

Abstract
Chronic short sleep duration has been linked to endothelial dysfunction and increased risk of cardiovascular disease. Circulating endothelial progenitor cells (EPCs) are vital to endogenous vascular repair processes and cardiovascular health. We tested the hypothesis that habitual short sleep duration is associated with impairment in EPC number and function. Cells with phenotypic EPC characteristics were isolated from 37 healthy, sedentary adults: 20 with normal sleep duration (13M/7F; age: 59±1 years; sleep duration: 7.7±0.1 h/night) and 17 with short sleep duration (9M/8F; 56±2 years; 6.0±0.2 h/night). EPC number was assessed by flow cytometric analysis of the percentage of peripheral blood mononuclear cells negative for CD45 and positive for CD34, VEGFR-2, and CD133 antigens. EPC colony-forming capacity was determined by colony-forming unit (CFU) assay; migration by Boyden chamber; and intracellular caspase-3 concentrations by immunoassay. There were no significant differences between groups in EPC number (0.001±0.0004 vs. 0.001±0.0003 %), colony-forming capacity (6.1±1.5 vs. 5.4±1.7 CFUs), or migration to VEGF (1410.1±151.2 vs. 1334.3±111.1 AU). Furthermore, there were no group differences in basal and staurosporine-stimulated intracellular concentrations of active caspase-3 (0.3±0.03 vs. 0.5±0.1 ng/mL; and 2.9±0.4 vs. 2.7±0.3 ng/mL), a marker of apoptotic susceptibility. Taken together, these data indicate that short sleep duration is not associated with EPC dysfunction in healthy adults. Numerical and functional impairment in circulating EPCs may not contribute to the increased cardiovascular risk with habitual short sleep duration.
Keywords: Endothelium, progenitors, sleep
INTRODUCTION
It has become increasingly apparent that chronic short sleep duration adversely affects cardiovascular health. Several recent epidemiological studies have reported that short sleep duration is associated with increased cardiovascular morbidity and mortality.[1] However, the biological mechanisms linking habitual short sleep duration and the development of cardiovascular diseases (CVDs) are not well-defined.
Endothelial damage and dysfunction is an important early event in the pathogenesis of atherosclerotic vascular disease and is independently associated with an increased risk of cardiovascular events.[2] Recent data from our laboratory[3] and others[4] suggest that acute and chronic sleep restriction is associated with pathogenic alterations in endothelial cell function. The capacity to repair the endothelium and restore its function after injury also plays a vital role in determining cardiovascular risk.[5] Circulating endothelial progenitor cells (EPCs) have recently been identified as an integral component of the endogenous vascular repair system.[5] Importantly, declines in both EPC number and function have been linked with accelerated atherosclerotic disease progression and poor CVD prognosis.[6–8] Thus, a potential mechanism contributing to the heightened cardiovascular risk with short sleep duration may be a reduction in EPC bioavailability and/or function. Accordingly, we tested the hypothesis that habitual short sleep duration is associated with impairment in EPC number and function.
MATERIALS AND METHODS
Subjects
Thirty-seven adults (age range: 43–65 years) were studied: 20 with normal habitual sleep duration (13 males/7 females) and 17 with short habitual sleep duration (9 males/8 females). All subjects were free of overt cardiometabolic disease as assessed by medical history, physical examination, and fasting blood chemistries. All female subjects were at least one year postmenopausal and had never taken or had discontinued use of hormone replacement therapy at least one year prior to the start of the study. None of the subjects smoked, were taking medications, or performed regular physical exercise for at least 6 months before the start of the study. Prior to participation, all of the subjects had the research study and its potential risks and benefits explained fully before providing written informed consent according to the guidelines of the University of Colorado at Boulder.
Sleep duration
Habitual sleep duration was measured as a component of the Stanford Physical Activity Questionnaire, as previously described.[3] Consistent with previous studies,[9] nightly average reported sleep duration was calculated as the weighted average of weeknights and weekend values [(5 × weekday sleep duration) + (2 × weekend sleep duration)/7]. Subjects were divided into two groups based upon their reported sleep duration: 7 to 9 h/night or “normal habitual sleep duration” and <7 h/night or “short habitual sleep duration”. These criteria were chosen based on published reports indicating that habitual sleep duration shorter than 7 h/night is associated with increased health risks, including hypertension, coronary artery disease, and stroke.[1] Although assessment of habitual sleep duration by self-report has the potential to introduce reporting bias and experimental error, previous investigations have demonstrated a robust correlation between self-reported sleep duration and that measured objectively through actigraphic monitoring.[10]
Body composition and metabolic measurements
Body mass was measured to the nearest 0.1 kg using a medical beam balance. Percent body fat was determined by dual-energy X-ray absorptiometry (Lunar, Madison, WI). Body mass index was calculated as mass (kilograms) divided by height (meters) squared. Fasting plasma lipid and lipoprotein, glucose, and insulin concentrations were determined using conventional methods by the clinical laboratory affiliated with the University of Colorado at Boulder Clinical and Translational Research Center.
EPC isolation, characterization, and numeration
Circulating putative EPCs were isolated from peripheral blood by Ficoll density–gradient centrifugation (Histopaque 1077, Sigma Aldrich, St Louis, MO, USA) as previously described by our laboratory.[11] Circulating EPC number was determined by fluorescence-activated cell sorting (FACS) analysis. Briefly, 2 × 106 cells were incubated at 4°C for 30 min with monoclonal antibodies for PC7-conjugated CD45 (Beckman Coulter, Fullerton, CA, USA), FITC-conjugated CD34 (Beckman Coulter), PE-conjugated VEGFR-2 (R&D Systems, Minneapolis, MN, USA) and APC-conjugated CD133 (Miltenyi Biotech, Auburn, CA, USA). Cells were gated for low expression of CD45 and then CD34+ cells were analyzed for events double-positive for VEGFR-2 and CD133 and presented as a percent of total viable mononuclear cells.
Colony-forming assay
EPC colony-forming capacity was determined as previously described.[11] Briefly, freshly isolated mononuclear cells were plated on six-well plates coated with human fibronectin (BD Biosciences, San Jose, CA, USA) for 48 h at 37°C. Thereafter, non-adherent cells were collected and 5 × 105 cells were replated onto 24-well fibronectin-coated plates (BD Biosciences). Growth medium was changed every three days, and the EPC colony-forming units (CFUs) were counted manually in four random wells on the seventh day by two independent investigators blinded to sample identification.
Migration assay
EPC migratory capacity was measured using a modified Boyden chamber.[12] Non-adherent cells (4 × 105) were resuspended in serum-free culture medium (Medium 199, GIBCO) and loaded in the upper compartment of a 24-well modified Boyden chamber coated with fibronectin (FluoroBlok, BD Biosciences). The upper chamber was placed in the lower chamber containing culture medium supplemented with vascular endothelial growth factor (2 ng/ml) and incubated for 22 h at 37°C. Following incubation, cells were labeled with calcein AM (Molecular Probes, Eugene, Oregon), and relative fluorescent units (RFUs) of migrated cells were determined in triplicate.
Caspase-3 activation
Activation of caspase-3 was induced by incubating EPCs with staurosporine (1 μmol/L) for 3 h; Sigma Aldrich, St. Louis, MO). Following staurosporine stimulation, cells were washed with PBS and incubated with 10 μmol biotin-ZVKD-fmk inhibitor for 1 h at 37°C. Thereafter, cells were washed and lysed with extraction buffer at a concentration of 1 × 107 cells/mL. The concentration of active caspase-3 in the supernatant was determined by enzyme immunoassay (R&D Systems, Minneapolis, MN).
Statistical analysis
Differences in subject baseline characteristics and the primary outcome variables were determined by analysis of variance (ANOVA). There were no significant gender interactions in any of the primary outcome variables; therefore, the data were pooled and presented together. Relations between sleep duration and EPC measures were assessed by linear regression analysis. Data are reported as mean ± SEM. Statistical significance was set at P<0.05.
RESULTS
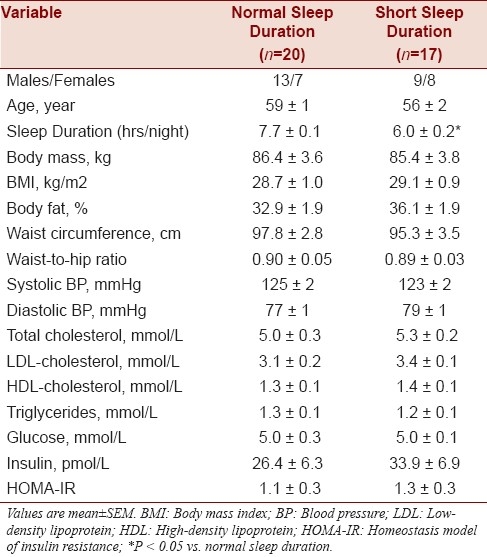
Selected subject characteristics are presented in Table 1. Anthropometric characteristics were similar between the groups. The short sleep duration group reported a significantly lower average sleep duration (6.0 ± 0.2 h/night; range: 4.0 to 6.9 h/night) compared with the normal sleep duration group (7.7 ± 0.1 h/night; range: 7.0 to 9.0 h/night). There were no differences between the groups in plasma lipid and lipoprotein, glucose, and insulin concentrations.
Table 1.
Selected subject characteristics
The number of CD45-/CD34+/VEGFR-2+/CD133+ cells was not different between the normal sleep (0.0011 ± 0.0003%) and short sleep duration (0.0014 ± 0.0004%) groups. There were no significant differences in the number of EPC CFUs between the normal sleep (6 ± 2) and short sleep duration (5 ± 2) groups [Figure 1]. EPC migratory activity was similar between adults reporting normal sleep duration (1410 ± 151 RFU) and adults reporting short sleep duration (1334 ± 111 RFU; Figure 1). Under basal conditions, there was no difference in intracellular active caspase-3 concentrations between the normal sleep (0.3 ± 0.03 ng/mL) and short sleep (0.4 ± 0.11 ng/mL) duration groups. Following treatment with the pro-apoptotic stimulus staurosporine, intracellular caspase-3 concentrations significantly increased in both groups. However, there was no significant difference in stimulated intracellular caspase-3 concentrations between the normal sleep duration (2.8 ± 0.3 ng/mL) and short sleep (2.7 ± 0.3 ng/mL) groups [Figure 1]. There were no significant correlations between sleep duration and any EPC measure.
Figure 1.
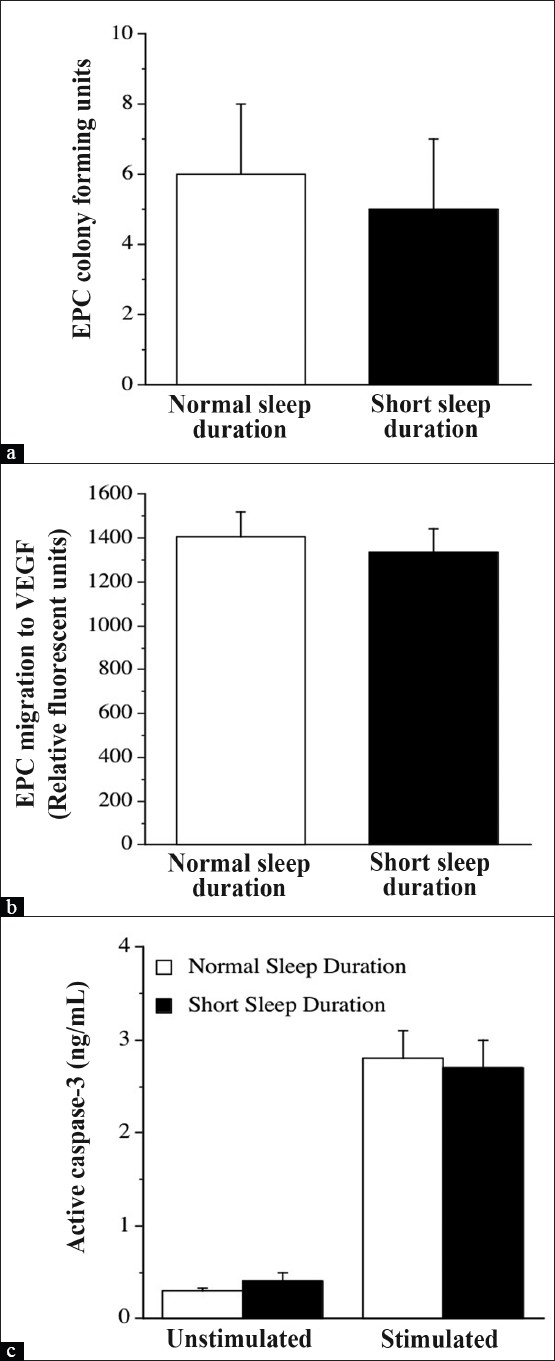
EPC colony-forming units (a), migratory activity (b), and intracellular concentrations of active caspase-3 under basal conditions and in response to staurosporine (c) in the normal sleep and short sleep duration groups. Values are mean ± SEM
DISCUSSION
The primary new findings of the present study are that EPC number, capacity to form colonies, migratory ability, and apoptotic susceptibility are not impaired in healthy adults who sleep less than 7 h/night. Collectively, these data suggest that a decrement in EPC number or function is not involved in the increased cardiovascular risk observed in this population.
Endothelial dysfunction has recently been identified as a potential link between short sleep duration and CVD.[3,4] Maintenance and repair of the endothelium is essential in preserving function and preventing the development and progression of atherosclerotic disease. EPCs play an important role in vascular repair and can promote both re-endothelialization and neovascularization.[5] Several studies have linked reduced number and function of circulating EPCs to endothelial dysfunction and CVD risk.[6–8] For example, Schmidt-Lucke and colleagues reported that lower number of circulating EPCs independently predicts atherosclerotic disease progression.[6] Moreover, reduced EPC colony-forming capacity and migratory ability have each been independently linked to an increased risk of CVD,[7,8] while heightened EPC susceptibility to apoptosis can underlie functional impairments and contribute to endothelial dysfunction and atherogenesis.[5]
In the present study, contrary to our hypothesis, habitual short sleep duration was not associated with impairments in EPC number or function. Circulating numbers of EPCs as well as their ability to form colonies, migrate and resist apoptosis was almost identical between middle-aged adults who chronically sleep ~6 h/night compared with those who sleep ~8 h/night. Although we observed no significant relation between sleep duration and any of the EPC measures, we studied a subpopulation (n=7) of adults within the short sleep duration group who reported ≤5 h of sleep per night to dismiss the possibility that the mean habitual sleep duration was not low enough in the short sleep group to induce EPC abnormalities. Similar to our findings in the overall study population, EPC number and functional characteristics were not different in the subpopulation with shorter chronic sleep duration compared with the normal sleep group. However, it remains possible that more severe chronic sleep restriction is associated with impairments in EPC number and/or function. This notion is supported by data indicating that ten days of severe sleep restriction (<4 h/night) elicits increased production of inflammatory mediators that have been shown to attenuate EPC survival, differentiation, and function.[13–15]
To our knowledge, this is the first study to examine the influence of habitual short sleep duration on EPC number and function. It is important to note, however, that we studied healthy individuals without co-existing cardiovascular risk factors. Considering obesity, type-2 diabetes, and the metabolic syndrome, common morbidities in middle-aged adults are associated with both short sleep duration[1] and EPC dysfunction;[11,16] the presence of these and other risk factors may alter our results. Nevertheless, these data suggest that numerical and functional impairment in EPCs, independent of other risk factors do not contribute to the increased cardiovascular risk associated with habitual short sleep duration.
Although there is currently no clear consensus on the most appropriate criteria for the isolation, culture or quantification of EPCs, in the present study we isolated and identified a putative EPC cell population that has been consistently shown to participate in vasculogenesis and vascular repair,[17] and is associated with both endothelial dysfunction and cardiovascular risk.[8] These cells were isolated with minimal time in culture to reduce phenotypic drift and exhibited several endothelial phenotypic characteristics.[17] In contrast, other investigators have argued that “outgrowth endothelial cells”, a proliferative endothelial phenotype derived from mononuclear cells following an extended period of in vitro culture (2–4 weeks), should be considered as true endothelial progenitors, given their morphological similarity to mature endothelial cells.[18] However, the culture of these cells involves untold phenotypic drift from the cells in circulation and their physiological and clinical relevance are unclear. Nevertheless, our results must be viewed within the context of our isolation, culture, and characterization methodology.
In conclusion, short sleep duration is not associated with impairment in either EPC number or function in otherwise healthy adults. Although recent data implicate endothelial damage and dysfunction as potential mechanisms underlying the increased cardiovascular risk associated with chronic short sleep duration, our results indicate that deficits in EPC number and function are not apparent in healthy individuals who sleep between four and seven hours per night. Future research is necessary to determine if more severe chronic sleep restriction is associated with impairment of endothelial repair processes.
Acknowledgments
We would like to thank all of the subjects who participated in the study, as well as Jared Greiner for his technical assistance. This study was supported by National Institutes of Health Awards HL076434, HL077450 and RR00051, and American Heart Association Award 0555678Z.
Footnotes
REFERENCES
- 1.Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51:294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lerman A, Zeiher AM. Endothelial function: Cardiac events. Circulation. 2005;111:363–8. doi: 10.1161/01.CIR.0000153339.27064.14. [DOI] [PubMed] [Google Scholar]
- 3.Weil BR, Mestek ML, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, et al. Short sleep duration is associated with enhanced endothelin-1 vasoconstrictor tone. Can J Physiol Pharmacol. 2010;88:777–81. doi: 10.1139/Y10-046. [DOI] [PubMed] [Google Scholar]
- 4.Sauvet F, Leftheriotis G, Gomez-Merino D, Langrume C, Drogou C, Van Beers P, et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol. 2010;108:68–75. doi: 10.1152/japplphysiol.00851.2009. [DOI] [PubMed] [Google Scholar]
- 5.Dimmeler S, Zeiher AM. Vascular repair by circulating endothelial progenitor cells: The missing link in atherosclerosis? J Mol Med. 2004;82:671–7. doi: 10.1007/s00109-004-0580-x. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt-Lucke C, Rossig L, Fichtlscherer S, Vasa M, Britten M, Kamper U, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: Proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–7. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 7.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 8.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 9.Hall MH, Muldoon MF, Jennings JR, Buysse DJ, Flory JD, Manuck SB. Self-reported sleep duration is associated with the metabolic syndrome in midlife adults. Sleep. 2008;31:635–43. doi: 10.1093/sleep/31.5.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lockley SW, Skene DJ, Arendt J. Comparison between subjective and actigraphic measurement of sleep and sleep rhythms. J Sleep Res. 1999;8:175–83. doi: 10.1046/j.1365-2869.1999.00155.x. [DOI] [PubMed] [Google Scholar]
- 11.MacEneaney OJ, Kushner EJ, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Endothelial progenitor cell number and colony-forming capacity in overweight and obese adults. Int J Obes (Lond) 2009;33:219–25. doi: 10.1038/ijo.2008.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacEneaney OJ, Kushner EJ, Westby CM, Cech JN, Greiner JJ, Stauffer BL, et al. Endothelial Progenitor Cell Function, Apoptosis, and Telomere Length in Overweight/Obese Humans. Obesity (Silver Spring) 2010;18:1677–82. doi: 10.1038/oby.2009.494. [DOI] [PubMed] [Google Scholar]
- 13.Meier-Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43:678–83. doi: 10.1016/j.jacc.2003.07.050. [DOI] [PubMed] [Google Scholar]
- 14.Verma S, Kuliszewski MA, Li SH, Szmitko PE, Zucco L, Wang CH, et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: Further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation. 2004;109:2058–67. doi: 10.1161/01.CIR.0000127577.63323.24. [DOI] [PubMed] [Google Scholar]
- 15.Suh W, Kim KL, Choi JH, Lee YS, Lee JY, Kim JM, et al. C-reactive protein impairs angiogenic functions and decreases the secretion of arteriogenic chemo-cytokines in human endothelial progenitor cells. Biochem Biophys Res Commun. 2004;321:65–71. doi: 10.1016/j.bbrc.2004.06.107. [DOI] [PubMed] [Google Scholar]
- 16.Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–57. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 17.Hur J, Yang HM, Yoon CH, Lee CS, Park KW, Kim JH, et al. Identification of a novel role of T cells in postnatal vasculogenesis: Characterization of endothelial progenitor cell colonies. Circulation. 2007;116:1671–82. doi: 10.1161/CIRCULATIONAHA.107.694778. [DOI] [PubMed] [Google Scholar]
- 18.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–9. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]