

Abstract
Myeloid cells provide important functions in low oxygen (O2) environments created by pathophysiological conditions, including sites of infection, inflammation, tissue injury and solid tumors. Hypoxia-inducible factors (HIFs) are principle regulators of hypoxic adaptation, regulating gene expression involved in glycolysis, erythropoiesis, angiogenesis, proliferation and stem cell function under low O2. Interestingly, increasing evidence accumulated over recent years suggests an additional important regulatory role for HIFs in inflammation. In macrophages, HIFs not only regulate glycolytic energy generation, but also optimize innate immunity, control pro-inflammatory gene expression, mediate bacterial killing and influence cell migration. In neutrophils, HIF-1α promotes survival under O2-deprived conditions and mediates blood vessel extravasation by modulating β2 integrin expression. Additionally, HIFs contribute to inflammatory functions in various other components of innate immunity, such as dendritic cells, mast cells and epithelial cells. This review will dissect the role of each HIF isoform in myeloid cell function and discuss their impact on acute and chronic inflammatory disorders. Currently, intensive studies are being conducted to illustrate the connection between inflammation and tumorigenesis. Detailed investigation revealing interaction between microenvironmental factors such as hypoxia and immune cells is needed. We will also discuss how hypoxia and HIFs control properties of tumor-associated macrophages and their relationship to tumor formation and progression.
Introduction
Tissue O2 concentrations are typically maintained by homeostatic mechanisms operating at the cellular, organ, and systemic levels. In vivo, O2 tension varies from 2.5% to 9% in most healthy tissues. However, inflamed or diseased tissues can be deprived of O2 due to vascular damage, intensive metabolic activity of bacteria and other pathogens, and large numbers of infiltrating cells, leading to O2 levels of less than 1% (Leek and Harris, 2002; Lewis et al., 1999). As the front line of a body’s defense, myeloid cells are required to function in this hypoxic microenvironment to combat infection, mediate inflammation, promote adaptive immunity and perform tissue repair functions (Lewis et al., 1999). These cells are unique in that they are well-adapted to hypoxia both metabolically and functionally. For example, neutrophils naturally operate under a proglycolytic program, and low O2 endows neutrophils with a survival advantage over normoxic conditions (Hannah et al., 1995; Walmsley et al., 2005a; Walmsley et al., 2005b). Macrophages specifically infiltrate hypoxic tissues, switch their metabolic program to glycolysis, resist apoptotic stimuli, and respond to O2 deprivation by altering gene expression to maximize their biological properties (Cramer et al., 2003; Murdoch et al., 2004).
Cells adapt to hypoxia by shifting their energy generation pathway from aerobic oxidative phosphorylation to anaerobic glycolysis, and recovering blood supply via stimulation of erythrocyte production and the generation of new blood vessels (Semenza, 2009). In addition to these established cellular responses, another aspect of hypoxia in connection with inflammation has been increasingly appreciated over recent years. A low O2 microenvironment appears to actually amplify myeloid cell-mediated inflammatory responses, contributing to a highly inflamed state. During O2 deprivation, many cellular responses are primarily regulated by hypoxia-inducible factors (HIFs). In pathological settings which involve inflammation or innate defense processes, HIFs are required to control programs associated with a broad range of myeloid cell functions (Cramer et al., 2003; Jantsch et al., 2008; Peyssonnaux et al., 2007; Peyssonnaux et al, 2005; Walmsley et al., 2005b). HIFs are therefore essential regulators of inflammation and innate immunity, as will be detailed below. The relationship between inflammation and cancer, and how hypoxia and HIFs contribute to these processes will also be discussed. Adaptation of tumor-associated macrophages (TAMs) to hypoxic tumor microenvironments and their influences on tumor phenotypes will be highlighted.
Regulation of HIF transcriptional pathways
Oxygen-dependent HIF activity
In mammalian cells, hypoxic adaptation is primarily regulated by master transcriptional factors, called HIFs, whose activity is based on the post-translational modification and stability of their α subunits (HIF-1α and HIF-2α). In O2 replete cells, prolyl hydroxylases (PHD-1, -2 and -3) modify the α subunit at two conserved prolines, resulting in polyubiquitylation via a specific von Hippel-Lindau (pVHL)-E3 ligase complex and subsequent degradation by proteasomes (Ivan et al., 2001; Jaakkola et al., 2001; Masson et al., 2001; Yu et al., 2001). Meanwhile, asparaginyl hydroxylation of HIF-α by factor inhibiting HIF (FIH) prevents its interaction with the co-activator p300/CBP, resulting in transcriptional inactivation under normoxia (Lando et al., 2002; Mahon et al., 2001; Sang et al., 2002). At sites of reduced O2 tension, PHD and FIH hydroxylase activities are reduced. Stabilized α subunits then translocate to the nucleus, form dimers with constitutive HIF-1β (also known as the aryl hydrocarbon receptor nuclear translocator [ARNT]) and bind to co-activators, permitting transcriptional activation of many hypoxia-response element (HRE)-bearing genes encoding metabolic, angiogenic and metastatic factors (Covello and Simon, 2004).
Inflammatory stimuli-induced HIF activity
Besides O2-dependent activation pathways, HIFs are also induced by inflammatory cytokines, growth factors and bacterial products at normoxic conditions, although the underlying molecular pathways are not fully revealed. Pro-inflammatory cytokines TNF-α and IL-1β have been shown to increase accumulation and transcriptional activity of HIF-1α. TNF-α-induced HIF-1α stimulation requires NF-κB at the level of HIF-1α protein stabilization without affecting its mRNA level (Jung et al., 2003a; Zhou et al., 2003). Similarly, IL-1β acts on HIF-1α protein stability by triggering NF-κB activity and inhibiting VHL-mediated protein degradation (Jung et al., 2003b). Moreover, TGF-β1 enhances HIF-1α protein stability by inhibiting PHD2 expression, via Smads (McMahon et al., 2006). The fact that HIF can be activated in response to inflammatory cytokines indicates HIF may play an important role in inflammation.
In addition to cytokines, bacteria and bacterial products such as LPS also stimulate HIF-1α activity under normal O2 levels. Several pathways have been reported to be involved in this process, including NF-κB (Fang et al., 2009; Frede et al., 2006; Nishi et al., 2008; Rius et al., 2008), ROS (Nishi et al., 2008), PHDs (Peyssonnaux et al., 2007) and p42/p44 mitogen-activated protein kinases (MAPKs) (Frede et al., 2006). The implication of NF-κB in this process has been controversial. Frede et al. reported that LPS induces HIF-1α mRNA expression in human monocytes through NF-κB binding to the promoter of the HIF-1α gene (Frede et al., 2006). Using IKK-β mutant macrophages, it was also shown that NF-κB is responsible for HIF-1α transcription and protein stability, and that IKK-β deficiency results in decreased expression of HIF targets such as glucose transporter-1 (Glut-1). In contrast, other studies demonstrated HIF-1α induction by LPS is not dependent on NF-κB activity (Fang et al., 2009; Nishi et al., 2008), but rather ROS generation (Nishi et al., 2008). Additionally, it has been shown that LPS increases HIF-1α protein accumulation through decreasing PHD2 and PHD3 levels in macrophages in a Toll-like receptor-4 (TLR4)-dependent manner (Peyssonnaux et al., 2007). Future studies elucidating the crosstalk between HIFs and NF-κB are required given the importance of these two transcriptional factors in regulating hypoxic response and inflammation, respectively.
Role of HIFs in myeloid cell functions
HIF regulates macrophage activity
Macrophages display a variety of functions depending upon the type of stimulus presented in the local environment (Gordon, 2003; Mosser and Edwards, 2008). Interestingly, these cells accumulate in large numbers within O2-deprived areas in various diseases such as bacterial infections, atherosclerosis, rheumatoid arthritis, wounds and solid tumors, suggesting that hypoxic responses regulate macrophage biological activities (Murdoch et al., 2005). Exposure to hypoxia markedly changes macrophage gene expression profiles, resulting in the up-regulation of surface receptors (e.g. CXCR4 and Glut-1) and pro-angiogenic factors (Fang et al., 2009). Two α subunits, HIF-1α and HIF-2α, have been demonstrated to promote the expression of most O2-regulated genes (Covello and Simon, 2004). Whereas HIF-1α appears to be expressed ubiquitously, HIF-2α is expressed in a more tissue-restricted manner (Covello and Simon, 2004). In macrophages, both HIF-1α and HIF-2α expression are induced in response to hypoxia in vitro (Burke et al., 2002; Griffiths et al., 2000). Moreover, HIF-1α appears to be required for macrophage maturation (Fang et al., 2009; Oda et al., 2006). Interestingly, HIF-2α protein is readily detected in vivo in bone marrow macrophages and has been shown to be highly expressed in TAMs found in various human cancers (Talks et al., 2000). To elucidate the relative contribution of each HIF-α in the regulation of hypoxia-induced macrophage gene expression, siRNA-mediated knockdown of individul HIF-α subunits were performed in human monocyte-derived macrophages (Fang et al., 2009). Whereas HIF-1α and HIF-2α regulate expression of multiple common genes such as CXCR4, Glut-1, adrenomedulin (ADM) and STAT-4, expression of certain genes such as adenosine A2a (ADORA2A) and ICAM1 was only modulated by HIF-2α. Furthermore, over-expression of HIF-2α, but not HIF-1α, in normoxic human macrophages leads to enhanced transcription of pro-angiogenic genes including VEGF, IL-8, platelet-derived growth factor β (PDGFB) and angiopoietin-like 4 (ANGPTL4) (White et al., 2004). Collectively, these studies suggest HIF isoforms may play overlapping, but also distinct, roles in macrophage adaptation to low O2.
To investigate macrophage biological properties, myeloid-specific ablation of the HIF-1α subunit in mice was created by crossing the floxed Hif-1α allele with a lysozyme M cre line (Cramer et al., 2003). This study demonstrated a dominant role for HIF-1α in regulating glycolysis in macrophages (Cramer et al., 2003) as HIF-1α deficiency results in a dramatically reduced ATP pool. This is consistent with other studies demonstrating that HIF-1α exclusively controls glycolysis (Hu et al., 2003). The metabolic defect in HIF-1α deletion in macrophages results in impairment of energy-demanding processes such as aggregation, migration and invasion (Cramer et al., 2003).
In addition to its key role in regulating metabolism and energy generation, fundamental work by Cramer et al. showed that HIF-1α mediates macrophage inflammatory responses. Compared to control mice, myeloid HIF-1α-null mice displayed reduced acute skin inflammation triggered by 12-O-tetradecanoylphorbol-13-acetate (TPA), as evidenced by decreased edema and leukocyte infiltration (Cramer et al., 2003). When induced to develop arthritis, these mice also showed compromised synovial infiltration, pannus formation and cartilage destruction, suggesting ameliorated chronic inflammatory responses mediated by HIF-1α-deficient macrophages.
Many of the pro-inflammatory cytokine/chemokine genes are activated by hypoxic treatment in human primary macrophages. Compromised expression of IL-1β, CXCL8 and VEGF was observed in cells exhibiting reduced expression of either HIF-1α or HIF-2α, indicating both HIFs are important for macrophage cytokine expression (Fang et al., 2009). Given these cytokines/chemokines are also known to be NF-κB targets, the role of NF-κB in inducing their expression under low O2 concentration has been evaluated. However, inactivation of NF-κB, either chemically or genetically, did not influence hypoxia-induced cytokine expression (Fang et al., 2009). This result suggests that HIFs, but not NF-kB, are important transcriptional effectors regulating the hypoxic gene expression of macrophages.
Innate immunity was also assessed in myeloid HIF-1α null mice by Peyssonnaux et al. (Peyssonnaux et al., 2005). The authors demonstrated that loss of myeloid HIF-1α resulted in decreased bacterial killing of group A Streptococcus and P. aeruginosa by macrophages in vitro and in vivo (Peyssonnaux et al., 2005), revealing the importance of myeloid HIF-1α in this process. Furthermore, exposure to these pathogens and LPS under normoxia induces HIF-1α activity in macrophages in a TLR-4 dependent fashion (Peyssonnaux et al., 2007). Finally, HIF-1α directly binds to the promoter of the Toll-like receptor 4 (TLR4) locus and up-regulates TLR4 expression during O2 deprivation (Kim et al., 2009). Thus, the interdependent relationship between HIF-1α and TLR4 activation may result in a positive feedback loop, amplifying HIF responses under hypoxia and infection. Collectively, these findings suggest hypoxic stress at sites of inflammation both enhances sensitivity to infection by strengthening TLR4 signaling and promotes the defense capacity of macrophages by increasing HIF-1α levels (Figure 1).
Figure 1. Possible mechanisms for maximal inflammatory responses elicited by LPS during hypoxia.
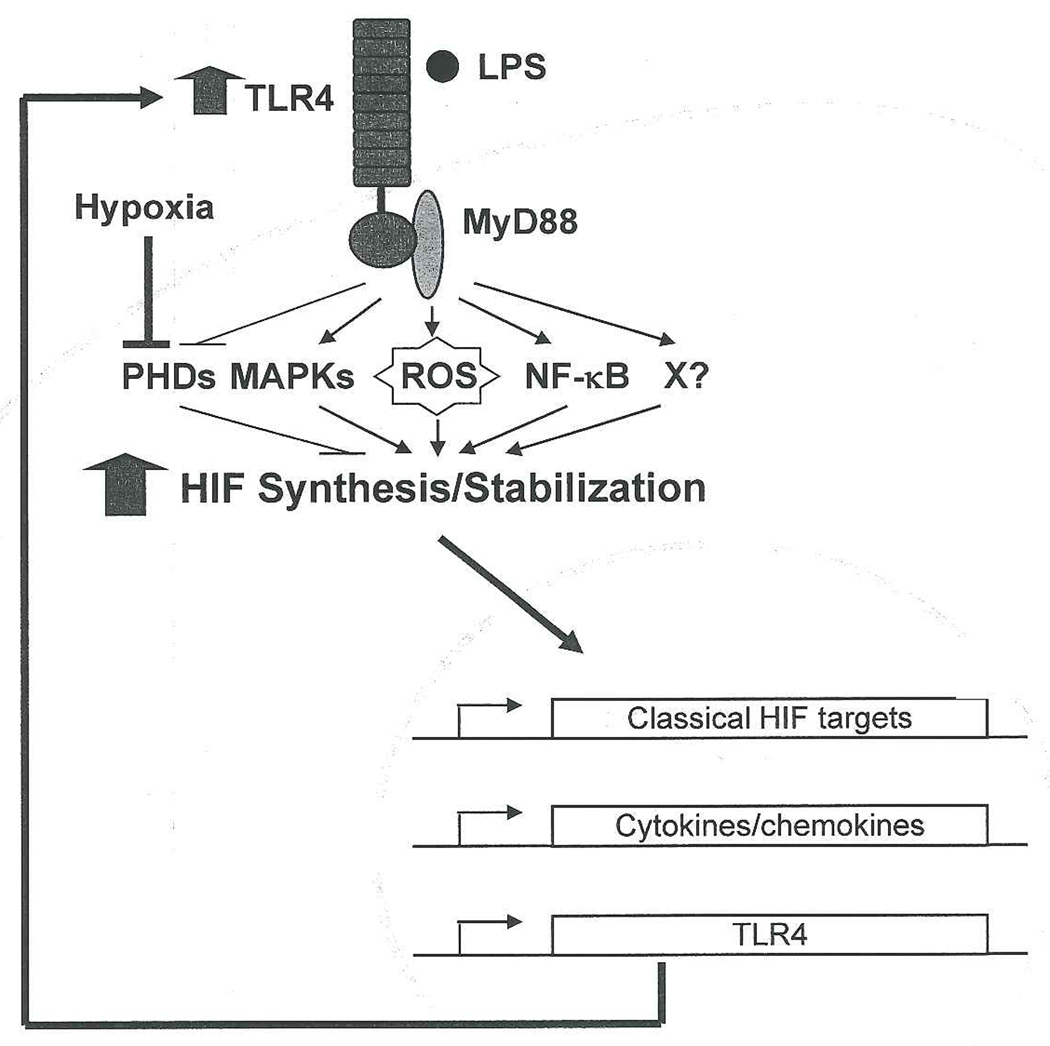
In macrophages, LPS induces HIF protein synthesis and stabilization in a TLR4/MyD88-dependent fashion. The signaling pathways for LPS-induced HIF stabilization possibly involve ROS, inhibition of PHDs, MAPK, NF-κB and/or unknown factors (X). Upon HIF protein accumulation, it translocates to the nucleus and stimulates expression of classical HIF target genes, inflammatory cytokines/chemokines, and TLR4. TLR4 further enhances HIF expression and transcriptional activity, resulting in a positive feedback loop. Together with the LPS-TLR4 pathway, hypoxia itself stabilizes HIF protein via inhibiting PHDs, serving to amplify HIF-mediated inflammatory responses during infection and O2 deprivation.
Given the importance of HIF-2α in macrophages implicated by gene expression studies (Fang et al., 2009; Griffiths et al., 2000; Talks et al., 2000; White et al., 2004), genetic experiments utilizing mouse models have now been performed to investigate the role of HIF-2α in various aspects of macrophage functions (Imtiyaz et. al., manuscript submitted). A clear division of labor exists between the two HIF-α isoforms in these cells where HIF-2α is clearly important for inflammatory cytokine expression, macrophage migration, and responses to inflammatory stimuli.
HIF function in other myeloid cells
Neutrophils are important phagocytes which clear invading pathogens and mediate acute inflammation (Bredetean et al., 2007). As they rely on glycolysis to generate ATP, these cells seem to be well-suited to function in the hypoxic microenvironment naturally present in inflammatory lesions (Walmsley et al., 2005a). Studies of HIF-1α-deficient neutrophils revealed that neutrophils require HIF-1α to perform glycolysis (Cramer et al., 2003). Using murine bone marrow-derived neutrophils and human peripheral blood neutrophils, Walmsley et al. demonstrated that cells deficient in HIF-1α failed to resist apoptosis under hypoxic conditions (Walmsley et al., 2005b). Moreover, this HIF-mediated survival effect is also dependent on the activity of NF-κB, and is eliminated with treatment of NF-κB inhibitors (gliotoxin and parthenolide). Migration of neutrophils from circulation to sites of infection or tissue damage involves a process of selectin-mediated rolling and β2 integrin-mediated adhesion to endothelium (Carlos and Harlan, 1994). It has been demonstrated that HIF-1α regulates β2 integrin (CD18 specifically) expression in these cells and thereby promotes neutrophil extravasation (Kong et al., 2004). HIF-2α is unlikely to play a role in neutrophil function as it is not expressed in this lineage (Walmsley et al., 2005b) (Imtiyaz et al., manuscript submitted).
Progress has been made in deciphering the role of HIF-1α in two other types of myeloid cells. As professional antigen presentation cells, dendritic cells (DCs) play a key role in linking innate and adaptive immunity. Recent work by Jantsch and collegues has revealed that hypoxia and HIF-1α modulate dendritic cell maturation, activation and antigen-presenting functions (Jantsch et al., 2008). Although hypoxia alone did not activate DCs, hypoxia combined with LPS led to marked increases in expression of co-stimulatory molecules, pro-inflammatory cytokine synthesis and induction of lymphocyte proliferation compared with LPS alone. This DC activation was accompanied by HIF-1α protein accumulation and enhanced glycolytic activity. Moreover, knockdown of HIF-1α significantly reduced glucose uptake, inhibited maturation and led to an impaired capacity to stimulate allogeneic T cells (Jantsch et al., 2008). Mast cells are granulocytes implicated in allergy, and evidence regarding their roles in both innate and adaptive immunity is now emerging. Activation of HIF-1α in mast cells stimulates expression of VEGF, CXCL8, IL-6 and TNF (Jeong et al., 2003; Lee et al., 2008). It will be interesting to determine the expression and possible function of HIF-2α in both DCs and mast cells in future experiments.
Hypoxia, HIFs and inflammatory diseases
Sepsis
Sepsis is an aberr ant host inflammatory response provoked by overwhelming infection or lipopolysaccharide (LPS). It leads to the potentially lethal systemic inflammatory response syndrome (SIRS) characterized by acute inflammation, hemodynamic compromise, multi-organ failure and even shock (Jean-Baptiste, 2007; Parrillo, 1993). Currently, sepsis is still the leading cause of mortality in intensive care units. The systemic effects of LPS are largely mediated by macrophages which produce a wide array of inflammatory cytokines (Jean-Baptiste, 2007; Ulloa and Tracey, 2005). Pro-inflammatory cytokines IL-1β, IL-12, TNF-α, and INF-γ have all been implicated in the toxic effects of endotoxemia, as neutralization of individual cytokines by specific antibodies protects mice from LPS-induced lethality (Dinarello, 1991; Doherty et al., 1992; Heinzel, 1990; Kumar et al., 1996; Tracey et al., 1987; Zisman et al., 1997). In contrast, the anti-inflammatory cytokine IL-10 has been proven to be beneficial (Howard et al., 1993; Nicoletti et al., 1997; Standiford et al., 1995). Studies of LPS-induced responses in myeloid HIF-targeted mice revealed that HIF-1α is important for the sepsis phenotype. HIF-1α deletion in myeloid cells led to reduced pro-inflammatory cytokines such as TNF-α, IL-1 and IL-12 (Peyssonnaux et al., 2007). In addition, HIF-1α contributes to the lethal effects of LPS as mice survived much longer when myeloid HIF-1α is absent. Moreover, HIF-1α deletion blocked LPS-induced hypotension and hypothermia caused by sepsis (Peyssonnaux et al., 2007). Whether HIF-2α ablation in macrophages results in a similar septic phenotype requires future study.
Atherosclerosis
Atherosclerosis is a chronic inflammatory response in the walls of arteries, in large part due to the accumulation of macrophages (known as foam cells) that take up oxidized low-density lipoproteins (Portugal et al., 2009). The build-up of “fatty streaks” by these fat-containing macrophages forms atherosclerotic plaques, leading to arterial stenosis and impaired perfusion (Zemplenyi et al., 1989). Using the hypoxia marker 7-(4'-(2-nitroimidazol-1-yl)-butyl)-theophylline, a previous study has shown zones of hypoxia are present in the plaques, probably due to impaired O2 diffusion into these lesions (Bjornheden et al., 1999). At least two macrophage-derived products have been implicated in the plaque development. The first one is very low-density lipoprotein receptor (VLDLR) which has been shown to be expressed by macrophages in atherosclerotic lesions (Nakazato et al., 1996). In vitro, VLDLR levels are increased by hypoxia in macrophages (Nakazato et al., 2001), although whether this is dependent on HIF expression is currently unclear. The second factor is CXCL-8 (i.e. IL-8), a potent chemoattractant for T lymphocytes and smooth muscle cells. Significant elevation in CXCL-8 production has been found in foam cells isolated from human atherosclerotic plaques compared with macrophages in culture (Liu et al., 1997). Of note, foam cells found in hypoxic zones displayed enhanced CXCL-8 levels in both rabbit and human atherosclerotic sites compared to the normal arterial wall (Rydberg et al., 2003). Moreover, hypoxia induces CXCL-8 expression in primary human macrophages, mediated by both HIF-1α and HIF-2β (Fang et al., 2009). Interestingly, expression of CXCR2, the receptor for CXCL-8, in macrophages significantly contributes to the progression of advanced atherosclerosis in mice (Boisvert et al., 2000), underscoring the importance of the CXCL8-CXCR2 signaling axis in this disease. It will be interesting to determine the genetic requirements for either HIF-1α or HIF-2α expression in macrophages using appropriate animal models of atherosclerosis.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is another type of chronic inflammatory disorder that primarily attacks the joints, producing a synovitis that often progresses to destruction of bone and cartilage (Muz et al., 2009). Although the cause of rheumatoid arthritis is unkown, hypoxia has been suggested to contribute to its pathology (Muz et al., 2009; Sivakumar et al., 2008). Using microelectrodes and the hypoxia marker pimonidazole (PIMO) staining, reduced O2 tension has been detected in the synovium of RA patients and animals (Peters et al., 2004; Sivakumar et al., 2008). The presence of hypoxia in RA joints is probably attributable to continuous synovial expansion which outstrips the blood-borne O2 supply. In RA synovial membrane cultures which contain macrophages, lymphocytes and fibroblasts, hypoxia appears to be a potent stimulus for VEGF induction, a classical hypoxia-responsive gene. Moreover, macrophages in RA joints express factors such as VEGF, IL-1, TNF-α, CXCL-8, CXCL-12, Cox-2 and MMP-1 (Muz et al., 2009), some of which are known hypoxia-regulated factors. The precise mechanism for how hypoxia regulates these molecules in RA joints is unclear and requires further investigation. Interestingly, several of these factors (e.g. VEGF, IL-1, TNF-α and CXCL-8) are known to promote angiogenesis, a characteristic of RA progression (Szekanecz et al., 1998). However, there appears to be a paradox in that abundant synovial vasculature is nonetheless associated with regions of synovial hypoxia. This may suggest that over-activation of the angiogenesis cascade by hypoxia results in formation of chaotic vessels with decreased blood flow, similar to that seen in many solid tumors.
As described above, in a mouse model of induced arthritis, it has been shown that myeloid HIF-1α activity is important for disease development (Cramer et al., 2003). To understand precisely how HIF-1α is involved in RA pathogenesis, it would be useful to isolate synovial macrophages to analyze HIF-dependent gene expression. It remains to be determined whether HIF-2α expression in myeloid cells (mainly macrophages) is also required in RA pathogenesis.
HIF activities in tumor-associated macrophages
Connecting inflammation and cancer
Since Rudolf Virchow’s observation in 1863 that leukocytes infiltrate malignant tissues, suggesting cancers arise at sites of chronic inflammation, a relationship between inflammation and cancer has emerged. Epidemiological studies clearly demonstrate that ~15% of human cancer deaths are associated with chronic viral or bacterial infections. For example, human papillomaviruses, hepatitis B virus (HBV) and hepatitis C virus (HCV), and the bacterium Helicobacter pylori cause cervical cancer, hepatocellular carcinoma and gastric cancer, respectively (Mantovani et al., 2008). This effect is attributed to inflammatory cells and cytokines thought to establish an inflammatory microenvironment in tumors (Balkwill and Mantovani, 2001). Interestingly, within tumors, macrophages represent a major component of infiltrating leukocytes (also including dendritic cells, neutrophils, mast cells and T cells) (Kelly et al., 1988; Leek et al., 1994), as well as the non-tumor stromal cell compartment.
Clinically, increased tumor-associated macrophage (TAM) density correlates with poor patient prognosis (Pollard, 2004; Bingle et al., 2002). Such correlative data are particularly convincing for breast (Leek et al., 1996), prostate (Lissbrant et al., 2000), cervical (Fujimoto et al., 2000) and ovarian cancers (Pollard, 2004). Using mouse models of macrophage colony stimulating factor (M-CSF) mutations, Lin et al. demonstrated that macrophage-deficient animals showed marked decreases in the rate of tumor metastasis, although primary tumor growth rate was normal (Lin et al., 2001). The authors concluded that TAM abundance potentiates tumor progression. Furthermore, using hepatocyte-specific NF-κB inactivation models, several groups have indicated that pro-inflammatory cytokines produced by Kupffer cells (namely IL-6, TNF-α and IL-1β) promote compensatory proliferation of hepatocytes, resulting in a significant increase in hepatocarcinogenesis (Luedde et al., 2007; Maeda et al., 2005; Naugler et al., 2007). Therefore, rather than limiting tumors, inflammation can actually promote tumor initiation, growth and metastasis.
Hypoxia and TAMs
Solid tumors contain large areas of hypoxia, exhibiting O2 tensions between 0.1–1%. The presence of increased hypoxic domains correlates with poor prognosis (Vaupel et al., 2001), due to the relative resistance of hypoxic cells to conventional cancer therapies (Hockel and Vaupel, 2001). Also, low O2 promotes rapid angiogenesis and exerts pressure for the selection of mutant tumor cells with survival or growth advantages (Brown and Giaccia, 1998). Interestingly, hypoxia and TAMs co-localize in tumor avascular or perinecrotic regions, indicating that TAMs specifically accumulate in O2-deprived regions within tumors. To accomplish this, tumor cells produce chemokines CCL2 and CCL5, and the cytokine M-CSF which serve to recruit monocytes from the local vasculature to tumors. Upon tumor infiltration, monocytes differentiate into TAMs and migrate along the chemoattractant gradient generated by hypoxia (Murdoch et al., 2004). Increased expression of macrophage chemoattractants such as VEGF, endothelins, IL-8 and endothelial monocyte activating polypeptide II (EMAP II) occurs in hypoxic tumor cells. Thereafter, due to down-regulation of adhesion markers and chemoattractant receptors, abrogation of chemotactic signal transduction, and the migration inhibitory actions of MIF, TAMs decrease their motility and are subsequently immobilized in these O2-deprived areas (Murdoch et al., 2004; Grimshaw and Balkwill, 2001).
Studies of breast cancer have revealed a positive correlation between numbers of TAMs in hypoxic sites and levels of angiogenesis, lymph node involvement and poor prognosis (Grimshaw and Balkwill, 2001). This suggests that O2 depletion promotes TAM responses leading to the development of pro-tumor phenotypes. Under hypoxia, TAMs up-regulate hypoxia-inducible transcription factors, and activate expression programs that appear to be pro-angiogenenic, pro-tumor growth, pro-metastatic and immunosuppressive (Lewis and Pollard, 2006; Pollard, 2004; Sica et al., 2006).
HIF-2α activity in TAMs
Although both HIF-1α and HIF-2α could be stabilized in hypoxic TAMs, work from Talks and collegues showed that HIF-2α, in particular, is strongly expressed in these cells across a wide range of human tumors (Talks et al., 2000). To elucidate the impact of high TAM HIF-2α expression on tumor phenotypes and prognosis, clinical studies have been performed on human breast cancer by Leek et al (Leek et al., 2002). This investigation revealed a positive correlation between the numbers of HIF-2α-expressing TAMs and poor prognosis. Moreover, high TAM HIF-2α levels are associated with increased tumor grade and tumor vascularity, suggesting HIF-2α expression in TAMs promotes tumor progression by improving angiogenesis (Leek et al., 2002). In another recent study, Kawanaka et al. investigated the significance of TAM HIF-2α expression in predicting survival and relapse on uterine cervical cancer patients undergoing radiotherapy (Kawanaka et al., 2008). Their results showed that increased numbers of HIF-2α-expressing TAMs are associated with poor disease-free survival and higher rate of local recurrence (Kawanaka et al., 2008). Given the clinical implications, studies determining the role of HIF-2α in TAMs using inflammation-associated tumor models and conditional knockout mouse lines is under investigation (Imtiyaz et al., manuscript submitted). It would be interesting to see how TAM HIF-2α affects tumor initiation, promotion and progression. Finally, determining whether HIF-1α is involved in TAM activities and tumorigenesis is certainly warranted.
Conclusions
Inflammation is a complex innate immune response elicited at sites experiencing infection, toxin exposure and injury. While proper inflammation helps to destroy infectious agents and restores tissue integrity, improper responses are harmful, leading to tissue destruction, vascular damage, and even organ failure in the case of sepsis. The connection between hypoxia and inflammation has become evident over the last decade, centering on the activity of HIFs. As ancient low-O2 adaptation regulators expressed in all metazoan species, HIFs also confer responses to immune stresses. This is manifested by their ability to regulate cytokine expression, myeloid cell migration and effector functions. HIFs also act to further amplify these responses under hypoxia. Dysregulation of HIFs has been shown to result in various diseases, as revealed above. HIFs therefore represent both drug candidates and targets dependent on disease types. In an immunodeficient scenario, boosting HIF activity is expected to improve inflammation and effector functions to defeat infection. This could be achieved by inhibiting the activities of PHDs and pVHL protein which negatively regulate HIFs. Given that many inflammatory disorders cause either prolonged or exaggerated inflammatory responses, targeting HIF activity or its downstream genes would be an attractive strategy for therapeutical intervention. Moreover, inhibition of VEGF leading to vessel normalization, and thus tissue re-oxygenation, may also be helpful to treat chronic inflammation.
References
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vase Biol. 1999;19:870–876. doi: 10.1161/01.atv.19.4.870. [DOI] [PubMed] [Google Scholar]
- Boisvert WA, Curtiss LK, Terkeltaub RA. Interleukin-8 and its receptor CXCR2 in atherosclerosis. Immunol Res. 2000;21:129–137. doi: 10.1385/ir:21:2-3:129. [DOI] [PubMed] [Google Scholar]
- Bredetean O, Ciochina AD, Mungiu OC. [The neutrophil in human pathology] Rev Med Chir Soc Med Nat Iasi. 2007;111:446–453. [PubMed] [Google Scholar]
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- Burke B, Tang N, Corke KP, Tazzyman D, Ameri K, Wells M, Lewis CE. Expression of HIF-1alpha by human macrophages: implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J Pathol. 2002;196:204–212. doi: 10.1002/path.1029. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Covello KL, Simon MC. HIFs, hypoxia, and vascular development. Curr Top Dev Biol. 2004;62:37–54. doi: 10.1016/S0070-2153(04)62002-3. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis. 1991;163:1177–1184. doi: 10.1093/infdis/163.6.1177. [DOI] [PubMed] [Google Scholar]
- Doherty GM, Lange JR, Langstein HN, Alexander HR, Buresh CM, Norton JA. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J Immunol. 1992;149:1666–1670. [PubMed] [Google Scholar]
- Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, Johnson RS, Imityaz HZ, Simon MC, Fredlund E, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114:844–859. doi: 10.1182/blood-2008-12-195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396:517–527. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto J, Sakaguchi H, Aoki I, Tamaya T. Clinical implications of expression of interleukin 8 related to angiogenesis in uterine cervical cancers. Cancer Res. 2000;60:2632–2635. [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Griffiths L, Binley K, Iqball S, Kan O, Maxwell P, Ratcliffe P, Lewis C, Harris A, Kingsman S, Naylor S. The macrophage - a novel system to deliver gene therapy to pathological hypoxia. Gene Ther. 2000;7:255–262. doi: 10.1038/sj.gt.3301058. [DOI] [PubMed] [Google Scholar]
- Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation--a potential mechanism. Eur J Immunol. 2001;31:480–489. doi: 10.1002/1521-4141(200102)31:2<480::aid-immu480>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Hannah S, Mecklenburgh K, Rahman I, Bellingan GJ, Greening A, Haslett C, Chilvers ER. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995;372:233–237. doi: 10.1016/0014-5793(95)00986-j. [DOI] [PubMed] [Google Scholar]
- Heinzel FP. The role of IFN-gamma in the pathology of experimental endotoxemia. J Immunol. 1990;145:2920–2924. [PubMed] [Google Scholar]
- Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–1208. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Glasner J, Warnecke C, Wiesener MS, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- Jean-Baptiste E. Cellular mechanisms in sepsis. J Intensive Care Med. 2007;22:63–72. doi: 10.1177/0885066606297123. [DOI] [PubMed] [Google Scholar]
- Jeong HJ, Chung HS, Lee BR, Kim SJ, Yoo SJ, Hong SH, Kim HM. Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem Biophys Res Commun. 2003;306:805–811. doi: 10.1016/s0006-291x(03)01073-8. [DOI] [PubMed] [Google Scholar]
- Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J. 2003a;370:1011–1017. doi: 10.1042/BJ20021279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/CoX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. Faseb J. 2003b;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- Kawanaka T, Kubo A, Ikushima H, Sano T, Takegawa Y, Nishitani H. Prognostic significance of HIF-2alpha expression on tumor infiltrating macrophages in patients with uterine cervical cancer undergoing radiotherapy. J Med Invest. 2008;55:78–86. doi: 10.2152/jmi.55.78. [DOI] [PubMed] [Google Scholar]
- Kelly PM, Davison RS, Bliss E, McGee JO. Macrophages in human breast disease: a quantitative immunohistochemical study. Br J Cancer. 1988;57:174–177. doi: 10.1038/bjc.1988.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS, Lee JY. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology. 2009 doi: 10.1111/j.1365-2567.2009.03203.x. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101:10440–10445. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Lee KS, Kim SR, Park SJ, Min KH, Lee KY, Choe YH, Park SY, Chai OH, Zhang X, Song CH, Lee YC. Mast cells can mediate vascular permeability through regulation of the PI3K-HIF-1alpha-VEGF axis. Am J Respir Crit Care Med. 2008;178:787–797. doi: 10.1164/rccm.200801-008OC. [DOI] [PubMed] [Google Scholar]
- Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. 2002;7:177–189. doi: 10.1023/a:1020304003704. [DOI] [PubMed] [Google Scholar]
- Leek RD, Harris AL, Lewis CE. Cytokine networks in solid human tumors: regulation of angiogenesis. J Leukoc Biol. 1994;56:423–435. doi: 10.1002/jlb.56.4.423. [DOI] [PubMed] [Google Scholar]
- Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]
- Leek RD, Talks KL, Pezzella F, Turley H, Campo L, Brown NS, Bicknell R, Taylor M, Gatter KC, Harris AL. Relation of hypoxia-inducible factor-2 alpha (HIF-2 alpha) expression in tumor-infiltrative macrophages to tumor angiogenesis and the oxidative thymidine phosphorylase pathway in Human breast cancer. Cancer Res. 2002;62:1326–1329. [PubMed] [Google Scholar]
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironrnents. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17:445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hulten LM, Wiklund O. Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arterioscler Thromb Vase Biol. 1997;17:317–323. doi: 10.1161/01.atv.17.2.317. [DOI] [PubMed] [Google Scholar]
- Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. Embo J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–24181. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175:6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- Muz B, Khan MN, Kiriakidis S, Paleolog EM. Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Ther. 2009;11:201. doi: 10.1186/ar2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato K, Ishibashi T, Nagata K, Seino Y, Wada Y, Sakamoto T, Matsuoka R, Teramoto T, Sekimata M, Homma Y, Maruyama Y. Expression of very low density lipoprotein receptor mRNA in circulating human monocytes: its up-regulation by hypoxia. Atherosclerosis. 2001;155:439–444. doi: 10.1016/s0021-9150(00)00580-3. [DOI] [PubMed] [Google Scholar]
- Nakazato K, Ishibashi T, Shindo J, Shiomi M, Maruyama Y. Expression of very low density lipoprotein receptor mRNA in rabbit atherosclerotic lesions. Am J Pathol. 1996;149:1831–1838. [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Mancuso G, Ciliberti FA, Beninati C, Carbone M, Franco S, Cusumano V. Endotoxin-induced lethality in neonatal mice is counteracted by interleukin-10 (IL-10) and exacerbated by anti-IL-10. Clin Diagn Lab Immunol. 1997;4:607–610. doi: 10.1128/cdli.4.5.607-610.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, Semenza GL, Shingu K, Hirota K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008;10:983–995. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- Oda T, Hirota K, Nishi K, Takabuchi S, Oda S, Yamada H, Arai T, Fukuda K, Kita T, Adachi T, et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am J Physiol Cell Physiol. 2006;291:C104–C113. doi: 10.1152/ajpcell.00614.2005. [DOI] [PubMed] [Google Scholar]
- Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- Peters CL, Morris CJ, Mapp PI, Blake DR, Lewis CE, Winrow VR. The transcription factors hypoxia-inducible factor 1alpha and Ets-1 colocalize in the hypoxic synovium of inflamed joints in adjuvant-induced arthritis. Arthritis Rheum. 2004;50:291–296. doi: 10.1002/art.11473. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178:7516–7519. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Portugal LR, Fernandes LR, Alvarez-Leite JI. Host cholesterol and inflammation as common key regulators of toxoplasmosis and artherosclerosis development. Expert Rev Anti Infect Ther. 2009;7:807–819. doi: 10.1586/eri.09.60. [DOI] [PubMed] [Google Scholar]
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg EK, Salomonsson L, Hulten LM, Noren K, Bondjers G, Wiklund O, Bjornheden T, Ohlsson BG. Hypoxia increases 25-hydroxycholesterol-induced interleukin-8 protein secretion in human macrophages. Atherosclerosis. 2003;170:245–252. doi: 10.1016/s0021-9150(03)00302-2. [DOI] [PubMed] [Google Scholar]
- Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 alpha is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol. 2002;22:2984–2992. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N. Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Hand Surg Am. 2008;33:49–58. doi: 10.1016/j.jhsa.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Standiford TJ, Strieter RM, Lukacs NW, Kunkel SL. Neutralization of IL-10 increases lethality in endotoxemia. Cooperative effects of macrophage inflammatory protein-2 and tumor necrosis factor. J Immunol. 1995;155:2222–2229. [PubMed] [Google Scholar]
- Szekanecz Z, Szegedi G, Koch AE. Angiogenesis in rheumatoid arthritis: pathogenic and clinical significance. J Investig Med. 1998;46:27–41. [PubMed] [Google Scholar]
- Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Kelleher DK, Hockel M. Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol. 2001;28:29–35. doi: 10.1016/s0093-7754(01)90210-6. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Cadwallader KA, Chilvers ER. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005a;26:434–439. doi: 10.1016/j.it.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005b;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, Beard GL, Mundy CR, Naylor S. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;53:1–8. doi: 10.1016/s0888-7543(03)00215-5. [DOI] [PubMed] [Google Scholar]
- Yu F, White SB, Zhao Q, Lee FS. Dynamic, site-specific interaction of hypoxia-inducible factor-1alpha with the von Hippel-Lindau tumor suppressor protein. Cancer Res. 2001;61:4136–4142. [PubMed] [Google Scholar]
- Zemplenyi T, Crawford DW, Cole MA. Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis. 1989;76:173–179. doi: 10.1016/0021-9150(89)90101-9. [DOI] [PubMed] [Google Scholar]
- Zhou J, Schmid T, Brune B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol Biol Cell. 2003;14:2216–2225. doi: 10.1091/mbc.E02-09-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisman DA, Kunkel SL, Strieter RM, Gauldie J, Tsai WC, Bramson J, Wilkowski JM, Bucknell KA, Standiford TJ. Anti-interleukin-12 therapy protects mice in lethal endotoxemia but impairs bacterial clearance in murine Escherichia coli peritoneal sepsis. Shock. 1997;8:349–356. doi: 10.1097/00024382-199711000-00006. [DOI] [PubMed] [Google Scholar]