

Abstract
The pfl_1841 gene from Pseudomonas fluorescens PfO-1 is the only gene in any of the three sequenced genomes of the Gram-negative bacterium Pseudomonas fluorescens that is annotated as a putative terpene synthase. The predicted Pfl_1841 protein, which harbors the two strictly conserved divalent metal binding domains found in all terpene cyclases, is closely related to several known or presumed 2-methylisoborneol synthases, with the closest match being to the MOL protein of Micromonaspora olivasterospora KY11048 that has been implicated as a 2-methylenebornane synthase. A synthetic gene encoding P. fluorescens Pfl_1841 and optimized for expression in Escherichia coli was expressed and purified as an N-terminal His6-tagged protein. Incubation of recombinant Pfl_1841 with 2-methylgeranyl diphosphate produced 2-methylenebornane as the major product accompanied by 1-methyl camphene as well as other minor, monomethyl-homomonoterpene hydrocarbons and alcohols. The steady-state kinetic parameters for the Pfl_1841-catalyzed reaction were KM = 110 ± 13 nM and kcat = 2.4 ± 0.1 × 10−2 s−1. Attempts to identify the P. fluorescens SAM-dependent 2-methylgeranyl diphosphate synthase have so far been unsuccessful.
2. Introduction
Pseudomonas fluorescens is a Gram negative, flagellated bacterium that is found colonizing plants, soil, and water.1–3 Strains of P. fluorescens, which are known for their production of fluorescent siderophores, or pyoverdines, that are produced in times of nutrient deprivation, also produce a variety of secondary natural products thought to be useful for bioremediation applications. Several non-ribosomal peptide synthase- and polyketide synthase-derived metabolites have been reported in Pseudomonads, although no terpenes or terpene cyclases have been identified in this genus to date.
The complete genome sequences of three strains of P. fluorescens, Pf-5, PfO-1 and SBW25, have recently been determined.2 Comparison shows that these three bacterial strains are genomically diverse, sharing only 61% of their gene cohort. A single gene, pfl_1841, which is found only in the PfO-1 strain, has been annotated as a putative terpene synthase. The predicted Pfl_1841 protein contains the aspartate-rich motif, 95DDHYCDD, and the “NSE” metal binding motif, 245NDLYSAYKE, variations of which are universally conserved in all terpene synthases.4 The aspartate-rich motif of Pfl_1841 is a variant of those found in most other terpene cyclases, which usually contain a “DDXXD” motif (where the X denotes any residue), but corresponds closely to that found in the sequences of a variety of confirmed and likely 2-methylisoborneol synthases (MIBS).5
2-Methylisoborneol (2-MIB) is a volatile homomonoterpene, known for its characteristic earthy odor, that is produced by a variety of actinomycete bacteria, cyanobacteria, and myxobacteria.6,7 There is considerable interest in the detection and remediation of 2-MIB and 2-MIB-producing organisms, due to the unpleasant off odor and taste that this metabolite imparts to water and food products. Recently, the two-step biosynthesis of 2-MIB from geranyl diphosphate (GPP) has been elucidated in several species of Streptomyces, in Myxobacteria, and in a cyanobacterium (Figure 1, lower pathway).5,6,8 In the first step of this pathway, 2-methylgeranyl diphosphate synthase (MGPPS) catalyzes the transfer of a methyl group from S-adenosy-L-methionine (SAM) to the C-2 position of GPP to form 2-methylgeranyl diphosphate (2-MGPP). MIB synthase then catalyzes the cyclization of 2-MGPP with capture of water to give 2-MIB.
Figure 1.
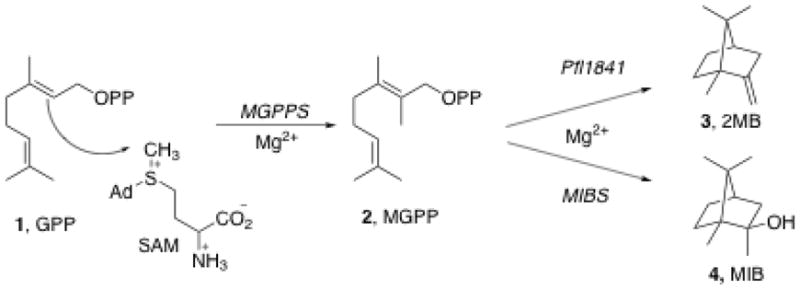
Biosynthesis of 2-methylenebornane (upper pathway) and 2-methylisoborneol (lower pathway).
In a 2008 study of the biosynthesis of 2-MIB, Komatsu et al. utilized a bioinformatics-based strategy to identify the genes responsible for the production of 2-MIB in actinomycetes.8 Using a hidden Markov model (HMM) based on the terpene synthase family metal binding domain (Pfam entry PF03906), the databases of all bacterial genome sequences, supplemented by several other Actinomycete genome sequences, were searched in order to harvest first the presumptive genes encoding predicted terpene synthases. More than 40 such sequences were identified and then classified into one of three distinct groups by phylogenetic analysis, with all the presumed monoterpene synthases falling into a single class, Group I, predicted to consist predominantly of MIB synthases. In the majority of organisms the predicted 2-MIB genes were located within a three-gene operon (Figure 2). The first gene in this operon typically encodes a predicted nucleotide binding protein of unknown function, with the second and third genes encoding a MIBS and MGPPS, respectively. The P. fluorescens Pfl_1841 protein was also found to be closely related to the general MIBS class, with the closest match being to the predicted monoterpene cyclase, MOL, from Micromonaspora olivasterospora KY11048. The 3-gene operon containing the MOL protein also harbors conserved genes encoding the upstream nucleotide binding protein and the downstream MGPP synthase. Although heterologous expression of the MOL operon in an engineered strain of Streptomyces avermitilis did not give any detectable product, cultures of M. olivasterospora were found to accumulate the monomethyl-monoterpene hydrocarbon 2-methylenebornane (2-MB) but no methylisoborneol (MIB) (Figure 1, upper pathway). A truncated recombinant form of the M. olivasterospora 2-MB synthase lacking a membrane anchor peptide catalyzed the formation of 2-MB from 2-MGPP (H. Ikeda, unpublished).
Figure 2.

Operon for the 2-MIB (S. coelicolor A3(2)) and 2-MB (M. olivasterospora) biosynthetic gene clusters. Grey, black and shaded arrows represent genes encoding a cyclic AMP nucleotide binding protein, a terpene synthase and a 2-methylgeranyl diphosphate synthase, respectively. Clear arrows represent the biosynthetically unrelated P. fluorescens PfO-1 hypothetical proteins Pfl_1840 (putative dehydrogenase) and Pfl_1842 (putative transport-related, membrane protein).
Although variations on the 2-MIB operon have also been found in which the order of the three genes is reversed or with additional inserted genes encoding proteins of unrelated function, the pfl_1841 gene locus is unique in that it lacks the organization found in either the usual 2-MIB operons or that for 2-methylenebornane in M. olivasterospora.7 Thus P. fluorescens PfO-1 has no apparent genes encoding a MGPP synthase or homologues of the cyclic nucleotide binding protein either flanking the pfl_1841 gene or clearly evident anywhere else in the genome. None of these three genes is present in either of the two other genome-sequenced P. fluorescens strains. The two genes surrounding pfl_1841 in P. fluorescens PfO-1 are annotated as a “putative dehydrogenase” (pfl_1840), and a “putative transport-related, membrane protein” (pfl_1842).
We describe below the expression and biochemical characterization of the recombinant Pfl_1841 protein of P. fluorescens PfO-1 and the demonstration that this enzyme is in fact a 2-methylenebornane synthase. This is the first terpene synthase to be identified in any Pseudomonad.
3. Results and Discussion
3.1 Cloning, expression and biochemical characterization of Pfl_1841
A synthetic pfl_1841 gene, optimized for expression in E. coli, was subcloned into the pET28a expression vector. The recombinant plasmid was introduced into E. coli BL21 (DE3) and the resultant transformants were used to produce N-terminal His6-tagged Pfl_1841 protein. Ni-NTA purification afforded pure Pfl_1841 protein (>95%).
Since the natural substrate for Pfl_1841 was initially unknown, we tested a number of acyclic allylic diphosphates as potential substrates. Thus farnesyl diphosphate (FPP), geranyl diphosphate (GPP) and 2-methylgeranyl diphosphate (2-MGPP) were individually incubated with purified Pfl_1841 protein. The enzymatic reaction mixtures were each extracted with pentane and analyzed for terpene products by GC-MS. Incubation of Pfl_1841 with FPP produced only trace amounts of the corresponding FPP elimination products, E,E-α-farnesene (1%) and E-β-farnesene (1%), along with the hydrolysis products, E-nerolidol (4%) and E,E-farnesol (1%) (Figure S13, SI). The very minor amounts of sesquiterpenes detected indicated that FPP was unlikely to be the natural substrate for Pfl_1841. Incubation of Pfl_1841 with GPP produced in low yield a complex mixture of 14 monoterpenes (C10H16, m/z 136) and monoterpene alcohols (C10H18O, m/z 154) (Figure S12, SI) including tricyclene (8) (1%), α-pinene (9) (4%), α-fenchene (7) (52%), α-phellandrene (10) (1%), β-phellandrene (11) (3%), γ-terpinene (12) (1%), terpinen-4-ol (13) (5%) and geraniol (6%), as well as minor amounts of as yet unidentified constituents. Geraniol is most likely generated by non-enzymatic, Mg2+-catalyzed solvolysis of GPP (Figure 3). The complexity of this mixture as well as the low yield of cyclization products suggested that GPP was also not the natural substrate of Pfl_1841, despite the production of α-fenchene as the predominant product. Notably, the S. coelicolor MIBS protein, SCO7701, which utilizes 2-MGPP as its natural substrate, can also catalyze the cyclization of GPP to a complex mixture of monoterpenes, but at a very low catalytic rate (kcat = 3.0 ± 0.2 × 10−5 s−1).5 A similar result was also observed in the cyclization of GPP by the recombinant MIBS protein of S. lasaliensis NRRL 3382R (H. Ikeda, unpublished
Figure 3.

Cyclization of GPP catalyzed by Pfl_1841.
Incubation of Pfl_1841 with 2-MGPP produced one major homomonoterpene product (C11H18, m/z 150) as well as several minor monomethyl-monoterpenes (Figure 4). The major product was identified by GC-MS as 2-methylenebornane (71%), with two other products shown to be 1-methylcamphene (1-MC, 3%) and 2-methylgeraniol (3%). The peaks for 2-MB and 1-MC were each identical in both retention time and mass spectrum to authentic reference standards in a mixture synthesized by the previously described acid-catalyzed dehydration of 2-MIB.9 None of the compounds in the enzymatic reaction mixture co-eluted with authentic 2-methyl-2-bornene, also formed by acid-catalyzed dehydration of 2-MIB, or with 2-methyllimonene, prepared by treatment of dihydrocarvone with methyllithium followed by dehydration with POCl3/Py. The minor product, 2-methylgeraniol, was likely formed by non-enzymatic, Mg2+-catalyzed solvolysis of 2-MeGPP. Other minor components in the enzymatic mixture were unidentified monomethyl-monoterpenes A (7%) and B (10%) and unknown homomonoterpene alcohols C (1%), D (3%) and E (2%). The mixture of compounds produced by the incubation of 2-MGPP with Pfl_1841 was less complex than that for the incubation of Pfl_1841 with GPP. Peaks A-E may correspond to monomethyl analogs of some of the minor products formed by the Pfl_1841-catalyzed cyclization of GPP.
Figure 4.

GC-MS trace of pentane extracts from the incubation of Pfl_1841 with 2-MeGPP. The major identified peak corresponds to 2-methylenebornane (3, 71%), with minor identified peaks corresponding to 1-methylcamphene (5, 3%) and 2-methylgeraniol (6, 2%). Unidentified minor products corresponding to monomethyl-monoterpenes (C11H18, m/z 150) are peaks A (7%) and B (10%) and to monomethyl-monoterpene alcohols (C11H20O, m/z 168) are C (1%), D (3%) and E (2%).
The steady-state kinetic parameters for the cyclization of 2-MGPP by Pfl_1841 were KM(2-MGPP) 110 ± 13 nM and kcat 2.4 ± 0.1 × 10−2 s−1, kcat/KM (2-MGPP) 2.14 × 105 M−1s−1, based on the cyclization of [1-3H]-2-MGPP (Figure 5).5 Both the KM and kcat determined for the Pfl_1841-catalyzed reaction were comparable to those reported for most other terpene cyclases, but contrast with the behavior of the S. coelicolor MIBS, which could not be saturated with 2-MGPP at concentrations up to the 100 μM solubility limit of 2-MGPP, with a calculated kcat/KM at low substrate concentrations of 0.8 × 103 M−1s−1.4,10–12 By comparison, the steady state kinetic parameters for incubation of Pfl_1841 with [1-3H]GPP were KM(GPP) 143 ± 12 nM and kcat 4.0 ± 0.1 × 10−3 s−1, kcat/KM (GPP) 1.9 × 104 M−1s−1 (Figure S17). The observed product distributions combined with the 10-fold greater kcat/KM for the cyclization of 2-MGPP compared with GPP are consistent with the conclusion that 2-MGPP is the natural substrate for Pfl_1841, which is therefore conclusively shown to be a 2-methylenebornane synthase.
Figure 5.

Michaelis-Menten plot of the reaction velocity for the Pfl_1841-catalyzed formation of 2-methylene bornane as a function of the concentration of 2-MGPP.
3.2 Search for the MGPPS in P. fluorescens PfO-1
The discovery that the Pfl_1841 protein is a 2-MB synthase prompted the search for the corresponding MGPPS protein that typically is encoded by a flanking gene in the MIBS biosynthetic operons as well as in the closely related cluster that harbors the MOL protein that also mediates the formation of 2-methylenebornane. As mentioned earlier, no gene encoding the MGPPS protein in P. fluorescens was found flanking pfl_1841. BLASTP searches of the predicted P. fluorescens proteome also did not reveal candidate proteins with significant sequence identity to any known MGPPS proteins. We therefore searched for candidate genes that might encode the MGPPS in P. fluorescens PfO-1 based on the presence of two motifs that are found in all of the actinomycete MGPP synthases, a methyltransferase motif PF08241 (methyltransf_11) and a cyclopropyl fatty acyl synthase motif PF02353. Two candidates in P. fluorescens PfO-1 that contained both motifs were encoded by the pfl_4741 and pfl_5666 genes. Although the corresponding predicted protein sequences both had very low (~20%) identity to the established MGPPS protein from S. coelicolor (SCO7701), no other candidates were as promising.
Synthetic genes encoding Pfl_4741 and Pfl_5666 optimized for expression in E. coli were subcloned into pET26b (Pfl_4741) and pET28a (Pfl_5666) vectors, respectively. Expression in E. coli BL21(DE3) followed by purification by ion-exchange (Pfl_4741) or Ni2+-NTA affinity chromatography (Pfl_5666) afforded the corresponding recombinant proteins. Individual overnight incubation of the Pfl_4741 or Pfl_5666 proteins with SAM and GPP was followed by treatment either with a mixture of acid phosphatase and apyrase to hydrolyze any 2-MGPP or with Pfl_1841 protein to convert eventual 2-MGPP to 2-MB. The resulting treated enzymatic reaction mixtures were extracted with pentane and analyzed by GC-MS to assay for the production of either 2-methylgeraniol or 2-MB. Unfortunately, the phosphatase-treated reaction mixtures produced only geraniol resulting from hydrolysis of the unreacted substrate GPP, while treatment with Pfl_1841 protein gave only GC-MS traces similar to those resulting from direct incubation of Pfl_1841 with GPP (supplemental information). It is therefore established that neither the pfl_4741 gene nor the pfl_5666 gene encodes the requisite MGPPS protein necessary for synthesis of the 2-MGPP substrate for the 2-MB synthase Pfl_1841, nor are any other candidate genes encoding an MGPPS protein evident in the P. fluorescens PfO-1 genome.
3.3 Screening of Pseudomonas fluorescens PfO-1 for terpene production
Detection of 2-MB from cell extracts of P. fluorescens PfO-1 would indicate that there is a still cryptic MGPPS that can provide endogeneous 2-MGPP substrate for the Pfl_1841 protein. P. fluoresecens PfO-1 was cultured on soy flour mannitol (SFM) agar plates, under conditions that produce monomethyl-monoterpenes in Actinomycetes.8 The SFM cultures were incubated for 1–7 days at 30°C and individual plates were extracted daily with pentane and analyzed for volatile secondary metabolites by GC-MS. Under these conditions no terpenoid compounds were observed in the extracts of in vivo surface cultures of P. fluorescens. It is still possible that the production of terpene compounds in P. fluorescens might require alternative culture conditions, as has been observed for expression of some terpenoid biosynthetic pathways in Streptomyces, but this remains to be demonstrated.12
4. Conclusion
We have established that the P. fluorescens PfO-1 pfl_1841 gene encodes the first terpene synthase (terpene cyclase) to be identified in any Pseudomonad species. The Pfl_1841 protein is a 2-methylenebornane synthase that catalyzes the cyclization of 2-MGPP to 2-MB, with steady-state kinetic parameters comparable to those of other previously characterized bacterial, fungal, and plant terpene synthases. Curiously, however, the associated 2-methylgeranyl diphosphate synthase (MGPPS) gene required for provision of the required substrate for 2-methylenebornane synthase, which would normally be expected to flank the 2-MB synthase gene, has not yet been identified among any of the predicted C-methyltransferases of P. fluorescens PfO-1, nor could any terpene products be detected in the organic extracts of surface cultures of this organism. At the moment it is a puzzle why P. fluorescens PfO-1 would harbor a fully functional homoterpene synthase, yet not appear to have any endogenous source of the requisite substrate for this 2-MB synthase. It is conceivable that the endogenous MGPPS gene differs significantly from known forms of this enzyme, or even that P. fluorescens might have an exogenous source of 2-MGPP or 2-methylgeraniol, such as from another bacterium in the same soil habitat. Alternatively, the absence of both the normally conserved flanking nucleotide binding protein and MGPPS genes may indicate that the standalone Pfl_1841 gene may be the artifactual residue of an earlier gene deletion, especially since there are no orthologs of any of the genes of the usual 3-gene homoterpene synthase operon in any of the three P. fluorescens genome sequences determined to date. Notably these three strains of nominally the same species share only 60% of their common gene complement.
Additional insights into the biosynthesis of 2-MB should be possible by studying the functional 2-MB biosynthetic genes that are found in other bacteria. A likely ortholog of both Pfl_1841 and the M. olivasteospora KY11048 proteins is the MCAG_05469 gene product of Micromonospora sp. ATCC39149. The predicted terpene synthase shows 38% sequence identity with the Pfl_1841 protein and the aspartate-rich binding motif of this protein (149DDYMVDE) is the variant found in both the 2-MB and MIB synthases. Genes encoding a predicted cyclic nucleotide binding protein and MGPPS protein are also present in the same operon.
5. Experimental
5.1 Materials
Geranyl diphosphate, S-adenosyl-L-methionine and 2-methylisoborneol were purchased from Sigma-Aldrich. Other reagents and solvents were purchased from Sigma-Aldrich or Fisher Scientific, were of the highest quality available, and were used without further purification. Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs and used according to manufacturer specifications. The pET28a+ and pET26b vectors were purchased from Novagen. E. coli BL21 (DE3) and E. coli XL-10 Gold cells and the QuikChange® II XL Site-Directed Mutagenesis Kit were obtained from Stratagene. Isopropylthio β-D-galactopyranoside (IPTG) was purchased from Invitrogen. Ni-NTA affinity resin was purchased from Qiagen. Amicon Ultra Centrifugal Filter Units (Amicon Ultra-15, 10,000 MWCO) were from Millipore. The acid catalyzed dehydration/rearrangement of 2-methylisoborneol, to produce the 2-methylenebornane, 1-methylcamphene, and 2-methyl-2-bornene standards, was performed according to the method of Schumann and Pendelton.9 2-Methylgeranyl diphosphate, E-2-methylgeraniol and [1-3H]-2-methylgeranyl diphosphate were synthesized according to the method of Wang and Cane.5 Pseudomonas fluorescens PfO-1 was a gift from Prof. Stuart B. Levy and was cultured according to previously published procedures.3
5.2 Methods
All DNA manipulations were performed following standard procedures.13 DNA sequencing was carried out at the U. C. Davis Sequencing Facility, Davis, CA. All proteins were handled at 4°C unless otherwise stated. Protein concentrations were determined according to the method of Bradford, using a Hewlett Packard 8452A Diode Array UV/Vis spectrophotometer with bovine serum albumin as the standard.14 Protein purity was estimated using SDS PAGE gel electrophoresis and visualized using Coomassie Blue stain according to the method of Laemmli.15
GC-MS analyses of in vitro-generated compounds were performed using a Hewlett-Packard Series 2 GC-MSD instrument (70 eV, Electron Impact, positive ion mode) and a 30 m × 0.25 mm HP5MS capillary column. The instrument method used was an injection volume of 1 μL, a solvent delay of 3 min and temperature program of 60°C for 2 min, followed by a temperature gradient of 60–280°C for 11 min (20°C min−1) and ending with a 2 min hold at 280°C. Comparisons to GC-MS detected compounds were done using the Mass Finder 4.0 Database (http://www.massfinder.com). LC-MS analyses of proteins were performed using a Thermo LXQ linear ion trap mass spectrometer and a Phenomenex C-4 column (2.1 mm × 150 mm, 5 μm pore size). The instrument method used was a linear gradient of 5% acetonitrile in water to 95% acetonitrile in water over 15 minutes at a flow rate of 200 μL/min.
5.3 Cloning of Pfl genes
Synthetic Pfl gene constructs were codon-optimized for expression in E. coli and synthesized by DNA2.0 into separate pJ201 vectors. The Pfl genes were flanked by 5′-NdeI and 3′-XhoI restriction sites, which were used in sub-cloning the genes into pET28a (Pfl_1841 and Pfl_5666) or pET26b (Pfl_4741) vectors.
5.4 Over-expression and purification of Pfl proteins
Recombinant Pfl plasmids were transformed into chemically competent E. coli BL21(DE3) cells. The transformed cells were used to inoculate 5 mL of Terrific Broth (TB) media (with 50 μg/mL of kanamycin sulfate), and incubated overnight at 37°C with shaking at 225 rpm. The 5 mL culture was then used to inoculate a 500 mL culture of TB media (with 50 μg/mL of kanamycin sulfate), which was grown to an OD600 of 0.6–0.9 at 37°C and 225 rpm. Over-expression was induced by the addition of 0.5 mM IPTG, and the cultures were allowed to grow at 28°C for an additional 12 h. The cells were harvested via centrifugation at 6000 X g for 30 min and the resulting cell pellet flash frozen in liquid nitrogen and stored at −80°C.
Protein purification of Pfl_1841 and Pfl_5666 involved thawing and resuspending a 500-mL cell pellet in 30 mL of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) containing pepstatin A (1 mg/L) and phenylmethylsulfonyl fluoride (1 mM). The cells were lysed with two passes through an ice-chilled French pressure cell (10 000 psi) and the resulting cell lysate was clarified by centrifugation at 13000 X g for 1 h before loading onto a 5 mL Ni-NTA column, pre-equilibrated with lysis buffer, at a flow rate of 2 mL/min. The column was washed with ten column volumes of lysis buffer, followed by ten column volumes of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) and the Pfl_1841 protein was eluted off with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 500 mM imidazole, pH 8.0), all at a flow rate of 2 mL min−1. All fractions containing protein were buffer exchanged into assay buffer (50 mM PIPES, 15 mM MgCl2, 100 mM NaCl, 5 mM β-mercaptoethanol, 20% glycerol, pH 6.7) using Amicon Ultra Centrifugal Filter units and used directly in experiments. LC-ESI-MS: His6-tag-Pfl_1841 39595 Da (predicted MD 39640 Da; His6-tag-Pfl_5666 46143 Da (predicted MD 46187 Da).
Protein purification of Pfl_4741 involved thawing a 500 mL cell pellet in 30 mL of lysis buffer (50 mM NaH2PO4, pH 7.0) containing pepstatin A (1 mg/L) and phenylmethylsulfonyl fluoride (1 mM). The cells were lysed with two passes through an ice-chilled French pressure cell and the cell lysate was clarified by centrifugation at 13000 x g for 1 h and the supernatant was loaded onto a 50 mL DEAE Sepharose fast flow column (GE healthcare) pre-equilibrated with lysis buffer, at a flow rate of 2 mL/min. The column was washed with 5 column volumes of lysis buffer, before eluting the protein with a linear gradient of 0 - 1 M NaCl in 50 mM of NaH2PO4 buffer, pH 7.0 (300 mL total volume). Fractions containing the Pfl_4741 protein were collected, buffer exchanged into assay buffer using Amicon Ultra centrifugal filter units and used directly in further experiments.
5.5 In vitro incubations of Pfl_1841 with FPP, GPP and 2-MGPP
To 10 mL of assay buffer (50 mM PIPES, 100 mM NaCl, 15 mM MgCl2, 5 mM β-mercaptoethanol and 20% glycerol, pH 6.7) and 60 μM of FPP, GPP or 2-MeGPP, was added 10 μM of purified Pfl_1841 protein. The enzymatic reaction was overlaid with 10 mL of pentane and incubated at 30°C for 12 h. Following the incubation, the enzymatic products were extracted with 3 × 10 mL of pentane and the organic extracts were combined, dried with Na2SO4 and concentrated in vacuo to 200 μL for GC-MS analysis.
5.6 In vitro incubations of Pfl_4741 and Pfl_5666 with SAM and GPP
To 10 mL of assay buffer (50 mM PIPES, 100 mM NaCl, 15 mM MgCl2, 5 mM β-mercaptoethanol and 20% glycerol, pH 6.7) containing 120 μM of SAM and 60 μM of GPP were added 10 μM of either Pfl_4741 or Pfl_5666 protein. The enzymatic reactions were overlaid with 10 mL of pentane and incubated at 30°C for 12 h. Following the incubation period, the enzyme reactions were split into two equal aliquots. To one aliquot was added 5 mL of phosphatase solution (containing 3 units of wheat germ acid phosphatase and 2 units of potato apyrase per 1.0 mL of 0.1 M sodium acetate, pH 5.0) and the reaction incubated at 30°C for 2 h. To the other aliquot was added 10 μM of purified Pfl_1841 and the reaction incubated at 30 °C for 12 h. At the end of the incubation period, each of the reaction mixtures was extracted with 3 × 5 mL of pentane and the organic layers from each were combined, dried over Na2SO4 and reduced in vacuo at 0°C to 200 μL for GCMS analysis.
5.7 Pfl_1841 Kinetic Assays with GPP and 2-MGPP
Kinetic parameters were measured in 1 mL of assay buffer (50 mM PIPES, 15 mM MgCl2, 100 mM NaCl, 5 mM β-mercaptoethanol, 20% glycerol, pH 6.7), with varying amounts of 2-methylGPP substrate (25 nM-2500 nM) and [1-3H]-2-methylGPP (5.5 Ci/mol). The reactions were initiated by the addition of 10.5 nM of Pfl_1841 protein, overlaid with 1 mL of pentane and incubated at 30°C for 5 min. The reactions were quenched by the addition of 75 μL of 500 mM EDTA (pH 8.0) and vortexing for 30 s. The pentane layer was loaded onto a silica plug (2 cm) in a Pasteur pipette and forced through with a stream of nitrogen into a scintillation vial containing 7 mL of Opti-Fluor. The enzymatic reaction was extracted 3 more times with 1 mL of ether, and all organic extracts were passed through the silica plug and collected. The combined extracts were counted via liquid scintillation counting using a Beckman-Coulter LS6500 scintillation counter. Kinetic constants were calculated using the program Kaleidagraph 4.0 and were fitted to the Michaelis-Menten equation, giving values of kcat 2.4 ± 0.1 × 10−2 s−1 and KM(MGPP) 110 ± 13 nM. Reported standard deviations in the steady-state kinetic parameters represent the calculated statistical errors in the non-linear, least squares regression analysis. To determine the steady-state kinetic parameters with GPP, incubations were carried out under the same conditions as above using varying concentrations of GPP (125 nM-2500 nM) and [1-3H]-GPP (170 Ci/mol). The calculated kinetic constants were kcat 4.0 ± 0.1 × 10−3 s−1 and KM(GPP) 143 ± 13 nM.
5.8 Growth of Pseudomonas fluorescens PfO-1 and analysis of volatile organic extracts
P. fluorescens PfO-1 was grown in soy flour mannitol (SFM) media on 90 mm agar plates at 30°C. The production of metabolites from P. fluorescens was assessed from 1–7 days of growth by adding 5 mL of methanol to a single culture, letting the culture sit for 30 min at room temperature and extracting the methanol layer with 3 × 5 mL of pentane. The pentane extracts were dried over Na2SO4, concentrated in vacuo at 0°C to 200 μL and analyzed by GC-MS.
5.9. Synthesis of 2-methyllimonene
To a stirred solution of 5 mL of (+)-dihydrocarvone (30.1 mmol) in 30 mL of THF at −78°C, was added dropwise 28 mL of a 1.6 M solution of methyllithium in diethyl ether (45 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 1 h before the addition of 100 mL of an ice-cold solution of saturated NH4Cl in water. The crude reaction mixture was extracted with 3 × 50 mL of diethyl ether, dried over Na2SO4, concentrated in vacuo and used directly in the next step.
To an ice-cold solution of 1.5 mL of pyridine and 200 μ (1.2 mmol) of the crude reaction mixture from above, was added 400 μ of POCI3 (4.4 mmol). The reaction mixture was warmed to room temperature and stirred for 1 h before the addition of 10 mL of ice-cold water. The reaction mixture was extracted with 3 × 10 mL of pentane, dried over Na2SO4 and r concentrated in vacuo. The crude reaction mixture was loaded onto a silica column pre-equilibrated with pentane (1 cm × 5 cm) and eluted with pentane to yield 127 mg of 2-methyllimonene as a colorless oil in 71% yield. 1H NMR (400 MHz, CDCl3) δ 4.71 – 4.66 (2H, m, CH2-8), 2.18 – 2.09 (1H, m, CH-4), 2.09 – 1.91 (2H, m, CH2-6), 1.96 – 1.85 (2H, m, CH2-3), 1.77 – 1.71 (1H, m, CHa-5), 1.72 (3H, s, CH3-9), 1.60 (3H, s, CH3-11), 1.59 (3H, s, CH3-10), 1.40 (1H, ddd, J 5.60, 11.50, 12.50 Hz, CHb-5); 13C NMR (100 MHz, CDCl3) 150.4 (C-7), 125.3 (C-2), 125.1 (C-1), 108.3 (C-8), 42.0 (C-4), 37.2 (C-3), 32.3 (C-6), 28.2 (C-5), 20.8 (C-9), 19.1 (C-11), 18.8 (C-10) ppm; HRMS (GC-MS, EI): M+, found 150.1416 (150.1409 theoretical).
Supplementary Material
Acknowledgments
We thank Dr. Matthew Mattana and Prof. Stuart R. Levy for their generous gift of Pseudomonas fluorescens PfO-1. We also thank Dr. Tun-Li Shen for assistance with mass spectrometry. This work was supported by National Institutes of Health Grant GM30301 to D. E. C. and by a Grant-in-Aid for Scientific Research on Innovative Areas from MEXT Japan, from JSPS 20310122 and from the Institute for Fermentation, Osaka, Japan (H.I.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gross H, Loper JE. Nat Prod Rep. 2009;26:1408. doi: 10.1039/b817075b. [DOI] [PubMed] [Google Scholar]
- 2.Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, Knight CG, Malone JG, Robinson Z, Spiers AJ, Harris S, Challis GL, Yaxley AM, Harris D, Seeger K, Murphy L, Rutter S, Squares R, Quail MA, Saunders E, Mavromatis K, Brettin TS, Bentley SD, Hothersall J, Stephens E, Thomas CM, Parkhill J, Levy SB, Rainey PB, Thomson NR. Genome Biol. 2009;10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compeau G, Al-Achi BJ, Platsouka E, Levy SB. Appl Environ Microbiol. 1988;54:2432. doi: 10.1128/aem.54.10.2432-2438.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christianson DW. Chem Rev. 2006;106:3412. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 5.Wang CM, Cane DE. J Am Chem Soc. 2008;130:8908. doi: 10.1021/ja803639g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz S, Dickschat JS. Nat Prod Rep. 2007;24:814. doi: 10.1039/b507392h. [DOI] [PubMed] [Google Scholar]
- 7.Giglio S, Chou WK, Ikeda H, Cane DE, Monis PT. Environ Sci Technol. 2011;45:992. doi: 10.1021/es102992p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komatsu M, Tsuda M, Omura S, Oikawa H, Ikeda H. Proc Natl Acad Sci U S A. 2008;105:7422. doi: 10.1073/pnas.0802312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumann R, Pendleton P. Water Research. 1997;31:1243. [Google Scholar]
- 10.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. Biochemistry. 2006;45:6179. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cane DE, He X, Kobayashi S, Omura S, Ikeda H. J Antibiot (Tokyo) 2006;59:471. doi: 10.1038/ja.2006.66. [DOI] [PubMed] [Google Scholar]
- 12.Takamatsu S, Lin X, Nara A, Komatsu M, Cane DE, Ikeda H. Microb Biotechnol. 2011;4:184. doi: 10.1111/j.1751-7915.2010.00209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambrook JFEF, Maniatis T. Molecular Cloning, A Laboratory Manual. 2. Cold Spring Harbor; NT: 1989. [Google Scholar]
- 14.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 15.Laemmli UK. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.