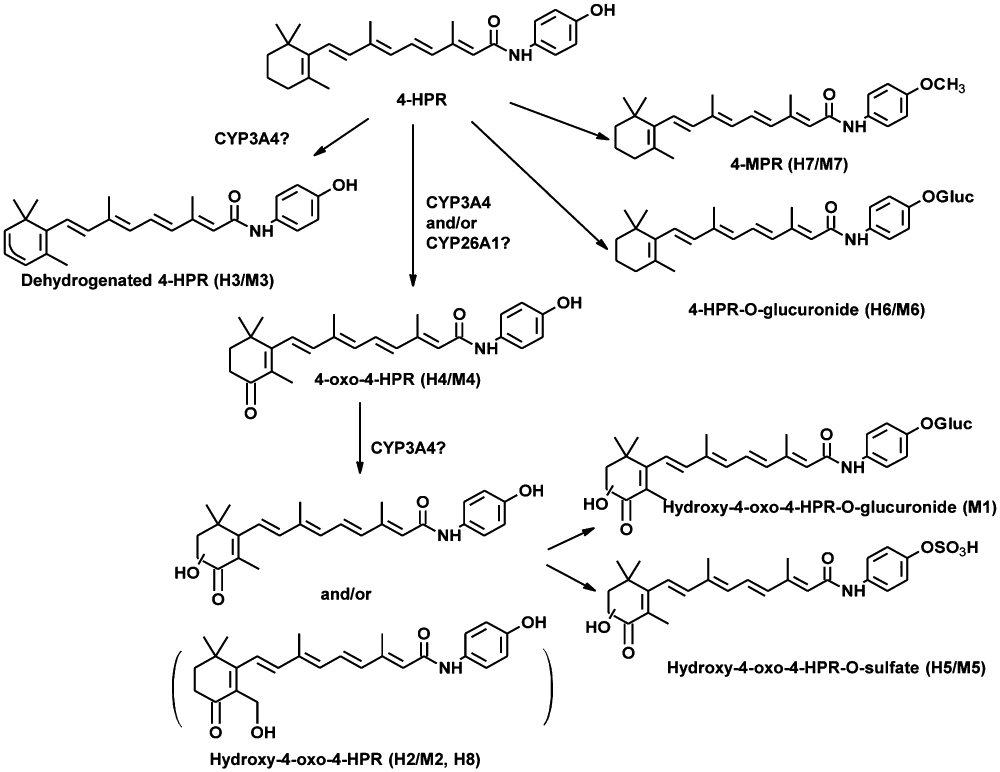
Proposed phase I and II biotransformation pathway for N-(4-hydroxyphenyl)retinamide (4-HPR). Potential pathways for the interconversion of one metabolite into another, that is, the conversion of N-(4-methoxyphenyl)retinamide (4-MPR) to 4-oxo-N-(4-hydroxyphenyl)retinamide (4-oxo-4-HPR), have been omitted due to the very low/no conversion kinetic parameters observed in HLM and MLM.
Our in vitro studies demonstrated that inhibitors of both CYP2C9 (fluconazole) and CYP3A4 (ketoconazole) significantly reduced 4-oxo-4-HPR formation in MLM, but that in HLM ketoconazole only partially inhibited 4-oxo-4-HPR formation. We then demonstrated that the co-administration of ketoconazole at human treatment-equivalent doses with 4-HPR in vivo increased 4-HPR plasma levels by at least twofold in two strains of mice, with NOD/SCID mice possibly being more sensitive to ketoconazole effects than nu/nu mice. These in vitro and in vivo data are compatible with results from a study by Illingworth et al. (2011), and support the role of CYP3A4 in the oxidation of 4-HPR. However, given that relatively high concentrations of ketoconazole were used to inhibit 4-oxo-4-HPR formation in HLM both in our study (10 µM) and in the study by Illingworth et al. (2011) (100 µM), it is likely that other CYP enzymes in addition to 3A4 contribute to 4-oxo-4-HPR formation in humans.
Based on the fact that we observed greater inhibition of 4-oxo-4-HPR formation by ketoconazole in MLM versus HLM (96.5% vs. 33.7% at 10 µM, see Figure 3A and B), the present results suggest that experiments in mice may overestimate the increase in 4-HPR plasma levels to be expected in humans when using concurrent administration of ketoconazole and 4-HPR, if such increases are based solely on the inhibition of metabolism of 4-HPR to 4-oxo-4-HPR. However, ketoconazole may inhibit other CYP enzyme isoforms in addition to 3A4 at concentrations above 1 µM (Jia and Liu, 2007). Thus ketoconazole co-treatment has the potential to decrease the conversion of 4-HPR to metabolites other than 4-oxo-4-HPR and could result in higher 4-HPR plasma levels in patients than would be suggested by analysis of 4-oxo-4-HPR formation alone in HLM. Also, importantly, although 4-oxo-4-HPR may obtain lower steady-state plasma levels than other 4-HPR metabolites in humans, the specific mass per time (flux) of 4-HPR through 4-oxo-4-HPR metabolism in humans is not known and, if great enough, could represent a useful target for pharmacological intervention by ketoconazole or other agents on this basis.
Based on microsomal results, in addition to increasing 4-HPR plasma levels we expected that co-administration of ketoconazole to mice would also result in a concomitant rise in 4-MPR plasma levels (as oxidation of 4-HPR was inhibited, more 4-HPR was available to the other metabolic pathways). However, in addition to a slight increase in 4-MPR levels, surprisingly, ketoconazole increased 4-oxo-4-HPR plasma levels in mice (Figure 4C), perhaps a counter-intuitive result. Considering that CYP3A4 may also mediate the formation of dehydrogenated 4-HPR and hydroxy-4-oxo-4-HPR, and that the metabolism of both ATRA and 4-oxo-4-ATRA has been reported to be inhibited by imidazole-containing P450 inhibitors such as ketoconazole and liarazole (Hultin et al., 1986; Van Wauwe et al., 1990; Van et al., 1994; Chithalen et al., 2002), it is possible that other CYP enzymes, including CYP26A1 whose expression has been shown to be inducible by ATRA and 4-HPR (Sonneveld et al., 1998; Ozpolat et al., 2002; 2004; Villani et al., 2004), may also contribute to the in vivo formation of 4-oxo-4-HPR. Another possible explanation of having slightly increased 4-oxo-4-HPR in mice is that ketoconazole inhibited further degradation of 4-oxo-4-HPR into hydroxyl-4-oxo-4-HPR (see Figure 5), and that the increases in 4-HPR concentration resulted from the inhibition of dehydrogenated 4-HPR formation which generated more 4-oxo-4-HPR in sequence.
As the metabolite 4-oxo-4-HPR is reported to exhibit more potent anti-tumor activity than 4-HPR and has also been shown to act synergistically with the parent compound (Villani et al., 2006), one may question whether the inhibition of 4-oxo-4-HPR formation might compromise the efficacy of 4-HPR. While the anti-tumor activity of 4-HPR has been demonstrated both with in vivo models and clinically (Maurer et al., 2007; Mohrbacher et al., 2007; Reynolds et al., 2007; Marachelian et al., 2009), only in vitro studies with 4-oxo-4-HPR are reported and it has not been investigated whether this translates to an anti-tumor role in vivo. Further, it is hypothesized that sufficiently increasing 4-HPR levels will result in increased anti-tumor activity that will more than adequately compensate for a possible partial decrease in 4-oxo-4-HPR levels. Indeed, in our in vivo mouse experiments, ketoconazole did not appreciably reduce 4-oxo-4-HPR levels in the plasma while 4-HPR concentrations approximately doubled, suggesting the further metabolism of 4-oxo-4-HPR to hydroxy-4-oxo-4-HPR might have been inhibited by ketoconazole as previously discussed.
Unfortunately, due to the lack of a sufficiently specific inhibitor of CYP26A1 and CYP26A1 isoenzymes, we could not address whether the formation of 4-oxo-4-HPR from 4-HPR is also catalyzed by CYP26A1 using liver microsome analyses or in vivo mouse studies. However, we assayed 4-oxo-4-HPR formation in seven human malignant lymphoid and neuroblastoma cell lines expressing CYP26A1 that were exposed to 4-HPR and 4-oxo-4-HPR was not measurable in any of the cell lines tested. These results in lymphoid cancer and neuroblastoma cell lines with basal levels of CYP26A1 expression differ from those reported by Villani et al., who employed forced overexpression of CYP26A1 in an ovarian cancer cell line (Villani et al., 2004), and may have therapeutic implications. Our results suggest that the limited amount of internal tumor 4-oxo-4-HPR formed by CYP26A1 and/or other enzymes in human malignant lymphoid and neuroblastoma cell types in vivo may not be responsible for the observed clinical activity of 4-HPR, although our studies did not address the possible effects of peripheral tissue metabolism of 4-HPR to 4-oxo-4-HPR and the effects of those 4-oxo-4-HPR sources on tumor responses. Conceivably, 4-HPR-resistant malignant cells may have increased expression of CYP26A1 and be capable of generating clinically significant local amounts of 4-oxo-4-HPR. Thus, it is difficult to directly predict the clinical impact, if any, that 4-HPR-resistant, 4-oxo-4-HPR-producing malignant cells may have on 4-HPR global pharmacokinetics as the relative contributions to 4-HPR metabolism of various tissues (liver, intestines, kidneys, etc.) and of varying tumor cell burdens remain to be determined.
Finally, we compared the levels of 4-HPR, 4-MPR and 4-oxo-4-HPR in the plasma of 4-HPR-treated patients and mice treated with human-equivalent doses of 4-HPR (Reagan-Shaw et al., 2008). When compared with 4-HPR and metabolite plasma concentrations observed in a phase I trial employing oral capsule 4-HPR in paediatric neuroblastoma patients at similar doses, the 4-HPR and 4-MPR levels measured in patients treated with 4-HPR/LXS oral powder appeared to be higher but the 4-oxo-4-HPR levels were approximately equivalent. Further, while the 4-HPR plasma concentrations measured in the present investigation were not appreciably different between patients and mice dosed on an equivalent basis, NOD/SCID mice appeared to convert more 4-HPR to 4-oxo-4-HPR while humans preferentially metabolized 4-HPR to 4-MPR (Table 1 and control mice in Figure 4C). This observed result seems to be at odds with the kinetic constants estimated by our in vitro studies, which suggested that humans and mice would form similar amounts of 4-oxo-4-HPR. Kinetic data, however, cannot be interpreted in isolation as other reactions may influence drug metabolism. It is also worth noting that our clinical and in vivo data are currently limited by small numbers of patients, a restricted patient age range (<26 years old), and by the number of strains of mice analyzed for 4-oxo-4-HPR. Further, the potentially greater responsiveness of NOD/SCID mice with respect to the extent to which ketoconazole increased 4-HPR levels (Figure 4A and B) suggests that not all mouse strains metabolize 4-HPR equivalently.
In summary, our evidence suggests that, overall, humans and mice metabolize 4-HPR to a common set of metabolites and the demonstration that the co-administration of ketoconazole increased 4-HPR plasma levels in mice strongly support the possibility that a similar effect may be observed in patients. The addition of a study arm to test the effect of the co-administration of ketoconazole on 4-HPR plasma levels and systemic toxicity in an on-going phase I trial of 4-HPR/LXS oral powder in paediatric neuroblastoma in the New Approaches to Neuroblastoma Therapy (NANT) Consortium (http://www.NANT.org) is in progress. Considering that relatively low human-equivalent doses of ketoconazole were sufficient to increase 4-HPR systemic exposure in mice, the combination of 4-HPR and ketoconazole is expected to be well tolerated in patients.
Acknowledgments
This work was funded by grants from the Cancer Prevention and Research Institute of Texas (CPRIT: RP10072), the National Institutes of Health (CA82830, CA100895, CA81403, CA49837), and by the Rapid Access to Intervention Development (RAID) Program, Developmental Therapeutics Program (DTP), National Cancer Institute (NCI). JPC was supported by a fellowship from the AT&T Foundation. The authors would like to acknowledge The New Approaches to Neuroblastoma Therapy (NANT) Consortium for providing patient samples for this study. NANT is supported by National Institutes of Health CA81403, The Children's Neuroblastoma Cancer Foundation (http://www.CNCFhope.org), the University of Southern California-Childrens Hospital Los Angeles Institute for Pediatric Research, Alex's Lemonade Stand Foundation, Dougherty Family Foundation, Evan T. J. Dunbar Neuroblastoma Foundation, and the Neuroblastoma Children's Cancer Society.
Glossary
Abbreviations
- 4-EPR
N-(4-ethoxyphenyl)retinamide
- 4-HPR (fenretinide)
N-(4-hydroxyphenyl)retinamide
- 4-MPR
N-(4-methoxyphenyl)retinamide
- 4-oxo-4-HPR
4-oxo-N-(4-hydroxyphenyl)retinamide
- ATRA
all-trans retinoic acid
- CYP
cytochrome P450
- HLM
human liver microsomes
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- MLM
mouse liver microsomes
- NOD/SCID
non-obese diabetic/severe combined immune deficiency
- RT-PCR
real-time reverse transcription-polymerase chain reaction
Conflict of interest
Certain intellectual property rights to 4-HPR/LXS™ oral powder, intravenous fenretinide emulsion and fenretinide in combination with ketoconazole, as cited here may be retained by The Childrens Hospital Los Angeles. BJM and CPR are co-inventors of this intellectual property and may potentially benefit financially from said intellectual property.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Appierto V, Cavadini E, Pergolizzi R, Cleris L, Lotan R, Canevari S, et al. Decrease in drug accumulation and in tumour aggressiveness marker expression in a fenretinide-induced resistant ovarian tumour cell line. Br J Cancer. 2001;84:1528–1534. doi: 10.1054/bjoc.2001.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Chiesa F, Tradati N, Grigolato R, Boracchi P, Biganzoli E, Crose N, et al. Randomized trial of fenretinide (4-HPR) to prevent recurrences, new localizations and carcinomas in patients operated on for oral leukoplakia: long-term results. Int J Cancer. 2005;115:625–629. doi: 10.1002/ijc.20923. [DOI] [PubMed] [Google Scholar]
- Chithalen JV, Luu L, Petkovich M, Jones G. HPLC-MS/MS analysis of the products generated from all-trans-retinoic acid using recombinant human CYP26A. J Lipid Res. 2002;43:1133–1142. doi: 10.1194/jlr.m100343-jlr200. [DOI] [PubMed] [Google Scholar]
- Delia D, Aiello A, Lombardi L, Pelicci PG, Grignani F, Grignani F, et al. N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993;53:6036–6041. [PubMed] [Google Scholar]
- Formelli F, Cleris L. Synthetic retinoid fenretinide is effective against a human ovarian carcinoma xenograft and potentiates cisplatin activity. Cancer Res. 1993;53:5374–5376. [PubMed] [Google Scholar]
- Formelli F, Cavadini E, Luksch R, Garaventa A, Villani M, Appierto V, et al. Pharmacokinetics of oral fenretinide in neuroblastoma patients: indications for optimal dose and dosing schedule also with respect to the active metabolite 4-oxo-fenretinide. Cancer Chemother Pharmacol. 2008;62:655–665. doi: 10.1007/s00280-007-0649-7. [DOI] [PubMed] [Google Scholar]
- Garaventa A, Luksch R, Lo Piccolo MS, Cavadini E, Montaldo PG, Pizzitola MR, et al. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res. 2003;9:2032–2039. [PubMed] [Google Scholar]
- Garcia AA, Morgan R, McNamara M, Scudder S, Tsao-Wei D, Groshen S, et al. Phase II trial of fenretinide (4-HPR) in recurrent ovarian and primary peritoneal carcinoma: a California cancer consortium trial. J Clin Oncol. 2004;22 abstr 5056. [Google Scholar]
- Hultin TA, May CM, Moon RC. N-(4-hydroxyphenyl)-all-trans-retinamide pharmacokinetics in female rats and mice. Drug Metab Dispos. 1986;14:714–717. [PubMed] [Google Scholar]
- Illingworth N, Boddy A, Daly A, Veal G. Characterization of the metabolism of fenretinide by human liver microsomes, cytochrome P450 enzymes and UDP-glucuronosyltransferases. Br J Pharmacol. 2011;162:989–999. doi: 10.1111/j.1476-5381.2010.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasti BR, LoRousso P, Parchment RE, Wozniak AJ, Flaherty LE, Shields LF, et al. Phase I clinical trial of fenretinide (NSC374551) in advanced solid tumors. Proc Am Soc Clin Oncol. 2001 abstr 485. [Google Scholar]
- Jia L, Liu X. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr Drug Metab. 2007;8:822–829. doi: 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalemkerian GP, Slusher R, Ramalingam S, Gadgeel S, Mabry M. Growth inhibition and induction of apoptosis by fenretinide in small-cell lung cancer cell lines. JNCI J Natl Cancer Inst. 1995;87:1674–1680. doi: 10.1093/jnci/87.22.1674. [DOI] [PubMed] [Google Scholar]
- Kang MH, Wan Z, Kang Y, Sposto R, Reynolds CP. Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst. 2008;100:580–595. doi: 10.1093/jnci/djn076. [DOI] [PubMed] [Google Scholar]
- Lee JS, Newman RA, Lippman SM, Fossella FV, Calayag M, Raber MN, et al. Phase I evaluation of all-trans retinoic acid with and without ketoconazole in adults with solid tumors. J Clin Oncol. 1995;13:1501–1508. doi: 10.1200/JCO.1995.13.6.1501. [DOI] [PubMed] [Google Scholar]
- Liu X, Maurer B, Frgala T, Page J, Noker P, Fulton R, et al. Preclinical toxicology and pharmacokinetics of intravenous lipid emulsion fenretinide. Mol Cancer Ther. 2007;12(Pt 2) abstr C159. [Google Scholar]
- Marachelian A, Kang MH, Hwang KH, Villablanca JG, Groshen S, Mathay KK, et al. Phase I study of fenretinide (4-HPR) oral powder in patients with recurrent or resistant neuroblastoma: New Approaches to Neuroblastoma Therapy (NANT) Consortium trial. J Clin Oncol. 2009;27:15s. (Suppl; abstr 10009) [Google Scholar]
- Mariotti A, Marcora E, Bunone G, Costa A, Veronesi U, Pierotti MA, et al. N-(4-hydroxyphenyl)retinamide: a potent inducer of apoptosis in human neuroblastoma cells. J Natl Cancer Inst. 1994;86:1245–1247. doi: 10.1093/jnci/86.16.1245. [DOI] [PubMed] [Google Scholar]
- Masters JR, Thomson JA, ly-Burns B, Reid YA, Dirks WG, Packer P, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci USA. 2001;98:8012–8017. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer BJ, Kalous O, Yesair DW, Wu X, Janeba J, Maldonado V, et al. Improved oral delivery of N-(4-hydroxyphenyl)retinamide with a novel LYM-X-SORB organized lipid complex. Clin Cancer Res. 2007;13:3079–3086. doi: 10.1158/1078-0432.CCR-06-1889. [DOI] [PubMed] [Google Scholar]
- Mehta RR, Hawthorne ME, Graves JM, Mehta RG. Metabolism of N-[4-hydroxyphenyl]retinamide (4-HPR) to N-[4-methoxyphenyl]retinamide (4-MPR) may serve as a biomarker for its efficacy against human breast cancer and melanoma cells. Eur J Cancer. 1998;34:902–907. doi: 10.1016/s0959-8049(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Mohrbacher A, Gutierrez M, Murgo AJ, Kummar S, Reynolds CP, Maurer BJ, et al. Phase I trial of fenretinide (4-HPR) intravenous emulsion for hematologic malignancies. J Clin Oncol. 2007;25:18s. (suppl; abstr 2851) [Google Scholar]
- Muindi JR, Young CW, Warrell RP., Jr Clinical pharmacology of all-trans retinoic acid. Leukemia. 1994;8:1807–1812. [PubMed] [Google Scholar]
- Nivoix Y, Leveque D, Herbrecht R, Koffel JC, Beretz L, Ubeaud-Sequier G. The enzymatic basis of drug-drug interactions with systemic triazole antifungals. Clin Pharmacokinet. 2008;47:779–792. doi: 10.2165/0003088-200847120-00003. [DOI] [PubMed] [Google Scholar]
- O'Donnell PH, Guo WX, Reynolds CP, Maurer BJ. N-(4-hydroxyphenyl)retinamide increases ceramide and is cytotoxic to acute lymphoblastic leukemia cell lines, but not to non-malignant lymphocytes. Leukemia. 2002;16:902–910. doi: 10.1038/sj.leu.2402485. [DOI] [PubMed] [Google Scholar]
- Oridate N, Lotan D, Xu XC, Hong WK, Lotan R. Differential induction of apoptosis by all-trans-retinoic acid and N-(4-hydroxyphenyl)retinamide in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 1996;2:855–863. [PubMed] [Google Scholar]
- Ozpolat B, Mehta K, Tari AM, Lopez-Berestein G. all-trans-Retinoic acid-induced expression and regulation of retinoic acid 4-hydroxylase (CYP26) in human promyelocytic leukemia. Am J Hematol. 2002;70:39–47. doi: 10.1002/ajh.10099. [DOI] [PubMed] [Google Scholar]
- Ozpolat B, Tari AM, Mehta K, Lopez-Berestein G. Nuclear retinoid receptors are involved in N-(4-hydroxyphenyl) retinamide (Fenretinide)-induced gene expression and growth inhibition in HL-60 acute myeloid leukemia cells. Leuk Lymphoma. 2004;45:979–985. doi: 10.1080/1042819031000151833. [DOI] [PubMed] [Google Scholar]
- Puduvalli VK, Yung WK, Hess KR, Kuhn JG, Groves MD, Levin VA, et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: a North American Brain Tumor Consortium study. J Clin Oncol. 2004;22:4282–4289. doi: 10.1200/JCO.2004.09.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Reynolds CP, Frgala T, Tsao-Wei D, Groshen S, Morgan R, McNamara M, et al. High plasma levels of fenretinide (4-HPR) were associated with improved outcome in a phase II study of recurrent ovarian cancer: a study by the California Cancer Consortium. J Clin Oncol. 2007;25:18s. (suppl; abstr 5555) [Google Scholar]
- Sabichi AL, Modiano MR, Lee JJ, Peng YM, Xu MJ, Villar H, et al. Breast tissue accumulation of retinamides in a randomized short-term study of fenretinide. Clin Cancer Res. 2003;9:2400–2405. [PubMed] [Google Scholar]
- Sabichi AL, Lerner SP, Atkinson EN, Grossman HB, Caraway NP, Dinney CP, et al. Phase III prevention trial of fenretinide in patients with resected non-muscle-invasive bladder cancer. Clin Cancer Res. 2008;14:224–229. doi: 10.1158/1078-0432.CCR-07-0733. [DOI] [PubMed] [Google Scholar]
- Scheen AJ. Pharmacokinetic interactions with thiazolidinediones. Clin Pharmacokinet. 2007;46:1–12. doi: 10.2165/00003088-200746010-00001. [DOI] [PubMed] [Google Scholar]
- Sonneveld E, van den Brink CE, van der Leede BM, Schulkes RK, Petkovich M, van der Burg B, et al. Human retinoic acid (RA) 4-hydroxylase (CYP26) is highly specific for all-trans-RA and can be induced through RA receptors in human breast and colon carcinoma cells. Cell Growth Differ. 1998;9:629–637. [PubMed] [Google Scholar]
- Thatcher N. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- Vaishampayan U, Heilbrun LK, Parchment RE, Jain V, Zwiebel J, Boinpally RR, et al. Phase II trial of fenretinide in advanced renal carcinoma. Invest New Drugs. 2005;23:179–185. doi: 10.1007/s10637-005-5864-7. [DOI] [PubMed] [Google Scholar]
- Van Wauwe JP, Coene MC, Goossens J, Van NG, Cools W, Lauwers W. Ketoconazole inhibits the in vitro and in vivo metabolism of all-trans-retinoic acid. J Pharmacol Exp Ther. 1988;245:718–722. [PubMed] [Google Scholar]
- Van Wauwe JP, Coene MC, Goossens J, Cools W, Monbaliu J. Effects of cytochrome P-450 inhibitors on the in vivo metabolism of all-trans-retinoic acid in rats. J Pharmacol Exp Ther. 1990;252:365–369. [PubMed] [Google Scholar]
- Van Wauwe JP, Coene MC, Cools W, Goossens J, Lauwers W, Le JL, et al. Liarozole fumarate inhibits the metabolism of 4-keto-all-trans-retinoic acid. Biochem Pharmacol. 1994;47:737–741. doi: 10.1016/0006-2952(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Veronesi U, de Palo G, Marubini E, Costa A, Formelli F, Mariani L, et al. Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J Natl Cancer Inst. 1999;91:1847–1856. doi: 10.1093/jnci/91.21.1847. [DOI] [PubMed] [Google Scholar]
- Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, Reynolds CP. Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children's Oncology Group (CCG 09709) J Clin Oncol. 2006;24:3423–3430. doi: 10.1200/JCO.2005.03.9271. [DOI] [PubMed] [Google Scholar]
- Villani MG, Appierto V, Cavadini E, Valsecchi M, Sonnino S, Curley RW, et al. Identification of the fenretinide metabolite 4-Oxo-fenretinide present in human plasma and formed in human ovarian carcinoma cells through induction of cytochrome P450 26A1. Clin Cancer Res. 2004;10:6265–6275. doi: 10.1158/1078-0432.CCR-04-0655. [DOI] [PubMed] [Google Scholar]
- Villani MG, Appierto V, Cavadini E, Bettiga A, Prinetti A, Clagett-Dame M, et al. 4-Oxo-fenretinide, a recently identified fenretinide metabolite, induces marked G2-M cell cycle arrest and apoptosis in fenretinide-sensitive and fenretinide-resistant cell lines. Cancer Res. 2006;66:3238–3247. doi: 10.1158/0008-5472.CAN-05-3362. [DOI] [PubMed] [Google Scholar]
- Vratilova J, Frgala T, Maurer BJ, Patrick RC. Liquid chromatography method for quantifying N-(4-hydroxyphenyl)retinamide and N-(4-methoxyphenyl)retinamide in tissues. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;808:125–130. doi: 10.1016/j.jchromb.2004.02.031. [DOI] [PubMed] [Google Scholar]
- William WN, Jr, Lee JJ, Lippman SM, Martin JW, Chakravarti N, Tran HT, et al. High-dose fenretinide in oral leukoplakia. Cancer Prev Res (Phila) 2009;2:22–26. doi: 10.1158/1940-6207.CAPR-08-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.