

Abstract
BACKGROUND AND PURPOSE
Diabetic cystopathy is one of the most common and incapacitating complications of diabetes mellitus. This study aimed to evaluate the functional, structural and molecular alterations of detrusor smooth muscle (DSM) in streptozotocin-induced diabetic mice, focusing on the contribution of Ca2+ influx through L-type voltage-operated Ca2+ channels (L-VOCC).
EXPERIMENTAL APPROACH
Male C57BL/6 mice were injected with streptozotocin (125 mg·kg−1). Four weeks later, contractile responses to carbachol, α,β-methylene ATP, KCl, extracellular Ca2+ and electrical-field stimulation were measured in urothelium-intact DSM strips. Cystometry and histomorphometry were performed, and mRNA expression for muscarinic M2/M3 receptors, purine P2X1 receptors and L-VOCC in the bladder was determined.
KEY RESULTS
Diabetic mice exhibited higher bladder capacity, frequency, non-void contractions and post-void pressure. Increased bladder weight, wall thickness, bladder volume and neural tissue were observed in diabetic bladders. Carbachol, α,β-methylene ATP, KCl, extracellular Ca2+ and electrical-field stimulation all produced greater DSM contractions in diabetic mice. The L-VOCC blocker nifedipine almost completely reversed the enhanced DSM contractions in bladders from diabetic animals. The Rho-kinase inhibitor Y27632 had no effect on the enhanced carbachol contractions in the diabetic group. Expression of mRNA for muscarinic M3 receptors and L-VOCC were greater in the bladders of diabetic mice, whereas levels of M2 and P2X1 receptors remained unchanged.
CONCLUSIONS AND IMPLICATIONS
Diabetic mice exhibit features of urinary bladder dysfunction, as characterized by overactive DSM and decreased voiding efficiency. Functional and molecular data suggest that overactive DSM in diabetes is the result of enhanced extracellular Ca2+ influx through L-VOCC.
Keywords: diabetes, detrusor smooth muscle, bladder dysfunction, L-type Ca2+ channels, Rho-kinase
Introduction
More than half of diabetic patients suffer with some type of diabetic bladder dysfunction (DBD) such as urgency, urinary incontinence and overactive bladder (Kaplan et al., 1995; Brown et al., 2005). Although this condition is not life-threatening, DBD affects many aspects of a patient's life, impairing social, physical and psychological activity, productivity at work and sexual health (Coyne et al., 2008; Irwin et al., 2008). Bladder dysfunction in diabetes is characterized by a large bladder capacity, decreased bladder sensation and poor bladder emptying, which lead to chronic urine retention and recurrent urinary tract infections. Bladder remodelling such as increases in total thickness, smooth muscle content and urothelial area has been observed in streptozotocin (STZ)-induced diabetic rats (Eika et al., 1994; Tammela et al., 1995) and mice (Poladia and Bauer, 2004). These changes may be a physical adaptation to increased urine production (Liu and Daneshgari, 2005; Daneshgari et al., 2006), as the requirement to void the excess urine would place extra physical stress on the bladder, and may be a reason for the onset of DBD.
The functions of the lower urinary tract to store and release urine are regulated by neural circuits located in the brain, spinal cord and peripheral ganglia (Abrams et al., 2006). The sacral parasympathetic outflow provides the major excitatory input to the urinary bladder via the release of cholinergic transmitters. Detrusor smooth muscle (DSM) expresses muscarinic M2 and M3 receptors in various animal species, but the latter are reported to be functionally more important for urinary bladder contractions (Matsui et al., 2000; Igawa et al., 2004). Muscarinic-mediated contractile responses are greater in the bladders of diabetic animals (Tammela et al., 1995), and this has been associated with up-regulation of M3 receptors (Cheng et al., 2007). However, other studies reported a decrease or no change in the muscarinic-dependent contractile responses in diabetes (Longhurst et al., 2004; Su et al., 2004). While ACh is the principal excitatory transmitter at the parasympathetic nerve terminals in DSM, there is good evidence that ATP, via P2X1 receptors, primarily mediates the atropine-resistant neurogenic bladder contractions, with variations among the mammalian species (Yoshimura et al., 2008). The purinergic atropine-resistant contraction is reported to contribute to some types of bladder dysfunction such as the overactive bladder secondary to outlet obstruction (Bayliss et al., 1999). However, the contribution of purine P2X receptors to the bladder dysfunction upon diabetic conditions remains poorly studied (Suadicani et al., 2009).
Smooth muscle contraction is regulated by an elevation of cytosolic Ca2+ via myosin light chain (MLC) phosphorylation, which is controlled by the balance between Ca2+ entry into the cell/release from intracellular stores, and Ca2+ sequestration/extrusion from the cell (Suzuki et al., 2010). It is well established that ACh stimulates muscarinic M3 receptors to cause DSM contractions via interaction with Gq to elicit phosphoinositide hydrolysis and generation of the second messenger inositol triphosphate (IP3), which activates the inositol triphosphate receptor to release Ca2+ from internal stores (Abrams et al., 2006). Activation of P2X receptors also leads to an increase in the intracellular Ca2+ concentration, with Ca2+ influx occurring through the pores of these channels (Koshimizu et al., 2000). Under physiological conditions, the muscarinic and purinergic-mediated bladder smooth muscle contractions are reported to also depend on extracellular Ca2+ influx secondary to L-type Ca2+ channel opening (Wegener et al., 2004; Rapp et al., 2005).
Although some of the reported data are contradictory, the contractile response of DSM in DBD is certainly altered. This could be a consequence of alterations in bladder autonomic innervation and/or changes at the level of receptor function and downstream intracellular second messenger systems. It is likely that alterations in the contractile properties of diabetic bladders are secondary to disturbances in Ca2+ handling/homeostasis, as these processes are so fundamental to regulation of contraction (Waring and Wendt, 2000). Despite their great importance in the regulation of receptor-mediated bladder contractions, little is known about the role of extracellular Ca2+ influx through voltage-operated L-type Ca2+ channels (L-VOCC) in DBD. The present study aimed to investigate the in vivo (cystometry), as well as the in vitro functional DSM alterations to receptor-dependent (muscarinic and purinergic responses) and -independent (responses to KCl and extracellular Ca2+) contractile responses in STZ-induced diabetic mice. This study also aimed to explore the contribution of Ca2+ sensitization and of Ca2+ influx through L-VOCC to the alterations of agonist-induced DSM contractions in the diabetic mice.
Methods
Animal model
All animal procedures and the experimental protocols were approved by the Ethical Principles in Animal Research adopted by Brazilian College for Animal Experimentation (COBEA). Male C57BL/6 mice (25–30 g) were housed at constant room temperature with 12 h light and dark cycles. Food and water were available ad libitum. The animals were divided into two groups, namely control and diabetic. Diabetic mice received a single i.p. injection of STZ at 125 mg·kg−1 dissolved in citrate buffer (20 mM, pH 4.5). Control mice were treated identically except that a similar volume of buffer was injected instead of STZ. Blood samples were taken from the tail 48 h after administration of STZ to confirm the induction of diabetes mellitus. Mice with serum glucose concentrations of ≥3 mg·mL−1 (as measured with the ACCUCHEK advantage blood glucose monitoring system; Roche Diagnostics, Indianapolis, IN, USA) were considered to be diabetic. All experimental comparisons of diabetic and non-diabetic mice were made 4 weeks following STZ administration.
Cystometry
Mice were anaesthetized with an i.p. injection of urethane (1.8 g·kg−1). Once surgical anaesthesia was reached, a 1 cm incision was made along the midline of the abdomen. The bladder was exposed and a butterfly cannula (25 G) was inserted into the bladder dome. The cannula was connected to a three-way tap, one port of which was connected to a pressure transducer and the other to the infusion pump through a catheter (PE50). Before the cystometry was started, the bladder was emptied via the third port. Continuous cystometry was carried out by infusing saline into the bladder at a rate of 0.6 mL·h−1. The following parameters were assessed: threshold pressure (TP; the intravesical pressure immediately before micturition); post-void pressure (PVP; the intravesical pressure immediately after micturition); peak pressure (PP; the peak pressure reached during micturition); capacity (CP; the volume of saline needed to induce the first micturition); compliance (CO; the ratio of CP to TP); frequency of voiding contractions (VC) and frequency of non-voiding contraction (NVCs). NVCs were defined as spontaneous bladder contractions greater than 4 mmHg from the baseline pressure that did not result in a void. Bladders from mice used in the cystometry were not used in the other experiments.
Histology
Control and diabetic mice were anaesthetized with isoflurane, and the urinary bladders were excised and fixed with phosphate-buffered formalin (10%) for 24 h. Bladders with intact urothelium were measured along perpendicular dimensions, and the volume was calculated according to the equation V = π(abc)/6, where a, b and c represent the three axes of the bladder. After macroscopic examination, the bladders were cut in half, and both halves were dehydrated, cleared in xylene and embedded in paraffin. Transverse sections (4 µm) were cut and stained with haematoxylin & eosin (H&E) and Masson trichrome.
Immunohistochemistry
Tissue sections (4 µm) were stained for smooth muscle actin and S100 protein (marker for neuronal cell protein) by an immunoperoxidase method. Briefly, two consecutive 4 µm thick sections were obtained, placed on silanized slides, deparaffinized in xylene and rehydrated. Endogenous peroxidase activity was quenched by incubating the slides with 3% H2O2 for 10 min. Antigen retrieval was performed by heating slides in citrate buffer (10 mM, pH 6.0) at 95°C for 30 min. As primary monoclonal antibodies, we used anti-smooth muscle actin (Clone1A4, Dako, diluted at 1:100) and anti-S100 (polyclonal rabbit, Dako, diluted at 1:400). Antigen-antibody binding was detected using the Advance system (Dako). Staining was achieved using 3,3-diamonobenzidine tetrahydrochloride (Sigma, St. Louis, MO, USA) and counterstaining using Mayer's haematoxylin. All reactions were performed using appropriate positive and negative controls. Sections of human and murine colon, known to contain smooth muscle and nerve fibres, respectively, expressing smooth muscle actin and S100 protein were used as positive controls in each batch of slides. Negative controls were obtained by omitting the primary antibody in adjacent sections.
Histomorphometry
Digital images from H&E, Masson trichrome and immunostained sections were obtained using a digital camera (Nikon Coolpix 995) connected to a bright field microscope (Nikon Eclipse E200). H&E images at low magnification (40×) were used to determine the bladder wall thickness. Collagen (C) content within bladder wall was determined using Masson's trichrome stained images, whereas smooth muscle (SM) and neural tissue (NT) contents were obtained from immunostained images (four random images from medium power fields – 100×), and expressed as SM, C and NT densities (% per mm2 of analysed tissue). The above-mentioned histomorphometric parameters were assessed using NIH's image analysis software (ImageJ 1.42).
In vitro functional studies
Mice were anaesthetized with isoflurane, and urinary bladders removed and sectioned horizontally at the level of the ureters. Two longitudinal DSM strips with intact urothelium were obtained from each bladder. Strips of DSM were mounted in 10 mL organ baths containing Krebs–Henseleit solution with the following composition (mM): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11 glucose, pH 7.4, at 37°C and bubbled with a gas mixture of 95% O2 and 5% CO2. Changes in isometric force were recorded using a Power Lab v.7.0 system (Colorado Springs, CO, USA). The resting tension was adjusted to 0.5 g at the beginning of the experiments. The equilibration period was 60 min and the bathing medium was changed every 15 min.
Concentration–response curves
To verify the viability of the preparations, high extracellular K+ solution (80 mM, achieved by replacement of NaCl in Krebs' buffer with an equimolar concentration of KCl) was added to the baths at the end of equilibration time. Cumulative concentration–response curves to the full muscarinic agonist carbachol (1 nM to 30 µM) were constructed by using one-half log unit. Concentration–response curves to carbachol were carried out in the absence and in the presence of either the Rho-kinase inhibitor Y27632 (10 µM) or the L-VOCC blocker nifedipine (3 nM). Non-cumulative concentration–response curves to the P2X receptor agonist α,β-methylene ATP (1–10 µM) were also obtained.
Nonlinear regression analysis to determine the pEC50 was carried out using GraphPad Prism (GraphPad Software, San Diego, CA, USA) with the constraint that Φ = 0. All concentration–response data were evaluated for a fit to a logistics function in the form:
E is the maximum response produced by agonists; c is the logarithm of the EC50, the concentration of drug that produces a half-maximal response; x is the logarithm of the concentration of the drug; the exponential term, n, is a curve-fitting parameter that defines the slope of the concentration–response line and Φ is the response observed in the absence of added drug. The values of pEC50 data represent the mean ± SEM maximal response (Emax); data were normalized to the wet weight of the respective urinary bladder strips, and the values of Emax were represented by mN mg-1 wet weight.
Concentration–response curves to extracellular CaCl2
To evaluate the direct effects of extracellular Ca2+ influx on the bladder contractions, cumulative concentration–response curves to CaCl2 (0.01–30 mM) in depolarizing conditions were constructed. The strips were prepared and mounted in 10 mL organ baths containing Krebs–Henseleit Ca2+-free solution containing EGTA (1 mM) to sequester Ca2+ ions, and cyclopiazonic acid (CPA, 1 µM) to deplete sarcoplasmic reticulum Ca2+ stores. Next, bath solution was removed and replaced by Krebs–Henseleit Ca2+-free solution containing KCl (80 mM) and CPA (1 µM). After an equilibration period of 15 min, the cumulative curve to CaCl2 was done (Lagaud et al., 1999).
Electrical-field stimulation-induced DSM contractions
Frequency–response curves (1–32 Hz) were elicited by stimulating the tissues for 10 s with pulses of 1 ms width at 80 V, with 3 min interval between stimulations. Subsequently, after incubation periods of 30 min, frequency–response curves were repeated in the presence of the muscarinic receptor antagonist atropine (1 µM) and/or the P2X receptor blocker suramin (100 µM), to confirm the responses were mediated by muscarinic (ACh) and P2X (ATP) receptor activation (Brading and Williams, 1990). Frequency–response curves were also repeated in the presence of the voltage-gated sodium channel blocker tetrodotoxin to confirm the neurogenic nature of the responses (1 µM).
Real-time RT-PCR
Total ribonucleic acid (RNA) was extracted from whole bladder samples with TRIzol (Gibco-BRL, Gaithersburg, MD, USA). RNA samples (3 µg) were incubated with 1 U DNaseI (Invitrogen Corp., Rockville, MD, USA) for 15 min at room temperature, and EDTA was added to a final concentration of 2 mM to stop the reaction. The DNaseI enzyme was subsequently inactivated by incubation at 65°C for 5 min. DNaseI-treated RNA samples obtained were then reverse transcribed with a Superscript III RT™ kit (Invitrogen, Life Technologies). cDNA syntheses were verified through polymerase chain reaction by amplification of β-actin gene. cDNA sample concentrations were quantified using a Nanodrop Spectrophotometer (ND-1000; Nanodrop Technologies Inc., Wilmington, DE, USA). Synthetic oligonucleotide primers were designed to amplify cDNA for the genes encoding the M2, M3 and P2X1 receptors, L-type Ca2+ channels, β-actin and GAPDH (PrimerExpress™; Applied Biosystems, Foster City, CA, USA). The primer sequences are listed in Table 1. The reactions were performed with 5 ng cDNA, 6 µL SYBR Green Master Mix® (Invitrogen Corp., Rockville, MD, USA) and the optimal primer concentration, in a total volume of 12 µL. Real-time RT-PCR was performed in equipment StepOnePlus™ Real-Time PCR System® (Applied Biosystems, Foster City, CA, USA). Reactions were prepared in MicroAmp Optical 96-well reaction plates (Applied Biosystems, Foster City, CA, USA). The reaction programme was: 95°C for 10 min, followed by 45 cycles of 95°C for 15 s then 60°C for 1 min. At the end of a normal amplification a degradation time was added, during which the temperature was increased gradually from 60°C to 95°C. Threshold cycle (Ct) was defined as the point at which the fluorescence rises appreciably above the background fluorescence. Gene expression was quantified using the Gnorm program (Vandesompele et al., 2002). Two replicas were run on the plate for each sample, and each sample was run twice independently. Results are expressed as mRNA levels of each gene studied, normalized according to β-actin and GAPDH expressions.
Table 1.
Sequence and size of amplified fragments for each primer pairs for muscarinic M2 and M3, L-type Ca2+ channel and purinergic P2X1
| Gene | Primer sequence | Tm |
|---|---|---|
| M2– F | 5′-ACACGGTTTCCACTTCCCTG-3′ | 82 |
| M2– R | 5′-TGCATGCGTCACCCTTTTG-3′ | |
| M3– F | 5′-CCCACAGGCAGTTCTCGAA-3′ | 81 |
| M3– R | 5′-CCTCCTAGATGACCGTTTCGT-3′ | |
| L-type Ca2+ channel – F | 5′-ACCCTCCTCCGTCGAATTC-3′ | 85 |
| L-type Ca2+ channel – R | 5′-GTGTGCCATCGCTGTTCAGA-3′ | |
| P2X1 – F | 5′-ATTCGCTTTGATATCCTTGTGG-3′ | 79 |
| P2X1 – R | 5′-GCCGATGGTAGTCATAGTAGGG-3′ | |
| GAPDH – F | 5′-TGCACCACCAACTGCTTA-3′ | 87 |
| GAPDH – R | 5′-GGATGCAGGGATGATGTTC-3′ | |
| β-actin – F | 5′-ACTGCCGCATCCTCTTCCT-3′ | 83 |
| β-actin – R | 5′-GAACCGCTCGTTGCCAATA-3′ |
Drugs
Urethane, STZ, carbachol, suramin, α,β-methylene ATP, atropine, nifedipine, CPA and Y27632 were obtained from Sigma Chem Co. (St. Louis, MO, USA).
Statistical analysis
Data are expressed as mean ± SEM of n experiments. In the cumulative concentration- and frequency–response curves data are expressed as mean of the contraction in mN·mg−1 of wet strip weight ± SEM of n experiments to control for differences in size between bladder strips. The program Instat (GraphPad Software) was used for statistical analysis. One-way analysis of variances (anova) followed by a Tukey's test was used. Student's paired t-test was performed when appropriate. P < 0.05 was accepted as significant.
Results
General characteristics of the diabetic model and morphometric analysis
The STZ-induced diabetic mice exhibited a marked increase in glucose blood levels compared with control group (3.44 ± 0.05 vs. 1.48 ± 0.08 mg·mL−1; P < 0.001; n = 7–10). The lower body weight and higher bladder weight seen in the diabetic mice (P < 0.05) confirmed the typical characteristics associated with STZ-induced diabetes (Table 2). In addition, morphometric analysis in the diabetic bladder mice revealed an increase in wall thickness, bladder volume and neural tissue density (Table 2, P < 0.05). The C content (13.9 ± 1.3 vs. 11.5 ± 1.5%) and smooth muscle density (85.9 ± 4.3 vs. 89.1 ± 1.9%) were not significantly modified in diabetic bladders compared with those from the control group. Figure 1 shows representative photomicrographs for bladder wall thickness, and smooth muscle, C and neural tissue densities.
Table 2.
Body weight, bladder weight and morphometric analysis in control and streptozotocin-induced diabetic mice
| Parameters | Control | Diabetic |
|---|---|---|
| Body weight (g) | 31.8 ± 0.3 | 23.0 ± 0.3** |
| Bladder weight (mg) | 26.3 ± 1.7 | 32.3 ± 1.9* |
| Bladder volume (mm3) | 15.82 ± 1.08 | 19.19 ± 1.10* |
| Bladder wall thickness (µm) | 348.37 ± 21.28 | 478.1 ± 29.11* |
| Smooth muscle density (%) | 89.14 ± 1.98 | 85.92 ± 4.29 |
| Collagen density (%) | 11.46 ± 1.46 | 13.88 ± 1.34 |
| Collagen/smooth muscle density ratio | 0.13 ± 0.02 | 0.15 ± 0.02 |
| Neural tissue density (%) | 3.88 ± 0.45 | 5.36 ± 0.73* |
Data represent the means ± SEM for 7–10 mice.
P < 0.05
P < 0.001 compared with control group (two-tailed t-test).
Figure 1.
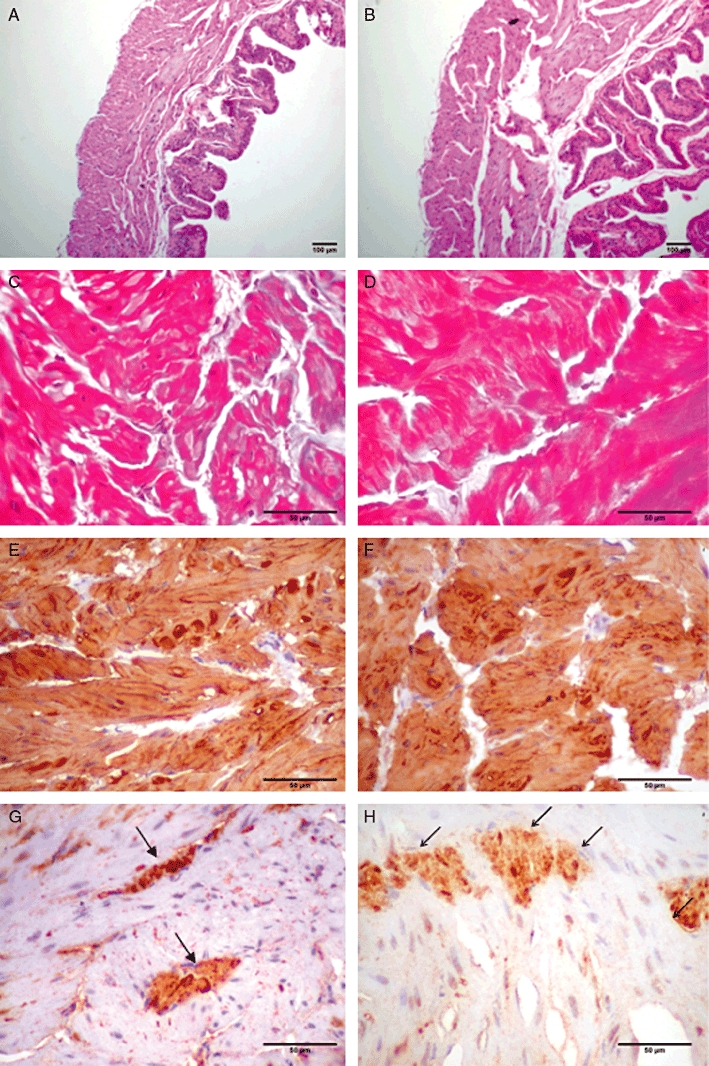
Photomicrographs illustrating the bladder wall thickness (A, control; B, diabetic); immunoexpression of smooth muscle actin in detrusor muscle fibres (C, control; D, diabetic); collagen fascicles (E, control; F, diabetic); and neural tissue density (G, control; H, diabetic). A/B: H&E (40× original magnification); C/D and G/H: immunoperoxidase (100× original magnification); E/F: Masson's trichrome (100× original magnification). Scale bar = 100 µm.
Cystometric studies
Figure 2 shows typical cystometric trace in control and STZ-induced diabetic mice. The micturition pattern in control mice was regular with rare NVCs (Figure 2A). Diabetic mice exhibited an irregular micturition pattern (Figure 2B), and a significant increase in bladder capacity, while TP remained unchanged (n = 6–7; Figure 3). Consequently, bladder compliance (CP/TP) was significantly higher (P < 0.05) in the diabetic group compared with control mice (0.09 ± 0.03 and 0.03 ± 0.007 mL·mmHg−1, respectively). The micturition frequency and amplitude of VCs, as well as the frequency and amplitude of NVCs were also significantly greater in the diabetic group (P < 0.01) compared with the control group (Figure 3). In addition, the PVP was significantly increased in diabetic animals (P < 0.001), and bladder never fully emptied in diabetic mice.
Figure 2.
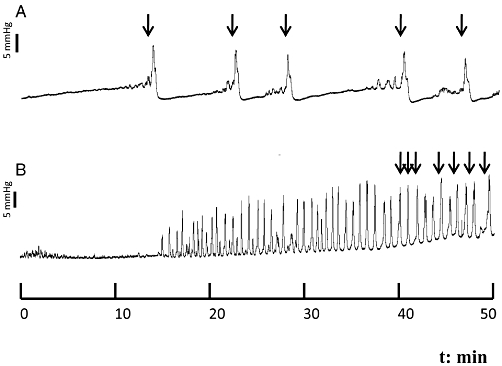
Representative cystometric recording from control (A) and streptozotocin-induced diabetic mice (B). Arrows in the cystometric trace indicate the micturition peaks.
Figure 3.
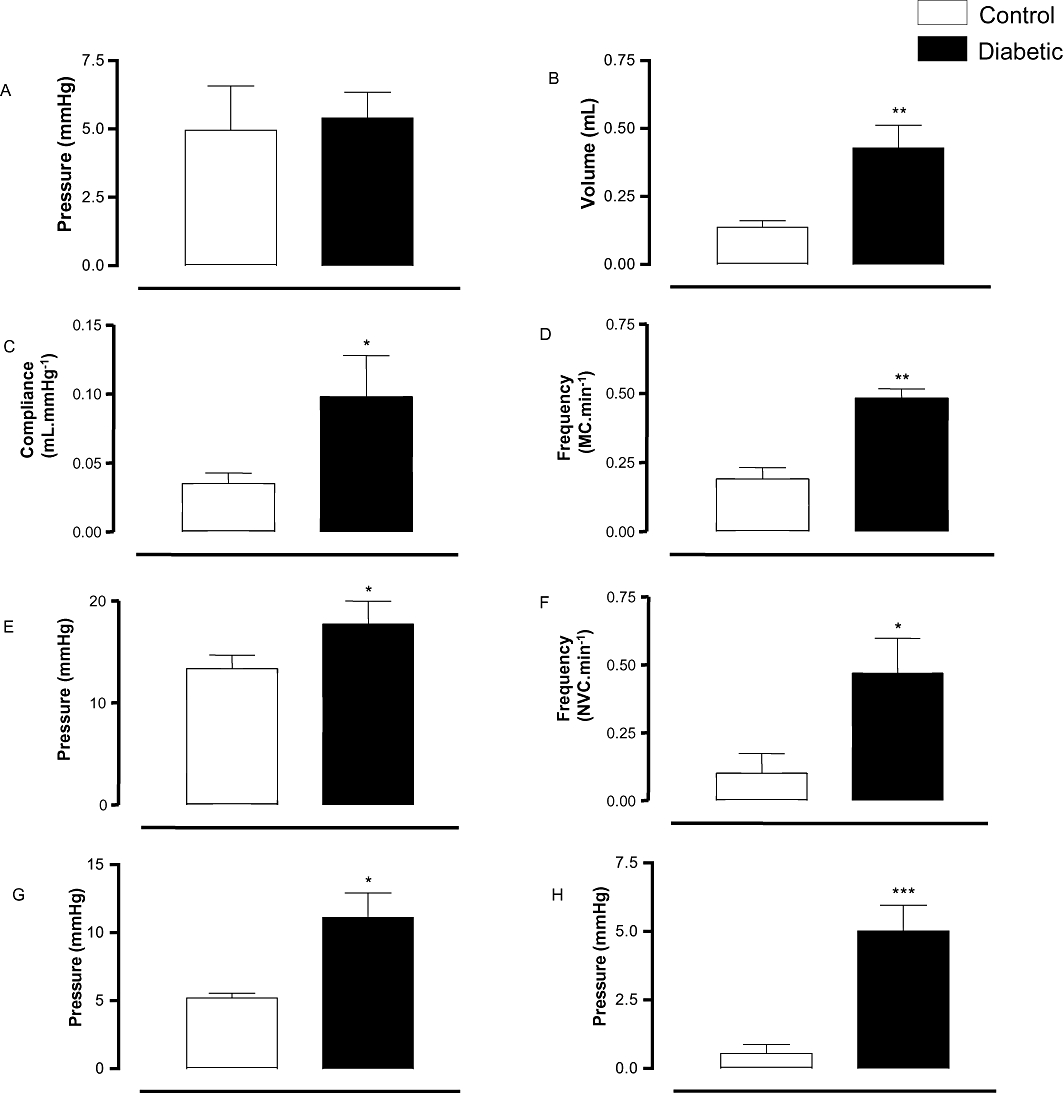
Cystometric study in control and streptozotocin-induced diabetic mice. (A) Threshold pressure, (B) capacity, (C) compliance, (D) micturition frequency, (E) peak pressure, (F) frequency of non-void contractions, (G) amplitude of non-void contractions and (H) post-void pressure. Data represent the means ± SEM for six to seven mice in each group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control group.
In vitro functional studies
Figure 4A shows that cumulative addition of the muscarinic agonist carbachol (1 nM–100 µM) produced concentration-dependent contractions in strips of isolated DSM. The maximal contractions were markedly higher in bladder muscle from the diabetic group (P < 0.01) compared with the control group (5.06 ± 0.62 and 2.04 ± 0.28 mN·mg−1, respectively; n = 8 each group). No differences in the pEC50 values for carbachol were found between control and diabetic groups (Table 3).
Figure 4.
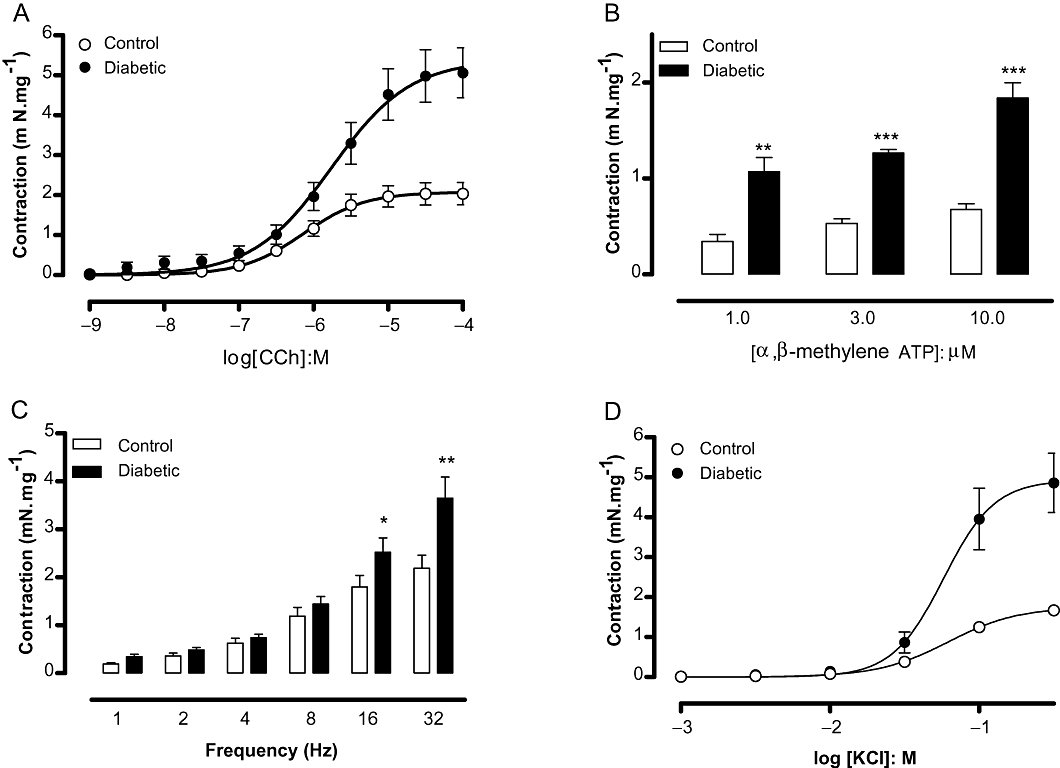
Detrusor smooth muscle contraction in response to the muscarinic agonist carbachol (A; n = 8), the P2X agonist α,β-methylene ATP (B; n = 4), electrical-field stimulation (C; n = 18) and potassium chloride (D; n = 7) in bladder strips from control and streptozotocin-induced diabetic mice. Data represent the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control group.
Table 3.
Potency (pEC50) and maximal responses (Emax) values obtained from concentration–response curves to carbachol and extracellular Ca2+ in detrusor smooth muscle from control and streptozotocin-induced diabetic mice
| Agonists | Groups | pEC50 | Emax (mN·mg·wet−1) |
|---|---|---|---|
| Carbachol | Control | 6.08 ± 0.09 | 2.04 ± 0.28 |
| Diabetic | 5.84 ± 0.10 | 5.06 ± 0.62** | |
| Control + nifedipine | 6.28 ± 0.06 | 1.55 ± 0.37 | |
| Diabetic + nifedipine | 6.04 ± 0.18 | 2.29 ± 0.43# | |
| Control + Y27632 | 5.84 ± 0.03 | 1.68 ± 0.31 | |
| Diabetic + Y27632 | 5.60 ± 0.05 | 4.41 ± 0.73* | |
| CaCl2 | Control | 2.06 ± 0.06 | 1.84 ± 0.23 |
| Diabetic | 2.13 ± 0.05 | 5.20 ± 0.66*** | |
| Control + nifedipine | 1.92 ± 0.07 | 0.95 ± 0.23 | |
| Diabetic + nifedipine | 1.75 ± 0.07 | 1.40 ± 0.26 |
Concentration–response curves to carbachol and Ca2+ were carried out in the absence or in the presence of either the Rho-kinase inhibitor Y27632 (10 µM) or the L-type calcium channel blocker nifedipine (3 nM). Potency is represented as −log of molar concentration to produce 50% of the maximal response. Data represent the mean ± SEM of five to seven mice.
P < 0.05
P < 0.01
P < 0.001 compared with respective control group.
P < 0.05 compared with untreated diabetic.
Similar to carbachol, the P2X receptor agonist α,β-methylene ATP (1–10 µM) caused concentration-dependent DSM contractions that were greater in bladder strips from diabetic mice compared with those from control mice (Figure 4B, n = 4 each group).
Electrical-field stimulation (EFS, n = 18) produced frequency-dependent DSM contractions in both groups, which were higher in the diabetic group at the highest frequencies employed (16 and 32 Hz; P < 0.05; Figure 4C). Pretreatment of DSM preparations with the muscarinic receptor antagonist atropine (1 µM) together with the purine receptor blocker suramin (100 µM) reduced the EFS-induced DSM contractions by approximately 60% (P < 0.01) in both control (2.18 ± 0.27 and 0.91 ± 0.24 mN·mg−1 for untreated and treated preparations, respectively) and diabetic mice (3.65 ± 0.42 and 1.29 ± 0.21 mN·mg−1 for untreated and treated preparations, respectively; n = 7), confirming that EFS-elicited contractions are mediated in part by ACh and ATP. In addition, pretreatment of DSM preparations with the voltage-gated sodium channel blocker tetrodotoxin (1 µM) nearly abolished the EFS-elicited contractions (>90% inhibition in all frequencies tested; n = 4).
In order to evaluate the DSM contractions independently of receptor stimulation, a concentration–response curve to KCl was constructed (n = 7; Figure 4D). Potassium chloride (10–30 mM) induced greater force development in bladder strips from diabetic mice (P < 0.001) than in those from the control group (Emax: 1.67 ± 0.11 and 4.86 ± 0.74 mN·mg−1 for control and diabetic, respectively).
Role of L-VOCC and Rho-kinase pathway in overactive DSM of diabetic mice
Pretreatment of DSM with the L-VOCC blocker nifedipine (3 nM; 30 min) did not significantly affect the carbachol-induced contractions in control bladder strips. At higher concentrations (30 and 300 nM), nifedipine greatly reduced carbachol-induced DSM contractions in control tissue (n = 4; data not shown). However, 3 nM nifedipine (n = 6) almost completely inhibited the enhanced carbachol-induced DSM contraction in the diabetic group, restoring the Emax to control levels (Figure 5A).
Figure 5.
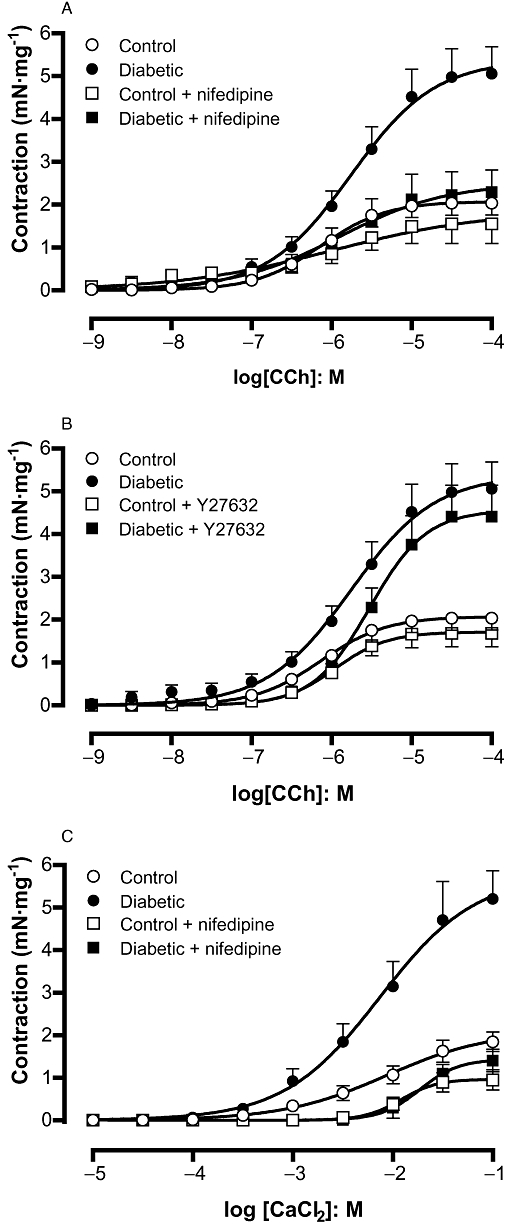
Detrusor smooth muscle contraction in response to the muscarinic agonist carbachol in the absence and in the presence of the L-type Ca2+ channel blocker nifedipine (A; 3 nM) or the Rho-kinase inhibitor Y27632 (B; 10 µM) in control and streptozotocin-induced diabetic mice. The effect of nifedipine on DSM contractions induced by extracellular calcium (in nominally Ca2+-free solution) in control and streptozotocin-induced diabetic mice is also shown (C). Data represent the means ± SEM for five to eight animals.
Pre-incubation of DSM with the Rho-kinase inhibitor Y27632 (10 µM) did not significantly affect the carbachol-induced DSM contractions either in diabetic or control DSM strips (Figure 5B). In separate experiments, DSM preparations from control mice were precontracted with KCl (80 mM), and concentration–relaxation curves to Y27632 were constructed. Cumulative addition of Y27632 produced concentration-dependent relaxations with a potency (pEC50) and maximum response (Emax) of 5.29 ± 0.20 and 79.6 ± 6.5% of KCl-induced contraction, respectively (n = 6).
Concentration–response curves to Ca2+ (0.1–100 mM, n = 5–7) were constructed in nominally Ca2+-free Krebs' solution containing KCl (80 mM) and CPA (1 µM) and supplemented with an appropriate concentration of CaCl2. DSM contractions to CaCl2 were higher in strips taken from diabetic DSM (P < 0.001) compared with those from control DSM (Figure 5C). Pretreatment of DSM with nifedipine reduced the CaCl2-evoked contractions in the diabetic group, restoring them to the same level as was seen in control tissue. In control DSM strips, nifedipine treatment significantly reduced the CaCl2-induced DSM contractions (Figure 5C).
No differences for the pEC50 values for carbachol and CaCl2 in the absence and the presence of either nifedipine or Y27632 were found between control and diabetic groups (Table 3).
Expression of mRNA for muscarinic M2 and M3 receptors, L-VOCC and P2X1 receptors
In bladders from control mice, the expression of muscarinic M2 receptor mRNA was greater than M3 receptor mRNA (Figure 6A,B, n = 5–7). In bladders from diabetic mice, the expression of M2 receptors did not change, whereas that of M3 receptors was significantly increased compared with control bladders (Figure 6A,B). Expression of L-VOCC mRNA was also significantly higher (P < 0.05) in diabetic bladders compared with control bladders (Figure 6D). STZ-induced diabetes did not modify the expression of mRNA for P2X1 receptors (Figure 6C).
Figure 6.
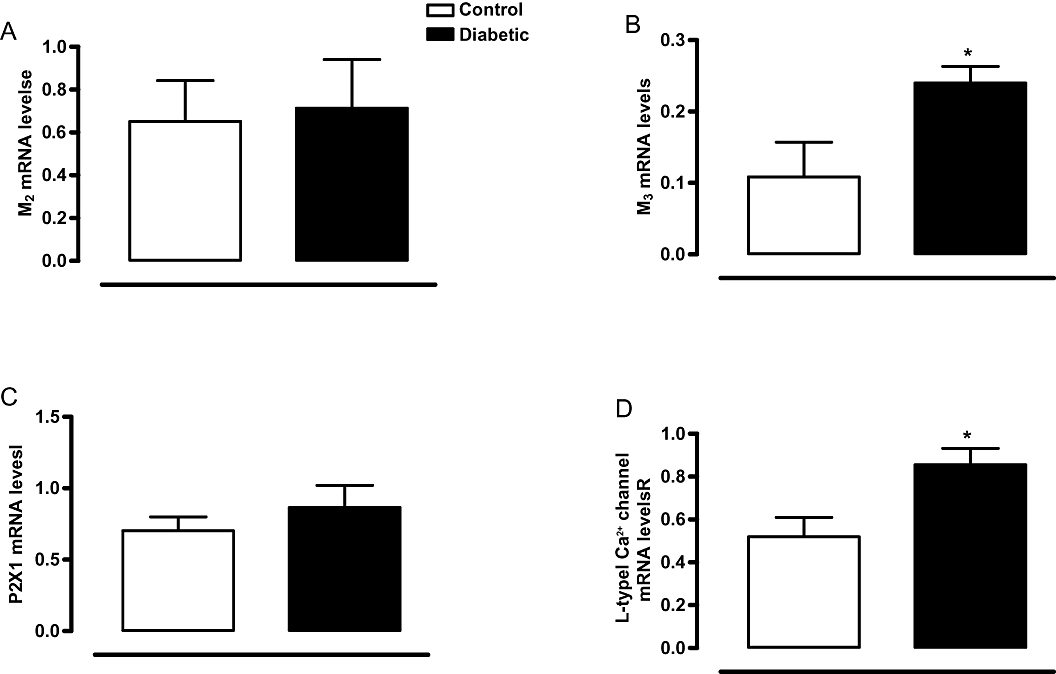
Relative bladder mRNA expression for muscarinic M2 (A) and M3 receptors (B), P2X1 receptors (C) and L-type voltage-operated Ca2+ channels (D) and in control and streptozotocin-induced diabetic mice. Values are expressed in arbitrary units. Data represent the means ± SEM for four to seven mice in each group. *P < 0.05 compared with control group.
Discussion
In this study we obtained data supporting previous findings that STZ-induced diabetic mice exhibit bladder dysfunction characterized by an overactive DSM. Furthermore, our findings suggest that the changes in smooth muscle activity are linked to increased expression and activity of L-VOCC and muscarinic M3 receptors.
Diabetes is associated with a number of cystopathic complications, including impaired bladder sensation, increase in residual volume, bladder overactivity and urge-incontinence (Yoshimura et al., 2005). STZ-induced diabetic rats or mice usually reproduce the main urodynamic alterations seen in clinical practice (Turner and Brading, 1999). However, discrepancies that possibly relate to animal species and strain used, and to the time-course of the diabetes, have been reported (Torimoto et al., 2004; Daneshgari et al., 2006; Melman et al., 2009). The results from our cystometry studies in STZ-induced diabetic mice confirmed that these mice exhibit most of the bladder complications seen in humans with diabetic cystopathies. These include increases in bladder compliance, micturition frequency and amplitude of VCs, as well as increases in amplitude and frequency of NVCs. Furthermore, the histomorphometric analysis revealed significant changes in diabetic bladder structure, which is in line with previous studies in STZ-induced diabetes (Liu and Daneshgari, 2005; Poladia and Bauer, 2005). The enhanced bladder wall thickness and volume in STZ-induced diabetic mice are consistent with the increases in bladder weight and capacity. These findings, along with cystometric data showing greater PVP in diabetic mice, point to an impaired bladder emptying, which is likely to reflect a failure to compensate for the enhanced diuresis and/or increased urethral resistance.
Autonomic neuropathy is a classical complication of diabetes in latter disease stages (Faerman et al., 1973; Vinik et al., 2003). In contrast, an enhancement of neural tissue density in diabetic mice bladder was found in our study, which is suggestive that 4 week STZ-induced diabetic mice did not progress to neuropathy, at least in the bladder. Of interest, increased expression of neurofilament protein has been reported in bladder dysfunction due to spinal cord injury (Yoshimura, 1999). It is likely that the increased neural tissue density contributes to the bladder dysfunction seen in the STZ-induced diabetic mice, as increased excitability of bladder afferent pathways, especially the C-fibre population, has been reported to be involved in the emergence of overactive bladder symptoms (Hirayama et al., 2003). Moreover, a previous study showed that EFS-induced contractions in rat bladders are higher at an early stage (7 days) of diabetes (Tammela et al., 1995). A cystometric study examined the temporal changes in bladder function from 3 to 20 weeks after induction of diabetes by STZ in male C57BL/6 mice (Daneshgari et al., 2006). These authors suggested that there is a transition from a compensated (increased bladder activity in response to elevated urine production) state to a decompensated bladder dysfunction (decreased bladder function), which would occur between 9–12 weeks after induction of diabetes. The neuropathy is believed to occur in this decompensated phase. In our model, therefore, we would be working with the compensated stage.
Functional studies in isolated bladder smooth muscle taken from diabetic rodents have generated contradictory data. Increased responsiveness of DSM strips to exogenous muscarinic agonists has been described in type 2 Goto-Kakizaki diabetic rats and diabetic ob/ob mice (Saito et al., 2008; Nobe et al., 2009), while DSM strips from STZ-induced diabetic rats and alloxan-induced diabetic rabbits instead showed a decrease or no changes in their muscarinic-mediated contractile responses (Longhurst et al., 2004; Su et al., 2004). These discrepancies are likely to reflect the animal species used, along with type and time of evaluation after induction of diabetes. The contribution of ATP as a parasympathetic co-transmitter activating P2X1 receptors and causing contraction, in healthy bladders, is suggested to be minor. However, in pathologically overactive bladders, the purinergic component accounts for up to half of the contraction (Palea et al., 1993; Bayliss et al., 1999). In a rabbit model of partial bladder outlet obstruction, a significant increase in the purinergic component of detrusor contraction was reported (Calvert et al., 2001). Nevertheless, the role of P2X1-induced contractions in DBD remains poorly explored. In our study, the contraction of DSM to exogenous muscarinic and purine receptor agonists (carbachol and α,β-methylene ATP, respectively) were markedly greater in diabetic mice compared with control animals. In agreement with these findings, the EFS-induced DSM contractions, which reflect the release of ACh and ATP from parasympathetic fibres (Tammela et al., 1994), were also higher in the diabetic group. These in vitro functional data are thus consistent with the cystometric alterations seen in anaesthetized diabetic mice. The hyperresponsiveness of DSM to carbachol, α,β-methylene ATP and EFS could reflect changes at the receptor level and/or beyond the receptor related to the downstream transducer signalling pathways. We thus decided to evaluate the expression of mRNA for muscarinic M2 and M3 as well as P2X1 receptors. In bladders from control mice, we found a higher expression of mRNA for muscarinic M2 than for M3 receptors, which is in agreement with previous studies (Matsui et al., 2000; Igawa et al., 2004). Moreover, in the diabetic mice bladder we detected a significantly greater expression of muscarinic M3 receptors than in control bladders, suggesting that up-regulation of such receptors contributes to overactive DSM in diabetes. At 8–9 weeks after diabetes induction, functional assays carried out in denuded bladder have recently revealed an increased M2 receptor function in comparison with control mice, suggesting that M2 receptor rescues impaired M3 receptor-mediated contraction in diabetes (Pak et al., 2010). Possibly the mice in their study are moving or have moved into the decompensated phase, so their bladders are behaving differently. If their bladders have decreased M3 function, and are not emptying properly, then there may be a subsequent compensatory increase in M2 receptors to enhance the muscle excitability. An increased expression of P2X1 receptor mRNA was reported in bladders from male human patients with detrusor instability caused by symptomatic bladder outlet obstruction (O'Reilly et al., 2001). However, in our study, despite the higher α,β-methylene ATP-induced DSM contractions in diabetic mice, no changes in mRNA expression for such receptors were found. Our data demonstrating increased expression of muscarinic M3 receptors, but no change in P2X1 receptors in diabetic mice, are consistent with previous studies using bladder tissue from diabetic rats (Cheng et al., 2007; Suadicani et al., 2009). It is important to mention that changes in mRNA expression may not correlate with the level of the protein, as it can vary independently of mRNA levels.
Over recent years there has been considerable interest in understanding the mechanisms by which Ca2+ mobilization can be modified in DSM in diabetes mellitus. Of particular interest is the [Ca2+]i-independent phosphorylation of MLC, which is regulated by the Rho-kinase signalling pathway (Chang et al., 2006; Christ and Andersson, 2007). The RhoA/Rho-kinase pathway modulates the level of phosphorylation of MLCs, mainly through inhibition of myosin phosphatase, and this process is reported to account for agonist-induced Ca2+ sensitization in smooth muscle contraction (Chitaley et al., 2001; Lee et al., 2004). The increased DSM contractions to carbachol in type 2 hyperglycaemic ob/ob mice were reduced by the Rho-kinase inhibitor fasudil (Nobe et al., 2009), whereas in this study using STZ-induced diabetic mice, the Rho-kinase inhibitor Y27632 had no significant effect on carbachol-induced contractions. The efficacy of Y27632 was confirmed by its ability to efficiently relax precontracted DSM preparations from control animals. Our data suggest that an increased activation of L-VOCC contributes to the enhanced carbachol-induced contractions in DSM strips from diabetic animals, and this is discussed in more detail below. Ca2+ entry through L-VOCC may elevate the cytosolic concentration to such an extent that contractions remain enhanced even when the presence of Y27632 reduces the Ca2+ sensitivity of the contractile proteins.
High levels of extracellular K+ depolarize the cell membrane and activate L-VOCC, leading to elevation of the intracellular Ca2+ concentration, which in turn activates contractile proteins (Andersson and Arner, 2004). In our study, KCl produced greater contractile responses in DSM strips from diabetic mice suggesting an enhancement of extracellular Ca2+ entry or greater Ca2+ sensitivity of the contractile proteins. In healthy conditions, Ca2+ entry through dihydropyridine-sensitive Ca2+ channels has been shown to be an important mechanism in generating the contractile response induced by EFS and carbachol in guinea-pig bladder (Rivera and Brading, 2006). Therefore, we next explored the contribution of extracellular Ca2+ influx via L-VOCC to the overactive DSM in diabetes. The enhanced DSM contractions to carbachol and extracellular Ca2+ in diabetic mice were normalized by the L-VOCC blocker nifedipine, at a concentration that had little effect on the DSM of control animals. Additionally, we found a significantly higher expression of L-VOCC mRNA in the bladders of diabetic mice. It is likely that in the diabetic state increased expression and/or sensitization of the L-type Ca2+ channel coupling to the M3 receptor facilitates the entry of Ca2+, which could contribute to contraction directly, or refill intracellular stores. Bladder outlet obstruction in rats leads to an increase in the cross-sectional area of the DSM cells and overactive DSM, and is suggested to be associated with a dysfunctional sarcoplasmic reticulum and with changes in the function of the L-VOCC (Stein et al., 2001). Whether the alteration in cell morphology causes the impaired Ca2+ handling or vice versa is unclear, but our finding of hyperplasia combined with enhanced L-VOCC activity supports the hypothesis that these are both key processes in the onset of DBD.
In conclusion, our data are consistent with the hypothesis that enhanced activation of L-VOCC downstream of muscarinic M3 receptors contributes to DBD in mice. This could be because more L-VOCC are present, or because the coupling is increased. The mechanisms by which this coupling occurs are not clear, but worthy of further investigation.
Acknowledgments
Luiz O.S. Leiria was a fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). Edson Antunes thanks Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
Glossary
Abbreviations
- C
collagen
- CO
compliance
- CP
capacity
- CPA
cyclopiazonic acid
- DBD
diabetic bladder dysfunction
- DSM
detrusor smooth muscle
- EFS
electrical-field stimulation
- EGTA
ethylene glycol-bis (2-amino-ethtlether)-N,N,N′,N′-tetra-acetic acid
- IP3
inositol trisphosphate
- L-VOCC
L-type voltage-operated Ca2+ channels
- MLC
myosin light chain
- NT
neural tissue
- NVC
non-voiding contraction
- PP
peak pressure
- PVP
post-void pressure
- SM
smooth muscle
- STZ
streptozotocin
- TP
threshold pressure
- VC
frequency of voiding contractions
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Abrams P, Andersson KE, Buccafusco JJ, Chapple C, de Groat WC, Fryer AD, et al. Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol. 2006;148:565–578. doi: 10.1038/sj.bjp.0706780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE, Arner A. Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev. 2004;84:935–986. doi: 10.1152/physrev.00038.2003. [DOI] [PubMed] [Google Scholar]
- Bayliss M, Wu C, Newgreen D, Mundy AR, Fry CH. A quantitative study of atropine-resistant contractile responses in human detrusor smooth muscle, from stable, unstable and obstructed bladders. J Urol. 1999;162:1833–1839. [PubMed] [Google Scholar]
- Brading AF, Williams JH. Contractile responses of smooth muscle strips from rat and guinea-pig urinary bladder to transmural stimulation: effects of atropine and a,-methylene ATP. Br J Pharmacol. 1990;99:493–498. doi: 10.1111/j.1476-5381.1990.tb12956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, Wessells H, Chancellor MB, Howards SS, Stamm WE, Stapleton AE, et al. Urologic complications of diabetes. Diabetes Care. 2005;28:177–185. doi: 10.2337/diacare.28.1.177. [DOI] [PubMed] [Google Scholar]
- Calvert RC, Thompson C, Khan MA, Mikhailidis DP, Morgan RJ, Burnstock G. Alterations in cholinergic and purinergic signalling in a model of the obstructed bladder. J Urol. 2001;166:1530–1533. [PubMed] [Google Scholar]
- Chang S, Hypolite JA, DiSanto ME, Changolkar A, Wein AJ, Chacko S. Increased basal phosphorilation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase β and CPI-17. Am J Physiol Renal Physiol. 2006;290:F650–F656. doi: 10.1152/ajprenal.00235.2005. [DOI] [PubMed] [Google Scholar]
- Cheng JT, Yu BC, Tong YC. Changes of M3-muscarinic receptor protein and mRNA expressions in the bladder urothelium and muscle layer of streptozotocin-induced diabetic rats. Neurosci Lett. 2007;423:1–5. doi: 10.1016/j.neulet.2007.05.062. [DOI] [PubMed] [Google Scholar]
- Chitaley K, Weber D, Webb RC. RhoA/Rho-kinase, vascular changes, and hypertension. Curr Hypertens Rep. 2001;3:139–144. doi: 10.1007/s11906-001-0028-4. [DOI] [PubMed] [Google Scholar]
- Christ GJ, Andersson KE. Rho-kinase and effects of rho-kinase inhibition on the lower urinary tract. Neurourol Urodyn. 2007;26:948–954. doi: 10.1002/nau.20475. [DOI] [PubMed] [Google Scholar]
- Coyne KS, Sexton CC, Irwin DE, Kopp ZS, Kelleher CJ, Milsom I. The impact of overactive bladder, incontinence and other lower urinary tract symptoms on quality of life, work productivity, sexuality and emotional well-being in men and women: results from the EPIC study. BJU Int. 2008;101:1388–1395. doi: 10.1111/j.1464-410X.2008.07601.x. [DOI] [PubMed] [Google Scholar]
- Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1728–R1735. doi: 10.1152/ajpregu.00654.2005. [DOI] [PubMed] [Google Scholar]
- Eika B, Levin RM, Longhurst PA. Comparison of urinary bladder function in rats with hereditary diabetes insipidus, streptozotocin-induced diabetes mellitus, and nondiabetic osmotic diuresis. J Urol. 1994;151:496–502. doi: 10.1016/s0022-5347(17)35001-2. [DOI] [PubMed] [Google Scholar]
- Faerman I, Glocer L, Celener D, Jadzinsky M, Fox D, Maler M, et al. Autonomic nervous system and diabetes. Histological and histochemical study of the autonomic nerve fibers of the urinary bladder in diabetic patients. Diabetes. 1973;22:225–237. doi: 10.2337/diab.22.4.225. [DOI] [PubMed] [Google Scholar]
- Hirayama A, Fujimoto K, Matsumoto Y, Ozono S, Hirao Y. Positive response to ice water test associated with high-grade bladder outlet obstruction in patients with benign prostatic hyperplasia. Urology. 2003;62:909–913. doi: 10.1016/s0090-4295(03)00588-0. [DOI] [PubMed] [Google Scholar]
- Igawa Y, Zhang X, Nishizawa O, Umeda M, Iwata A, Taketo MM, et al. Cystometric findings in mice lacking muscarinic M2 or M3 receptors. J Urol. 2004;172:2460–2464. doi: 10.1097/01.ju.0000138054.77785.4a. [DOI] [PubMed] [Google Scholar]
- Irwin DE, Milsom I, Reilly K, Hunskaar S, Kopp Z, Herschorn S, et al. Overactive bladder is associated with erectile dysfunction and reduced sexual quality of life in men. J Sex Med. 2008;5:2904–2910. doi: 10.1111/j.1743-6109.2008.01000.x. [DOI] [PubMed] [Google Scholar]
- Kaplan SA, Te AE, Blaivas JG. Urodynamic findings in patients with diabetic cystopathy. J Urol. 1995;153:342–344. doi: 10.1097/00005392-199502000-00013. [DOI] [PubMed] [Google Scholar]
- Koshimizu TA, Van Goor F, Tomić M, Wong AO, Tanoue A, Tsujimoto G, et al. Characterization of calcium signaling by purinergic receptor-channels expressed in excitable cells. Mol Pharmacol. 2000;58:936–945. doi: 10.1124/mol.58.5.936. [DOI] [PubMed] [Google Scholar]
- Lagaud GJ, Randriamboavonjv V, Roul G, Stoclet JC, Andriantsitohaina R. Mechanism of Ca2+ release and entry during contraction elicited by norepinephrine in rat resistance arteries. Am J Physiol. 1999;276:H300–H308. doi: 10.1152/ajpheart.1999.276.1.H300. [DOI] [PubMed] [Google Scholar]
- Lee DL, Webb RC, Jin L. Hypertension and RhoA/Rho-kinase signaling in the vasculature: highlights from the recent literature. Hypertension. 2004;44:796–799. doi: 10.1161/01.HYP.0000148303.98066.ab. [DOI] [PubMed] [Google Scholar]
- Liu G, Daneshgari F. Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am J Physiol Renal Physiol. 2005;288:F1220–F1226. doi: 10.1152/ajprenal.00449.2004. [DOI] [PubMed] [Google Scholar]
- Longhurst PA, Levendusky MC, Bezuijen MWF. Diabetes mellitus increases the rate of development of decompensation in rats with outlet obstruction. J Urol. 2004;171:933–937. doi: 10.1097/01.ju.0000093561.95283.df. [DOI] [PubMed] [Google Scholar]
- Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, et al. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci USA. 2000;97:9579–9584. doi: 10.1073/pnas.97.17.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman A, Zotova E, Kim M, Arezzo J, Davies K, DiSanto M, et al. Longitudinal studies of time-dependent changes in both bladder and erectile function after streptozotocin-induced diabetes in Fischer 344 male rats. BJU Int. 2009;104:1292–1300. doi: 10.1111/j.1464-410X.2009.08573.x. [DOI] [PubMed] [Google Scholar]
- Nobe K, Yamazaki T, Tsumita N, Hashimoto T, Honda K. Glucose-dependent enhancement of diabetic bladder contraction is associated with a Rho kinase-regulated protein kinase C pathway. J Pharmacol Exp Ther. 2009;28:940–950. doi: 10.1124/jpet.108.144907. [DOI] [PubMed] [Google Scholar]
- O'Reilly BA, Kosaka AH, Chang TK, Ford AP, Popert R, McMahon SB. A quantitative analysis of purinoceptor expression in the bladders of patients with symptomatic outlet obstruction. BJU Int. 2001;87:617–622. doi: 10.1046/j.1464-410x.2001.02179.x. [DOI] [PubMed] [Google Scholar]
- Pak KJ, Ostrom RS, Matsui M, Ehlert FJ. Impaired M3 and enhanced M2 muscarinic receptor contractile function in a streptozotocin model of mouse diabetic urinary bladder. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:441–454. doi: 10.1007/s00210-010-0509-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palea S, Artibani W, Ostardo E, Trist DG, Pietra C. Evidence for purinergic neurotransmission in human urinary bladder affected by interstitial cystitis. J Urol. 1993;150:2007–2012. doi: 10.1016/s0022-5347(17)35955-4. [DOI] [PubMed] [Google Scholar]
- Poladia DP, Bauer JA. Oxidant driven signaling pathways during diabetes: role of Rac1 and modulation of protein kinase activity in mouse urinary bladder. Biochimie. 2004;86:543–551. doi: 10.1016/j.biochi.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Poladia DP, Bauer JA. Functional, structural, and neuronal alterations in urinary bladder during diabetes: investigations of a mouse model. Pharmacology. 2005;74:84–94. doi: 10.1159/000083962. [DOI] [PubMed] [Google Scholar]
- Rapp DE, Lyon MB, Bales GT, Cook SP. A role for the P2X receptor in urinary tract physiology and in the pathophysiology of urinary dysfunction. Eur Urol. 2005;48:303–308. doi: 10.1016/j.eururo.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Rivera L, Brading AF. The role of Ca2+ influx and intracellular Ca2+ release in the muscarinic-mediated contraction of mammalian urinary bladder smooth muscle. BJU Int. 2006;98:868–875. doi: 10.1111/j.1464-410X.2006.06431.x. [DOI] [PubMed] [Google Scholar]
- Saito M, Okada S, Kazuyama E, Satoh I, Kinoshita Y, Satoh K. Pharmacological properties, functional alterations and gene expression of muscarinic receptors in young and old type 2 Goto-Kakizaki diabetic rat bladders. J Urol. 2008;180:2701–2705. doi: 10.1016/j.juro.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Stein R, Hutcheson JC, Gong C, Canning DA, Carr MC, Zderic SA. The decompensated detrusor IV: experimental bladder outlet obstruction and its functional correlation to the expression of the ryanodine and voltage operated calcium channels. J Urol. 2001;165:2284–2288. doi: 10.1016/S0022-5347(05)66185-X. [DOI] [PubMed] [Google Scholar]
- Su X, Changolkar A, Chacko S, Moreland RS. Diabetes decreases rabbit bladder smooth muscle contraction while increasing levels of myosin light chain phosphorylation. Am J Physiol Renal Physiol. 2004;287:F690–F699. doi: 10.1152/ajprenal.00027.2004. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Urban-Maldonado M, Tar MT, Melman A, Spray DC. Effects of ageing and streptozotocin-induced diabetes on connexin43 and P2 purinoceptor expression in the rat corpora cavernosa and urinary bladder. BJU Int. 2009;103:1686–1693. doi: 10.1111/j.1464-410X.2008.08337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Inoue T, Ra C. L-type Ca2+ channels: a new player in the regulation of Ca2+ signaling, cell activation and cell survival in immune cells. Mol Immunol. 2010;47:640–648. doi: 10.1016/j.molimm.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Tammela TL, Briscoe JA, Levin RM, Longhurst PA. Factors underlying the increased sensitivity to field stimulation of urinary bladder strips from streptozotocin-induced diabetic rats. Br J Pharmacol. 1994;113:195–203. doi: 10.1111/j.1476-5381.1994.tb16193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela TLJ, Leggett RE, Levin RM, Longhurst PA. Temporal changes in micturition and bladder contractility after sucrose dieresis and streptozotocin-induced diabetes mellitus in rats. J Urol. 1995;153:2014–2021. [PubMed] [Google Scholar]
- Torimoto K, Fraser MO, Hirao Y, de Groat WC, Chancellor MB, Yoshimura N. Urethral dysfunction in diabetic rats. J Urol. 2004;171:1959–1964. doi: 10.1097/01.ju.0000121283.92963.05. [DOI] [PubMed] [Google Scholar]
- Turner WH, Brading AF. Smooth muscle of the bladder in the normal and the diseased state: pathophysiology, diagnosis and treatment. Pharmacol Ther. 1999;75:77–110. doi: 10.1016/s0163-7258(97)00038-7. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH 0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes Care. 2003;26:1553–1579. doi: 10.2337/diacare.26.5.1553. [DOI] [PubMed] [Google Scholar]
- Waring JV, Wendt IR. Effects of streptozotocin-induced diabetes mellitus on intracellular calcium and contraction of longitudinal smooth muscle from rat urinary bladder. J Urol. 2000;163:323–330. [PubMed] [Google Scholar]
- Wegener JW, Schulla V, Lee TS, Koller A, Feil S, Feil R, et al. An essential role of Cav1.2 L-type calcium channel for urinary bladder function. FASEB J. 2004;18:1159–1161. doi: 10.1096/fj.04-1516fje. [DOI] [PubMed] [Google Scholar]
- Yoshimura N. Bladder afferent pathway and spinal cord injury: possible mechanisms inducing hyperreflexia of the urinary bladder. Prog Neurobiol. 1999;57:583–606. doi: 10.1016/s0301-0082(98)00070-7. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Chancellor MB, Andersson KE, Christ GJ. Recent advances in understanding the biology of diabetes-associated bladder complications and novel therapy. BJU Int. 2005;95:733–738. doi: 10.1111/j.1464-410X.2005.05392.x. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Kaiho Y, Miyazato M, Yunoki T, Tai C, Chancellor MB, et al. Therapeutic receptor targets for lower urinary tract dysfunction. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:437–448. doi: 10.1007/s00210-007-0209-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.