

Abstract
Diabetic retinopathy (DR) is one of the most common complications of diabetes and is a leading cause of blindness in people of the working age in Western countries. A major pathology of DR is microvascular complications such as non-perfused vessels, microaneurysms, dot/blot hemorrhages, cotton-wool spots, venous beading, vascular loops, vascular leakage and neovascularization. Multiple mechanisms are involved in these alternations. This review will focus on the role of inflammation in diabetic retinal microvascular complications and discuss the potential therapies by targeting inflammation.
Keywords: Diabetic retinopathy, inflammation, microvascular complications
INTRODUCTION
Diabetic retinopathy (DR) is one of the most common complications of diabetes and is a leading cause of blindness in people of the working age in industrialized countries.[1] Approximately 25% of type 1 diabetic patients may have signs of retinopathy after 5 years of diabetes, increasing to 60% after 10 years. Eventually, after 25 years, almost all (97%) type 1 diabetic patients will develop retinopathy.[2,3] Type 2 diabetic patients may already have background retinopathy at the time of diagnosis, and over 60% will develop some form of retinopathy after 20 years.[4] According to the International Diabetes Federation (IDF-Atlas, 4th ed., 2009), diabetes currently affects nearly 285 million people worldwide, and this number is expected to reach 438 million by 2030. It is expected that DR will have a growing impact on large populations.
Tremendous efforts have been made to identify mechanisms and develop therapies for DR. These studies have led to the recognition of hyperglycemia, hypertension and dyslipidemia as major risk factors for DR. Consequently, tight glycemic control, blood pressure control and lipid-lowering therapy have shown proven benefits in reducing the incidence and progression of DR.[5] In clinic, laser photocoagulation and vitrectomy remain the two conventional approaches to treat sight-threatening conditions such as macular edema and proliferative DR.[6] Vascular endothelial growth factor (VEGF) blockers (Pegaptanib, Ranibizumab and Bevacizumab) in combination with laser photocoagulation represents an emerging novel therapy to reduce macular edema and induce neovascular regression.[5–7] In spite of this progress, DR remains a major clinical challenge, and the number of patients keeps growing due to difficulty in achieving tight glycemic control, metabolic memory, unresponsive to the current therapeutic approaches and significant side-effects from therapies.[5,6,8–10] There is a great need to develop new therapeutic approaches for this devastating disease.
INFLAMMATORY FEATURES IN DIABETIC RETINOPATHY
DR is characterized as a microvascular complication of diabetes.[2,11] Vascular alterations in the early stage of the disease include alterations in blood flow, death of retinal pericytes (perivascular contractile cells), basement membrane thickening and subtle increases in vascular permeability. With the progression of disease, obvious alterations in the vascular structure can be seen upon ophthalmoscopic examination. These include non-perfused vessels, microaneurysms, dot/blot hemorrhages, cotton-wool spots, venous beading, vascular loops and significant vascular leakage. The above alternations happen in the non-proliferative stage of DR, in which vision loss is mainly caused by macular edema as the consequence of increased vascular permeability. In the proliferative stage of DR, neovascularization happens on the retinal surface after vascular function is impaired by capillary occlusion, non-perfusion and degeneration. In this stage, severe vision loss or even blindness may be caused by bleeding, hemorrhage and subsequent retinal detachment because of the newly formed fragile vessels.[12] The mechanisms by which diabetes causes microvascular complications and disease progression in the retina are not fully understood. However, studies in patient samples and animal models have shown that DR has features of chronic, subclinical inflammation.
Inflammation is the body's defense against pathogens and is also a critical step in wound healing. This process involves multiple mediators such as pro-inflammatory cytokines, chemokines and adhesion molecules that initiate the interaction between leukocytes and the endothelium and guide directional leukocyte migration toward infected or injured tissue. Pro-inflammatory cytokines (such as tumor necrosis factor [TNF]—α and interleukins) and chemokines (such as CCL2 and CCL5) released from infected/injured tissue activate the endothelium to increase expression of adhesion molecules (such as E-selectin, intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule [VCAM]-1) and chemokines. Mediated by adhesion molecules and chemokines, leukocytes attach to the vessel wall, transmigrate through the endothelium and move to the infected or injured tissue.[13] While normal inflammation is beneficial, excessive or uncontrolled inflammation can cause tissue injury and result in diseases.[13]
Although there are no pathogens in DR, analysis of inflammatory molecules in vitreous, serum and retina form diabetic patients or experimental animals indicate that DR is associated with significant increases in pro-inflammatory cytokines, chemokines and adhesion molecules. High TNF-α levels have been observed in vitreous, serum and ocular fibrovascular membranes from patients with DR and in retinas from rodent model of diabetes mellitus.[14–16] Interleukin-1β (IL-1β) and its downstream signaling molecule caspase 1 are significantly increased in vitreous, retinas and serum from diabetic patients and rats.[15,17,18] Chemokines such as CCL2, CCL5, CXCL8, CXCL10 and CXCL12 are also upregulated in vitreous samples from DR patients.[19–21] Increases in IL-6, ICAM-1 and VCAM-1 have been shown to be related to the progression of DR.[21–24]
Correlated with increases in inflammatory molecules, DR is associated with recruitment of leukocytes. The numbers of neutrophils are significantly elevated in both retinal and choroidal vessels from diabetic patients and monkeys. The accumulation of neutrophils is correlated with upregulation of ICAM-1 immunoreactivity in the vessels and is associated with capillary closure.[25,26] In diabetic rodent models, there is a cumulative and sustained increase in leukocyte adherence to the retinal vasculature (leukostasis) along with the progression of DR.[27] The increase in leukostasis may be attributable to diabetes-induced increases in ICAM-1 and integrins in endothelial cells and leukocytes, respectively, in that blockade of ICAM-1 or deletion of CD18 (ICAM-1 receptor subunit on leukocytes) prevents diabetes-induced leukostasis.[27–29] In addition to leukocytes in the circulation, microglia that are scattered throughout the retina are likely to be involved in DR.[30] In DR, they are rapidly activated to release inflammatory cytokines such as TNF-α.[31] A2A adenosine receptor agonist effectively blocks TNF-α production in microglia and markedly decreases hyperglycemia-induced retinal cell death (Ibrahim et al., 2011).
In summary, this evidence demonstrates that DR has all the characters of inflammatory disease, and suggests involvement of low-grade chronic inflammation in this disease. Tremendous efforts have been made to understand how diabetes causes inflammatory reactions in DR in the absence of pathogens.
MECHANISMS OF RETINAL INFLAMMATION
Risk factors of DR include hyperglycemia, hypertension and dyslipidemia. These factors have been shown to induce inflammation by a variety of mechanisms, including oxidative stress, NF-kB activation, dysregulation of nitric oxide synthase (NOS) and formation of advanced glycation endproducts (AGEs).
Oxidative stress happens when ROS are overproduced or when endogenous anti-oxidant systems are impaired. Mitochondria can generate ROS by leakage of electrons to molecular oxygen at electron transport chain complexes I, II and III.[32] In diabetes, the metabolism of glucose-derived pyruvate through the ETC complexes is increased because of high glucose concentration within cells, resulting in superoxide overproduction by mitochondria.[33] This pathway not only produces more ROS by itself but also initiates other pathways, leading to a breakdown in the balance between pro-oxidant and endogenous anti-oxidant systems, such as increases in glucose flux through the aldose reductase pathway, formation of AGEs and activation of protein kinase c (PKC).[34] All these changes can lead to oxidative stress by decreasing the activities of anti-oxidant enzymes[35,36] or further activating the ROS-generating machinery inside the cells.[37–41] In addition to mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is another important source of ROS in diabetes, and studies in other models have suggested a positive reciprocal regulation between mitochondria and NADPH oxidase-derived ROS.[42,43]
NADPH oxidase is the major source of ROS in the cardiovascular system. It consists of a NOX catalytic subunit, 4 phox subunits (p22phox, p40phox, p47phox and p67phox) and the low-molecular weight G protein Rac.[44] The p47phox and p67phox subunits are essential for activity. Assembly of the complex is initiated upon phosphorylation of p47phox, which is regulated by many intracellular signaling kinases such as PKC, mitogen-activated protein kinases and Akt.[45–49] The p67phox subunit mediates direct binding of the complex with activated Rac, which initiates the electron transfer reaction that produces superoxide.[50] These regulatory mechanisms contribute to the unique feature of NADPH oxidase-derived ROS as signaling mediator in many cellular responses. However, when these mechanisms are activated because of diabetes, they can cause NADPH oxidase to overproduce ROS. These mechanisms in fact allow NADPH oxidase to function as a key link between hyperglycemia and hypertension. Increased NAPDH oxidase activity has been found in diabetic patients and animals and high glucose-treated endothelial cells.[51–54] NADPH oxidase is also potently activated by angiotensin II, a product of the renin–angiotensin system that plays a key role in hypertension.[55,56] Overall, these changes lead to a net increase in ROS, and diabetes-induced sustained oxidative stress is a major cause of retinal inflammation.
ROS are important intracellular signaling molecules in the inflammatory cascade. ROS play a key role in inflammatory gene expression by activation of redox-sensitive transcription factors such as NF-kB, signal transducers and activators of transcription proteins and activator protein 1.[57,58] The activity of NF-kB is increased in high glucose-treated retinal endothelial cells, pericytes or glial cells and in diabetic retinas from patients or animal models.[59–64] This activation is significantly blocked by inhibiting redox systems by blockade of NADPH oxidase or antioxidant treatment,[63,64] indicating a cause-and-effect relationship. Activation of NF-kB has an essential role in diabetes-induced retinal inflammation, in that inhibition of NF-kB blocks both high glucose and diabetes-induced production of inflammatory molecules in retinal cells and retinal tissue, and attenuates leukostasis.[59–61] Similarly, studies in animal and tissue culture models have demonstrated the involvement of NADPH oxidase in diabetes-induced inflammation and breakdown of the blood–retinal barrier (BRB).[65,66] The results showed that diabetes-induced increases in retinal ROS, VEGF expression and vascular permeability are accompanied by increases in the NADPH oxidase catalytic subunit NOX2.[65–67] The experiments further demonstrated that deleting the NOX2 gene or inhibiting NADPH oxidase prevented diabetes-induced increases in retinal permeability and ICAM-1 expression and leukostasis, indicating that the NOX2 NADPH oxidase is critically involved in the pathology of DR.[65,66,68] Correlated with the involvement of oxidative stress in diabetes-induced retinal inflammation, studies have shown that supplement of antioxidants such as vitamins C and E attenuates the development of acellular capillaries and decreases the number of pericyte ghosts in diabetic rats.[69] Such effects are further enhanced when a more comprehensive mixture of anti-oxidant diet is applied.[69] Clinical studies also show that high doses of vitamin E reverse some of the changes in the retinal vessels of diabetic patients.[70]
In addition to oxidative stress, diabetes-induced vascular inflammation is very closely related to nitric oxide (NO), which is an important second messenger that regulates many physiological and pathological events, including vascular dilation and vascular inflammation. Studies indicate that NO has a biphasic role in vascular and inflammatory diseases, depending on the specific source and the amount produced.[71] The constitutively expressed NOSs, endothelial NOS (eNOS) and neuronal NOS (nNOS), are Ca2+-dependent and regulated to produce low levels of NO. NO from nNOS is involved in neural signaling and is also expressed in smooth muscle cells, where it has a key role in regulating vascular responses to tissue hypoxia. NO from eNOS maintains blood flow and prevents platelet aggregation and leukostasis.[72] In fact, deleting eNOS results in elevated expression of inducible NOS (iNOS) (an NF-kB-mediated inflammatory molecule) in retina that is associated with accelerated development and increased severity of DR.[73] In contrast with eNOS and nNOS, iNOS is Ca2+-independent, constitutively active and produces large amounts of NO.[71] It is not expressed in normal retinas but is induced in retinal glial and microglial cells during inflammatory conditions, including DR. NO from iNOS plays a role in causing tissue damage and inflammation. Inhibition of iNOS by inhibitor or gene deletion prevents ICAM-1 expression, leukostasis and vascular permeability in the diabetic retina.[74] The NOS pathway is dysregulated by diabetes and contributes to the pathogenesis of DR. On the one hand, ROS generated from NADPH oxidase or mitochondrial oxidase can rapidly react with NO to form RNOS and reduce bioavailable NO, which reduces vessel dilation and increases leukocyte adhesion and platelet aggregation, leading to inflammation.[71,75] On the other hand, NOS can be an important source of ROS due to “NOS uncoupling,” in which the enzyme generates superoxide rather than NO when its substrate L-arginine is limited by increased arginase activity[71] or when the co-factor tetrahydrabiopterin is oxidized. Consistently, inhibition of arginase is shown to increase bioavailable NO, reduce superoxide formation and block inflammatory reactions during retinal inflammation.[68]
Although hyperglycemia has a direct impact to the pathology of DR, the byproducts of hyperglycemia, particularly AGEs, also have a significant role in retinal inflammation in DR. AGEs are formed by non-enzymatic glycation and oxidation of amino groups of proteins, lipids and DNA.[76] This process is accelerated in diabetes in the presence of high glucose and oxidative stress.[76–78] In the diabetic retina, the level of AGEs is prominently increased and AGE immunoreactivity is localized in vitreous, internal limiting membrane and retinal vasculature.[76] The receptor for AGEs (RAGE) is expressed in numerous cells in the retina, including Muller cells, endothelial cells and neurons.[76] The AGE-RAGE pathway can activate many downstream signaling molecules, including NF-kB and ROS, to induce inflammatory reactions such as ICAM-1 expression and leukostasis in the retina and retinal cells.[78,79] Blocking AGE with soluble RAGE or inhibiting AGE formation with LR-90 significantly reduces diabetes-induced ICAM-1 expression, leukostasis and VEGF expression.[80,81]
In summary, multiple pathways are involved in diabetes-induced retinal inflammation [Figure 1]. However, it should be noted that inflammation is not an endpoint of the above inflammatory mechanisms. Inflammation, in turn, further activates these inflammatory mechanisms. For example, oxidative stress is both a cause and a consequence of inflammation. Many inflammatory cytokines induce oxidative stress and use the NADPH oxidase pathway to induce expression of other inflammatory molecules such as CCL2.[82,83] Leukocytes also release ROS in response to inflammatory stimuli. Blockade of IL-6 not only prevents angiotensin II-induced leukostasis in the retina but also abolishes NADPH oxidase-mediated ROS formation.[84] Thus, inflammation serves as a key mediator in the positive feedback loop of inflammatory cascades in DR.
Figure 1.
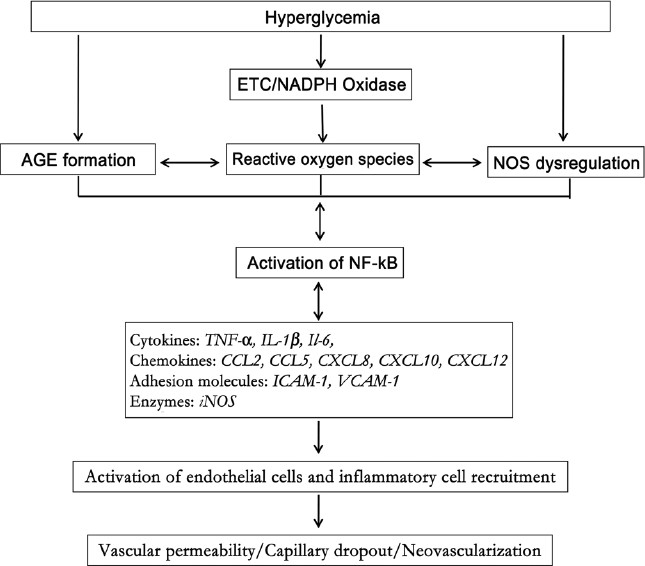
Cascade of events that contribute to the inflammatory response in diabetic retinopathy. Hyperglycemia induces formation of advanced glycation end products, generation of reactive oxygen species from multiple sources, mainly nicotinamide adenine dinucleotide phosphate oxidase and mitochondrial electron transport chain, and dysregulation of nitric oxide synthase (NOS) pathway. These changes activate NF-kB and, in turn, upregulation of cytokines, chemokines and iNOS. This leads to upregulation of adhesion molecules and subsequent leukocyte/endothelial interaction
INFLAMMATION IN THE DEVELOPMENT OF DIABETIC RETINOPATHY
Because diabetes is found to cause retinal inflammation by divergent mechanisms, significant efforts have been made to understand how inflammation is involved in the microvascular complications in the disease. These studies have demonstrated important roles of inflammatory cytokines and leukocytes in vascular leakage, vessel occlusion and degeneration and pathological neovascularization [Figure 1].
Inflammation is a cause of retinal vascular leakage. The microvascular endothelium forms an effective barrier to control the movement of blood fluid and proteins across the vessel wall.[85] This barrier is formed by tight junctions and adhesion junctions that join endothelial cells to each other. Signaling molecules, including pro-inflammatory cytokines and chemokines, which induce disorganization/redistribution of junction proteins in the retinal endothelium may lead to breakdown of the BRB, resulting in an abnormal extravasation of blood components and retinal edema. TNF-α is known to cause significant retinal endothelial permeability within a few hours by PKCζ-mediated downregulation of tight junction proteins.[86] Intravitreal injection of TNF-α leads to increased retinal vascular permeability, which is prevented by PKCζ inhibitor.[86] Moreover, blocking TNF-α with Etanercept, a soluble TNF-α receptor, prevents BRB breakdown in the diabetic rat model.[14] Similar increases in endothelial permeability are also observed when endothelial cells are treated with other cytokines such as CCL2 or CXCL8.[87,88] BRB breakdown can also be caused by cytokine-induced vascular cell death and by leukocyte-mediated vascular alternations. Leukocytes recruited in inflammation have a key role in causing vascular leakage by mechanisms including inducing junction protein alteration, releasing cytokines or increasing ROS as well as inducing vessel occlusion and injury.[27,28,89]
Inflammation induces retinal vessel occlusion and capillary dropout. Retinal vessel occlusion and degeneration is a typical feature of DR. Mechanisms leading to capillary degeneration may involve inflammatory cytokine-induced endothelial cell death because inflammatory cytokines such as TNF-α and IL-1β are also known to increase caspase 3 activity and potently induce endothelial cell apoptosis.[86,90] Alternatively, leukostasis in DR may cause capillary occlusion when leukocytes block blood flow because of their large cell volume and high rigidity. This notion is supported by studies showing that transient leukocyte attachment to the vessel wall correlates with capillary non-perfusion, and capillary reperfusion can occur when leukocytes detach and move on.[27] In addition to blocking blow flow, leukocytes induce endothelial death during leukostasis via Fas ligand (FasL) and Fas-mediated apoptosis.[91] The level of Fas is increased in retinal vascular endothelial cells during diabetes, while FasL is increased on neutrophils. Blocking Fas or inhibiting ICAM-1/CD18-mediated leukostasis significantly reduces retinal endothelial cell injury, apoptosis and retinal capillary degeneration.[28,91]
Inflammation is also involved in pathological neovascularization in DR. Chronic inflammation is known as a key player in angiogenesis in many diseases such as rheumatoid arthritis and cancer. Leukocytes, recruited by inflammatory cytokines produced in the disease tissue, enhance the formation of new vasculature by releasing angiogenic factors and increasing the activity of matrix metallopeptidase.[92] Direct investigation of inflammation in proliferative DR is not feasible because there are no small animal models that can develop reproducible proliferative DR, as occurs in humans. Therefore, studies of retinal neovascularization are carried out in rodent models of oxygen-induced retinopathy, in which retinal vessel obliteration is induced with hyperoxia. These studies have shown that inflammatory genes are upregulated at the onset of the hypoxia as well as during the period of neovascularization.[93,94] Blockade of inflammatory cytokines, such as TNF-α, attenuates neovascularization.[95,96] In addition to inflammatory cytokines, monocytes/macrophages are found to be present in the neovascular tufts, and depleting the monocyte lineage with clodronate-liposomes leads to the suppression of pathological but not physiological retinal angiogenesis.[97]
CONCLUSIONS AND PROSPECTIVE
Although there is no pathogen in the diabetic retina, DR has all the common features of inflammation, such as increase in inflammatory molecules in retina, vitreous and plasma, leukocyte recruitment and tissue edema. These changes are caused by diabetes-induced disorders such as oxidative stress, dysregulation of the NOS pathway, AGEs formation and NF-kB activation [Figure 1]. Inflammation is now recognized as an important player in the pathogenesis of DR, and a number of studies have been designed to address whether blockade of specific inflammatory molecules can be beneficial for DR. Subcutaneous administration of TNF-α trap (a soluble TNF-α receptor/Fc construct, Etanercept) significantly blocks retinal inflammation, retina cell injury and vessel leakage in diabetic rats.[14,98] In one clinical study with four patients, Infliximab, a TNF-α-neutralizing antibody, was shown to improve visual acuity and reduce macular thickness in patients who failed to improve in response to laser photocoagulation treatment.[99] Leukocyte function-associated antigen-1 (LFA-1, an integrin) expressed in leukocytes is important for leukocyte-endothelial cell interaction by binding to ICAM. Topical delivery of SAR 1118, a small antagonist of LFA-1, dose-dependently reduced leukostasis and retinal vascular leakage in a diabetic rat model.[100] In addition to ICAM-1, blockade of VCAM-1-mediated leukocyte attachment with anti-CD49a neutralizing antibody significantly attenuated the diabetes-induced leukostasis and vascular leakage.[101] In spite of the rapid progress in this field, there are many issues that remain to be addressed before effective anti-inflammatory therapy can be designed to treat DR. For example, inflammation is involved in almost all diseases and, therefore, it is unclear why inflammatory reactions in most diseases do not affect retinal vasculature. Is there crosstalk between diabetes-induced inflammation and other chronic disease-induced inflammation in relation to the pathogenesis of DR? Although inflammation is involved in the pathogenesis of DR, there is a higher incidence of DR in type 1 diabetic patients than in type 2 diabetic patients. Is it because of the fact that type 1 and type 2 diabetes induce different inflammatory reactions? Retinal inflammation might be more severe in type 1 diabetes as this disease is associated with aberrant autoimmunity. However, there is no study to address such difference at this moment. Given that diabetes induces the production of inflammatory molecules in both retinal local cells and blood leukocytes, what is the individual role of retinal local cells and leukocytes in inflammatory reactions and vascular injury in DR? What are the specific subtypes of leukocytes that are involved in DR? Many inflammatory molecules have redundant functions and cause activation of similar downstream targets such as NF-kB. What is their specific role and what are their reciprocal interactions in DR? These mysteries have led to challenges when considering any of the inflammatory molecules as a drug target as blockade of any one of them may be effective if the molecule is indispensible. However, the blockade can also be ineffective, or even worse, if compensation happens or goes in an unwanted direction. Further studies may resolve these issues and bring novel therapies for DR by controlling inflammatory reactions precisely and at the right time.
Acknowledgments
This work was generously supported by the American Heart Association 11SDG4960005 and the Juvenile Diabetes Foundation JDRF 10-2009-575 (to W. Zhang), by NIH Grants EY04618 and VA Merit Award (to R.B. Caldwell), by NIH Grant HL70215 (to R.W. Caldwell), by NIH Grant EY11766 (to R.B. Caldwell and R.W. Caldwell) and by the American Heart Association SDG00104 and Vision Discovery Institute (GHSU) (to M. Al-Shabrawey).
Footnotes
Source of Support: The American Heart Association 11SDG4960005 and SDG00104, the Juvenile Diabetes Foundation JDRF 10-2009-575, NIH Grants EY11766, EY04618, HL70215 and VA Merit Award and Vision Discovery Institute (GHSU)
Conflict of Interest: None declared.
REFERENCES
- 1.Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, et al. Retinopathy in diabetes. Diabetes Care. 2004;27(Suppl 1):S84–7. doi: 10.2337/diacare.27.2007.s84. [DOI] [PubMed] [Google Scholar]
- 2.Aiello LP, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, 3rd, et al. Diabetic retinopathy. Diabetes Care. 1998;21:143–56. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- 3.Klein R, Knudtson MD, Lee KE, Gangnon R, Klein BE. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XXII the twenty-five-year progression of retinopathy in persons with type 1 diabetes. Ophthalmology. 2008;115:1859–68. doi: 10.1016/j.ophtha.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams R, Airey M, Baxter H, Forrester J, Kennedy-Martin T, Girach A. Epidemiology of diabetic retinopathy and macular oedema: A systematic review. Eye (Lond) 2004;18:963–83. doi: 10.1038/sj.eye.6701476. [DOI] [PubMed] [Google Scholar]
- 5.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376:124–36. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 6.Yam JC, Kwok AK. Update on the treatment of diabetic retinopathy. Hong Kong Med J. 2007;13:46–60. [PubMed] [Google Scholar]
- 7.Arevalo JF, Sanchez JG, Lasave AF, Wu L, Maia M, Bonafonte S, et al. Intravitreal Bevacizumab (Avastin) for Diabetic Retinopathy at 24-months: The 2008 Juan Verdaguer-Planas Lecture((R)) Curr Diabetes Rev. 2010;6:313–22. doi: 10.2174/157339910793360842. [DOI] [PubMed] [Google Scholar]
- 8.Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: Role of inflammatory mediators in retinal pericytes. Exp Eye Res. 2010;90:617–23. doi: 10.1016/j.exer.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol. 2010;299:F14–25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein R, Klein BE. Diabetic eye disease. Lancet. 1997;350:197–204. doi: 10.1016/S0140-6736(97)04195-0. [DOI] [PubMed] [Google Scholar]
- 12.Davidson JA, Ciulla TA, McGill JB, Kles KA, Anderson PW. How the diabetic eye loses vision. Endocrine. 2007;32:107–16. doi: 10.1007/s12020-007-0040-9. [DOI] [PubMed] [Google Scholar]
- 13.Barreiro O, Martin P, Gonzalez-Amaro R, Sanchez-Madrid F. Molecular cues guiding inflammatory responses. Cardiovasc Res. 2010;86:174–82. doi: 10.1093/cvr/cvq001. [DOI] [PubMed] [Google Scholar]
- 14.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. Faseb J. 2002;16:438–40. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 15.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (Lond) 2006;20:1366–9. doi: 10.1038/sj.eye.6702138. [DOI] [PubMed] [Google Scholar]
- 16.Limb GA, Chignell AH, Green W, LeRoy F, Dumonde DC. Distribution of TNF alpha and its reactive vascular adhesion molecules in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol. 1996;80:168–73. doi: 10.1136/bjo.80.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–30. doi: 10.2337/db06-0427. [DOI] [PubMed] [Google Scholar]
- 18.Kowluru RA, Odenbach S. Role of interleukin-1beta in the development of retinopathy in rats: Effect of antioxidants. Invest Ophthalmol Vis Sci. 2004;45:4161–6. doi: 10.1167/iovs.04-0633. [DOI] [PubMed] [Google Scholar]
- 19.Murugeswari P, Shukla D, Rajendran A, Kim R, Namperumalsamy P, Muthukkaruppan V. Proinflammatory cytokines and angiogenic and anti-angiogenic factors in vitreous of patients with proliferative diabetic retinopathy and eales’ disease. Retina. 2008;28:817–24. doi: 10.1097/IAE.0b013e31816576d5. [DOI] [PubMed] [Google Scholar]
- 20.Maier R, Weger M, Haller-Schober EM, El-Shabrawi Y, Wedrich A, Theisl A, et al. Multiplex bead analysis of vitreous and serum concentrations of inflammatory and proangiogenic factors in diabetic patients. Mol Vis. 2008;14:637–43. [PMC free article] [PubMed] [Google Scholar]
- 21.Meleth AD, Agron E, Chan CC, Reed GF, Arora K, Byrnes G, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:4295–301. doi: 10.1167/iovs.04-1057. [DOI] [PubMed] [Google Scholar]
- 22.Mocan MC, Kadayifcilar S, Eldem B. Elevated intravitreal interleukin-6 levels in patients with proliferative diabetic retinopathy. Can J Ophthalmol. 2006;41:747–52. doi: 10.3129/i06-070. [DOI] [PubMed] [Google Scholar]
- 23.Funatsu H, Yamashita H, Ikeda T, Mimura T, Eguchi S, Hori S. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology. 2003;110:1690–6. doi: 10.1016/S0161-6420(03)00568-2. [DOI] [PubMed] [Google Scholar]
- 24.Adamiec-Mroczek J, Oficjalska-Mlynczak J. Assessment of selected adhesion molecule and proinflammatory cytokine levels in the vitreous body of patients with type 2 diabetes--role of the inflammatory-immune process in the pathogenesis of proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2008;246:1665–70. doi: 10.1007/s00417-008-0868-6. [DOI] [PubMed] [Google Scholar]
- 25.McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–53. [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SY, Johnson MA, McLeod DS, Alexander T, Hansen BC, Lutty GA. Neutrophils are associated with capillary closure in spontaneously diabetic monkey retinas. Diabetes. 2005;54:1534–42. doi: 10.2337/diabetes.54.5.1534. [DOI] [PubMed] [Google Scholar]
- 27.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA. 1999;96:10836–41. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18:1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 29.Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, et al. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci. 2000;41:1153–8. [PubMed] [Google Scholar]
- 30.Chen L, Yang P, Kijlstra A. Distribution, markers, and functions of retinal microglia. Ocul Immunol Inflamm. 2002;10:27–39. doi: 10.1076/ocii.10.1.27.10328. [DOI] [PubMed] [Google Scholar]
- 31.Yang LP, Sun HL, Wu LM, Guo XJ, Dou HL, Tso MO, et al. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:2319–27. doi: 10.1167/iovs.08-2642. [DOI] [PubMed] [Google Scholar]
- 32.Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 35.Merzouk S, Hichami A, Madani S, Merzouk H, Berrouiguet AY, Prost J, et al. Antioxidant status and levels of different vitamins determined by high performance liquid chromatography in diabetic subjects with multiple complications. Gen Physiol Biophys. 2003;22:15–27. [PubMed] [Google Scholar]
- 36.Bhatia S, Shukla R, Venkata Madhu S, Kaur Gambhir J, Madhava Prabhu K. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin Biochem. 2003;36:557–62. doi: 10.1016/s0009-9120(03)00094-8. [DOI] [PubMed] [Google Scholar]
- 37.Thornalley PJ. Glycation in diabetic neuropathy: Characteristics, consequences, causes, and therapeutic options. Int Rev Neurobiol. 2002;50:37–57. doi: 10.1016/s0074-7742(02)50072-6. [DOI] [PubMed] [Google Scholar]
- 38.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 39.Curtis TM, Scholfield CN. The role of lipids and protein kinase Cs in the pathogenesis of diabetic retinopathy. Diabetes Metab Res Rev. 2004;20:28–43. doi: 10.1002/dmrr.431. [DOI] [PubMed] [Google Scholar]
- 40.Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14:S227–32. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 41.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–96. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 43.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–16. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 45.Heyworth PG, Curnutte JT, Nauseef WM, Volpp BD, Pearson DW, Rosen H, et al. Neutrophil nicotinamide adenine dinucleotide phosphate oxidase assembly.Translocation of p47-phox and p67-phox requires interaction between p47-phox and cytochrome b558. J Clin Invest. 1991;87:352–6. doi: 10.1172/JCI114993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoyal CR, Gutierrez A, Young BM, Catz SD, Lin JH, Tsichlis PN, et al. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc Natl Acad Sci USA. 2003;100:5130–5. doi: 10.1073/pnas.1031526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dang PM, Fontayne A, Hakim J, El Benna J, Perianin A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol. 2001;166:1206–13. doi: 10.4049/jimmunol.166.2.1206. [DOI] [PubMed] [Google Scholar]
- 48.Korchak HM, Kilpatrick LE. Roles for beta II-protein kinase C and RACK1 in positive and negative signaling for superoxide anion generation in differentiated HL60 cells. J Biol Chem. 2001;276:8910–7. doi: 10.1074/jbc.M008326200. [DOI] [PubMed] [Google Scholar]
- 49.El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, Babior BM. Activation of p38 in stimulated human neutrophils: Phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys. 1996;334:395–400. doi: 10.1006/abbi.1996.0470. [DOI] [PubMed] [Google Scholar]
- 50.Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, Sumimoto H. Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem. 1999;274:25051–60. doi: 10.1074/jbc.274.35.25051. [DOI] [PubMed] [Google Scholar]
- 51.Ellis EA, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000;28:91–101. doi: 10.1016/s0891-5849(99)00216-6. [DOI] [PubMed] [Google Scholar]
- 52.Inoguchi T, Tsubouchi H, Etoh T, Kakimoto M, Sonta T, Utsumi H, et al. A possible target of antioxidative therapy for diabetic vascular complications-vascular NAD(P)H oxidase. Curr Med Chem. 2003;10:1759–64. doi: 10.2174/0929867033457133. [DOI] [PubMed] [Google Scholar]
- 53.Sonta T, Inoguchi T, Tsubouchi H, Sekiguchi N, Kobayashi K, Matsumoto S, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–23. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 55.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–74. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 56.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302:148–58. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turpaev KT. Reactive oxygen species and regulation of gene expression. Biochemistry (Mosc) 2002;67:281–92. doi: 10.1023/a:1014819832003. [DOI] [PubMed] [Google Scholar]
- 58.Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 59.Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, et al. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci. 2007;48:4342–50. doi: 10.1167/iovs.06-1473. [DOI] [PubMed] [Google Scholar]
- 60.Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–8. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 61.Harada C, Okumura A, Namekata K, Nakamura K, Mitamura Y, Ohguro H, et al. Role of monocyte chemotactic protein-1 and nuclear factor kappa B in the pathogenesis of proliferative diabetic retinopathy. Diabetes Res Clin Pract. 2006;74:249–56. doi: 10.1016/j.diabres.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 62.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–7. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 63.Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–80. doi: 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- 64.Tawfik A, Sanders T, Kahook K, Akeel S, Elmarakby A, Al-Shabrawey M. Suppression of retinal peroxisome proliferator-activated receptor gamma in experimental diabetes and oxygen-induced retinopathy: Role of NADPH oxidase. Invest Ophthalmol Vis Sci. 2009;50:878–84. doi: 10.1167/iovs.08-2005. [DOI] [PubMed] [Google Scholar]
- 65.Al-Shabrawey M, Rojas M, Sanders T, Behzadian A, El-Remessy A, Bartoli M, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008;49:3239–44. doi: 10.1167/iovs.08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Al-Shabrawey M, Bartoli M, El-Remessy AB, Ma G, Matragoon S, Lemtalsi T, et al. Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:3231–8. doi: 10.1167/iovs.08-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Al-Shabrawey M, Bartoli M, El-Remessy AB, Platt DH, Matragoon S, Behzadian MA, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Baban B, Rojas M, Tofigh S, Virmani SK, Patel C, et al. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am J Pathol. 2009;175:891–902. doi: 10.2353/ajpath.2009.081115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–42. doi: 10.2337/diabetes.50.8.1938. [DOI] [PubMed] [Google Scholar]
- 70.Bursell SE, King GL. Can protein kinase C inhibition and vitamin E prevent the development of diabetic vascular complications? Diabetes Res Clin Pract. 1999;45:169–82. doi: 10.1016/s0168-8227(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 71.Caldwell RB, Zhang W, Romero MJ, Caldwell RW. Vascular dysfunction in retinopathy-an emerging role for arginase. Brain Res Bull. 2010;81:303–9. doi: 10.1016/j.brainresbull.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kubes P, Suzuki M, Granger DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–5. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Q, Verma A, Han PY, Nakagawa T, Johnson RJ, Grant MB, et al. Diabetic eNOS knockout mice develop accelerated retinopathy. Invest Ophthalmol Vis Sci. 2010;51:5240–6. doi: 10.1167/iovs.09-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leal EC, Manivannan A, Hosoya K, Terasaki T, Cunha-Vaz J, Ambrosio AF, et al. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2007;48:5257–65. doi: 10.1167/iovs.07-0112. [DOI] [PubMed] [Google Scholar]
- 75.Nagaoka T, Kuo L, Ren Y, Yoshida A, Hein TW. C-reactive protein inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Invest Ophthalmol Vis Sci. 2008;49:2053–60. doi: 10.1167/iovs.07-1387. [DOI] [PubMed] [Google Scholar]
- 76.Barile GR, Schmidt AM. RAGE and its ligands in retinal disease. Curr Mol Med. 2007;7:758–65. doi: 10.2174/156652407783220778. [DOI] [PubMed] [Google Scholar]
- 77.Stitt AW, Curtis TM. Advanced glycation and retinal pathology during diabetes. Pharmacol Rep. 2005;57(Suppl):156–68. [PubMed] [Google Scholar]
- 78.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 79.Moore TC, Moore JE, Kaji Y, Frizzell N, Usui T, Poulaki V, et al. The role of advanced glycation end products in retinal microvascular leukostasis. Invest Ophthalmol Vis Sci. 2003;44:4457–64. doi: 10.1167/iovs.02-1063. [DOI] [PubMed] [Google Scholar]
- 80.Kaji Y, Usui T, Ishida S, Yamashiro K, Moore TC, Moore J, et al. Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with a soluble form of a receptor for advanced glycation end products. Invest Ophthalmol Vis Sci. 2007;48:858–65. doi: 10.1167/iovs.06-0495. [DOI] [PubMed] [Google Scholar]
- 81.Bhatwadekar A, Glenn JV, Figarola JL, Scott S, Gardiner TA, Rahbar S, et al. A new advanced glycation inhibitor, LR-90, prevents experimental diabetic retinopathy in rats. Br J Ophthalmol. 2008;92:545–7. doi: 10.1136/bjo.2007.127910. [DOI] [PubMed] [Google Scholar]
- 82.Zhang W, Rojas M, Lilly B, Tsai NT, Lemtalsi T, Liou GI, et al. NAD(P)H oxidase-dependent regulation of CCL2 production during retinal inflammation. Invest Ophthalmol Vis Sci. 2009;50:3033–40. doi: 10.1167/iovs.08-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang G, Lucas R, Caldwell R, Yao L, Romero MJ, Caldwell RW. Novel mechanisms of endothelial dysfunction in diabetes. J Cardiovasc Dis Res. 2010;1:59–63. doi: 10.4103/0975-3583.64432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rojas M, Zhang W, Lee DL, Romero MJ, Nguyen DT, Al-Shabrawey M, et al. Role of IL-6 in angiotensin II-induced retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2010;51:1709–18. doi: 10.1167/iovs.09-3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–23. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 86.Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–82. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379–88. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- 88.Gavard J, Hou X, Qu Y, Masedunskas A, Martin D, Weigert R, et al. A role for a CXCR2/phosphatidylinositol 3-kinase gamma signaling axis in acute and chronic vascular permeability. Mol Cell Biol. 2009;29:2469–80. doi: 10.1128/MCB.01304-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, et al. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br. J. Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Joussen AM, Poulaki V, Mitsiades N, Cai WY, Suzuma I, Pak J, et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. Faseb J. 2003;17:76–8. doi: 10.1096/fj.02-0157fje. [DOI] [PubMed] [Google Scholar]
- 92.Costa C, Incio J, Soares R. Angiogenesis and chronic inflammation: Cause or consequence? Angiogenesis. 2007;10:149–66. doi: 10.1007/s10456-007-9074-0. [DOI] [PubMed] [Google Scholar]
- 93.Ishikawa K, Yoshida S, Kadota K, Nakamura T, Niiro H, Arakawa S, et al. Gene expression profile of hyperoxic and hypoxic retinas in a mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2010;51:4307–19. doi: 10.1167/iovs.09-4605. [DOI] [PubMed] [Google Scholar]
- 94.Sato T, Kusaka S, Hashida N, Saishin Y, Fujikado T, Tano Y. Comprehensive gene-expression profile in murine oxygen-induced retinopathy. Br J Ophthalmol. 2009;93:96–103. doi: 10.1136/bjo.2008.142646. [DOI] [PubMed] [Google Scholar]
- 95.Connor KM, SanGiovanni JP, Lofqvist C, Aderman CM, Chen J, Higuchi A, et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–73. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gardiner TA, Gibson DS, de Gooyer TE, de la Cruz VF, McDonald DM, Stitt AW. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am J Pathol. 2005;166:637–44. doi: 10.1016/s0002-9440(10)62284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ishida S, Usui T, Yamashiro K, Kaji Y, Amano S, Ogura Y, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198:483–9. doi: 10.1084/jem.20022027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–28. [PMC free article] [PubMed] [Google Scholar]
- 99.Sfikakis PP, Markomichelakis N, Theodossiadis GP, Grigoropoulos V, Katsilambros N, Theodossiadis PG. Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care. 2005;28:445–7. doi: 10.2337/diacare.28.2.445. [DOI] [PubMed] [Google Scholar]
- 100.Rao VR, Prescott E, Shelke NB, Trivedi R, Thomas P, Struble C, et al. Delivery of SAR 1118 to Retina Via Ophthalmic Drops and its Effectiveness in Reduction of Retinal Leukostasis and Vascular Leakiness in Rat Streptozotocin (STZ) Model of Diabetic Retinopathy (DR) Invest Ophthalmol Vis Sci. 2010;51:5198–204. doi: 10.1167/iovs.09-5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iliaki E, Poulaki V, Mitsiades N, Mitsiades CS, Miller JW, Gragoudas ES. Role of alpha 4 integrin (CD49d) in the pathogenesis of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:4898–904. doi: 10.1167/iovs.08-2013. [DOI] [PubMed] [Google Scholar]