

Abstract
We confirm that FSH stimulates osteoclast formation, function and survival to enhance bone resorption. It does so via the activation of a pertussis toxin-sensitive Gi-coupled FSH receptor that we and others have identified on murine and human osteoclast precursors and mature osteoclasts. FSH additionally enhances the production of several osteoclastogenic cytokines, importantly TNFα, likely within the bone marrow microenvironment, to augment its pro-resorptive action. FSH levels in humans rise before estrogen falls, and this hormonal change coincides with the most rapid rates of bone loss. On the basis of accumulating evidence, we reaffirm that FSH contributes to the rapid perimenopausal and early post-menopausal bone loss, which might thus be amenable to FSH blockade.
Introduction
Since Fuller Albright first discovered the action of estrogen on bone [1], it has become dictum that hypogonadal osteoporosis is caused solely to low circulating estrogen levels. We recently challenged this view, and suggested that high FSH levels, which normally accompany ovarian failure, offer a contribution [2]. We found that mice devoid of either FSH or its receptor, FSHR, failed to display hyper-resorption, despite severe hypogonadism [2]. This was associated with dysfunctional osteoclasts observed both in vivo upon histomorphometry, and ex vivo, in isolated osteoclast cultures [2]. While the absence of hyper-resorption in chronic FSHR deficiency can be attributed, in part, due to the accompanying hyperandrogenemia [3], this does not exclude the possibility that high FSH levels per se contribute to hypogonadal bone loss. This view is buttressed by estrogen-independent correlations between perimenopausal bone loss and rising FSH levels noted over 4 years [4], and from observations that women with high FSH levels suffer greater bone loss than those with low-to-normal FSH levels, estrogen levels being the same [5].
Consistent with our hypothesis that FSH does directly cause bone loss in vivo, we have shown that that FSH stimulates the formation and resorptive activity of osteoclasts in vitro [2]. This action, we found, is exerted via FSHRs coupled to Gi2α that appears upstream of the pro-osteoclastogenic MAP kinase and NF-κB pathways, as well as through the enhanced production of tumor necrosis factor-α (TNFα), which expands the osteoclast precursor pool [2, 6]. The current study provides further, more compelling, evidence for a direct action of FSH on both murine and human osteoclasts, as has been examined in terms of effects on their genesis, function and fate. The accompanying manuscript studies in depth the effects of FSH on TNFα expression.
Methods
For the osteoclastogenesis experiments, murine bone marrow cells were extracted from femurs and tibias and washed and cultured in α-MEM with FBS (10%), M-CSF (30 ng/ml) and penicillin/streptomycin (1%) for 24 hours. Non-adherent hematopoetic stem cell precursors were removed for purification with Ficoll-Paque Plus (Amersham Biotech). The interface cell layer was isolated for culture in α-MEM with FBS (10%), M-CSF (50 ng/ml) and RANK-L (60 ng/ml) at 5×104 cells/well in 96-well plates. A tartrate-resistant acid phosphatase (TRAP) kit (Sigma-Aldrich) was used to stain osteoclasts for counting. RAW264.7 and RAW-C3 cells were also cultured with RANK-L (30 ng/ml) for 5 to 8 days to form TRAP-positive osteoclasts.
For osteoclast survival experiments, RAW264.7 cells were cultured at 5×105/6 cm dish for 24 hours in α-MEM ad FBS (10%). Cells were treated with camptothesin (0.1 μM) with and without FSH or M-CSF for 24 hrs, and live cells detected by trypan blue uptake. In separate experiments, apoptotic cells were detected by flow cytometry (FACS Calibur) using Annexin V FITC Apoptosis Detection Kit per manufacturer's directions (BD Biosciences, San Diego, CA), and results analyzed with FlowJo software (Treestar, Inc) as described previously [20].
Whole cell lysates and/or nuclear sub-fractions were obtained as described previously with modifications [25]. Western blotting was performed using enhanced chemiluminscence detection (Active Motif, or Amersham, Piscataway NJ). Antibodies for c-fos, c-jun were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), and antibodies against phospho-Erk1/2, phospho-JNK, and phospho-Akt were from Cell Signaling Technologies (Beverley, MA). In separate experiments cAMP levels were measured in RAW264.7 cell extracts exposed to FSH and/or pertussis toxin using a method described previously [2].
CD14 monocytes were isolated as described [2] plated at 3.75×106 cells/cm2 in human M-CSF (25 ng/ml), human RANKL (30 ng/ml) (Research Diagnostics, Flanders, NJ), vitamin D3 (10-8 M) (Sigma), dexamethasone (2×10-8M) (Sigma), heat inactivated FBS (10%) (HyClone) in DMEM (Cambrex, East Rutherford, NH). Cultures were maintained for 7 days and fixed in citrate/acetate buffer, pH 4.5 with acetone (40%). TRAP was determined using napthol AS-BI phosphate (12.5 mg/ml) in the presence of tartaric acid (0.67 mM) with fast garnet GBC to produce red precipitate. For pit forming activity, CD14 monocytes were plated on dentine slices for 15 days in the presence of human M-CSF (25 ng/ml) and human RANKL (30 ng/ml). The slices were then sonicated (ammonium hydroxide, 1 M, 2 minutes) and stained with toluidine blue (0.5%), pH 7.4, for 2 minutes. Phalloidin labeling and acid secretion assays were performed as described [26].
Results
Confirming our previous results [2] with new cell preparations and reagents, we show that graded concentrations of FSH stimulate, between 50% and 2-fold, the genesis of TRAP-positive osteoclasts from cloned RAW264.7 and RAW-C3 cells, as well as murine bone marrow osteoclast precursors (Figure 1A-C). Figure 1D further illustrates that pertussis toxin, which eliminates receptor-Gi coupling, prevents the increase in TRAP-positive osteoclast formation from RAW264.7 cells, consistent with data using Gi2α-deficient murine osteoclast precursors [2]. Additionally, the expected decrements in cAMP with FSH due to Gi2α coupling noted in murine osteoclast precursors [2] were also fully reversed by pertussis toxin (Figure 1E). Overall, therefore, the findings provide further, more definitive evidence using cloned RAW cells that the FSH-induced osteoclastogenesis arises through a pertussis-toxin-sensitive Gi protein.
Figure 1.
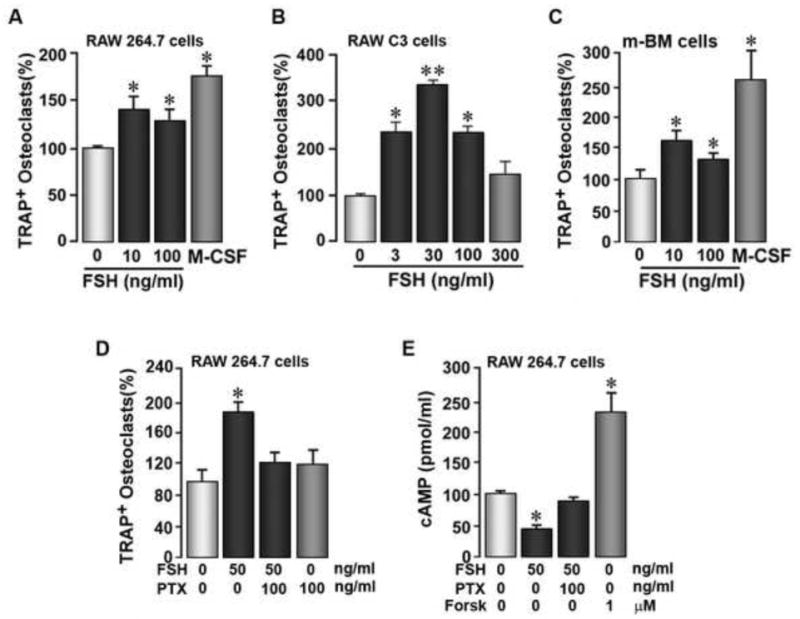
The effect of FSH on tartrate-resistant acid phosphatase-positive (TRAP+) osteoclast formation from, and cyclic AMP (cAMP) levels in, RAW264.7 or RAW-C3 cells, or primary mouse bone marrow (m-BM) cells, as indicated, in the presence or absence of pertussis toxin (PTX). Abbrev: For – forskolin. Statistics by Student's t-test, *p < 0.05, **p < 0.01, 3-8 wells/treatment for TRAP+ osteoclast formation assays and n = 5 wells/treatment for the cAMP assay.
We next utilized RAW264.7 cells to explore, in considerable depth, the effect of FSH on two signaling molecules, the MAP kinase Erk and Akt kinase, which we have previously shown are activated in murine osteoclast precursors [2]. Western immunoblotting of RAW264.7 cell extracts displayed increased phosphorylation of Akt at 60 minutes (Figure 2Aa), in stark contrast to that seen at 10 minutes in primary cells [2]. However, this increase was impressively inhibited by pertussis toxin (Figure 2Ab) indicating that the effect of FSH on Akt was also downstream of the pertussis toxin-sensitive Gi. Phosphorylation of Erk1/2 was also enhanced at 30 minutes (Figure 2Ba), again in contrast to an earlier effect noted in primary cells [2]. The effect of FSH was, however, less profound than that due to RANK-L, and no effects of FSH on JNK phosphorylation were observed in RAW264.7 cells (Figure 2Ba). Importantly, however, the effect of maximal (50 ng/ml) and sub-maximal (12.5 ng/ml) concentrations of RANK-L were additive to those of FSH (30 ng/ml), suggesting common mechanisms for downstream activation. This was confirmed by examining the nuclear localization of c-fos in response to FSH and RANK-L. FSH stimulated c-fos localization in RAW264.7 cells, as before with murine cells [2], but with a more protracted time course, peaking at 120 minutes (Figure 2Ca). The effects of sub-maximal (15 and 30 ng/ml) and maximal (60 and 60 ng/ml) of RANK-L and FSH, respectively, were again additive (Figure 2Cb).
Figure 2.
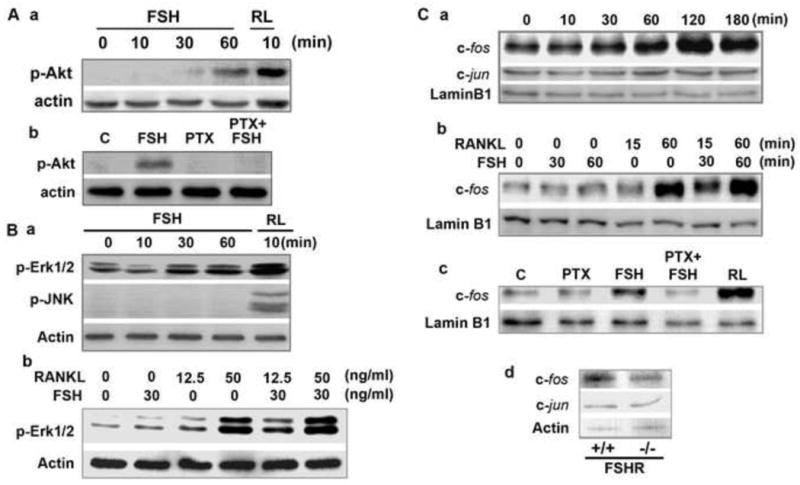
Western immunoblots showing the effect of FSH and/or RANK-L on the phosphorylation of Akt (p-Akt), Erk (p-Erk) or JNK (p-JNK) measured in total cell lysates (panels: A and B) (loading control: actin), or on c-fos and c-jun measured in nuclear extracts (loading control: lamin B1) in RAW264.7 cells (panels: Ca to Cc) in the presence or absence of pertussis toxin (PTX). Western immunoblots for c-fos and c-jun in cells isolated from bone marrow precursors derived from FSH receptor null mice (FSHR-/-) and wild type littermates (FSHR+/+) (panel: Cd).
That a Gi protein was upstream of FSH-induced c-fos nuclear localization was confirmed using pertussis toxin. The effect of FSH in inducing nuclear localization of c-fos was strongly attenuated in the presence of pertussis toxin (Figure 2Cc). Notably, consistent with a lack of effects of FSH on JNK activation (Figure 2Ba), the nuclear localization of c-jun remained unaltered up to 180 minutes following exposure to FSH (Figure 2Ca). Likewise, c-jun levels remained unaffected in FSHR-/- osteoclast precursors, whereas wild type cells displayed lower c-fos levels (Figure 2Cd). The latter observation attests to the direct regulation of c-fos nuclear localization by FSH.
In addition to stimulating osteoclast formation by activating the aforementioned pathways, we also confirmed and extended previous results on the direct action of FSH on the resorptive function of mature human osteoclasts [2]. In the pit assay, where resorptive lacunae positive for toluidine blue are counted, FSH (30 ng/ml) stimulated resorption (Figure 3A). This was consistent with a substantial change in the phalloidin-labeled actin rings in osteoclasts on plastic coverslips (Figure 3B), suggesting that FSH enhances osteoclast activation. Typically, osteoclasts on artificial matrix show punctate attachments (Fig 3B, top frame), while on bone a complete attachment ring occurs; with FSH enhancement, the cells on plastic show this resorptive ring (Fig 3B, bottom frame). Accompanied with this, there was a concentration-dependent increase in lysotracker-positive acid lakes under osteoclast surfaces, indicative of increased acid secretion (Figure 3Ca). When analysed by image intensity comparison in a second experiment (Figure 3Cb), where GnRH was used as an additional control, considerable variation in lysotracker activity is apparent, although 20-100 cells per group were analysed and the differences without and with FSH were significant. Overall, therefore, FSH stimulated bone resorption by mature osteoclasts by increasing substrate adhesion and extracellular acid secretion.
Figure 3.
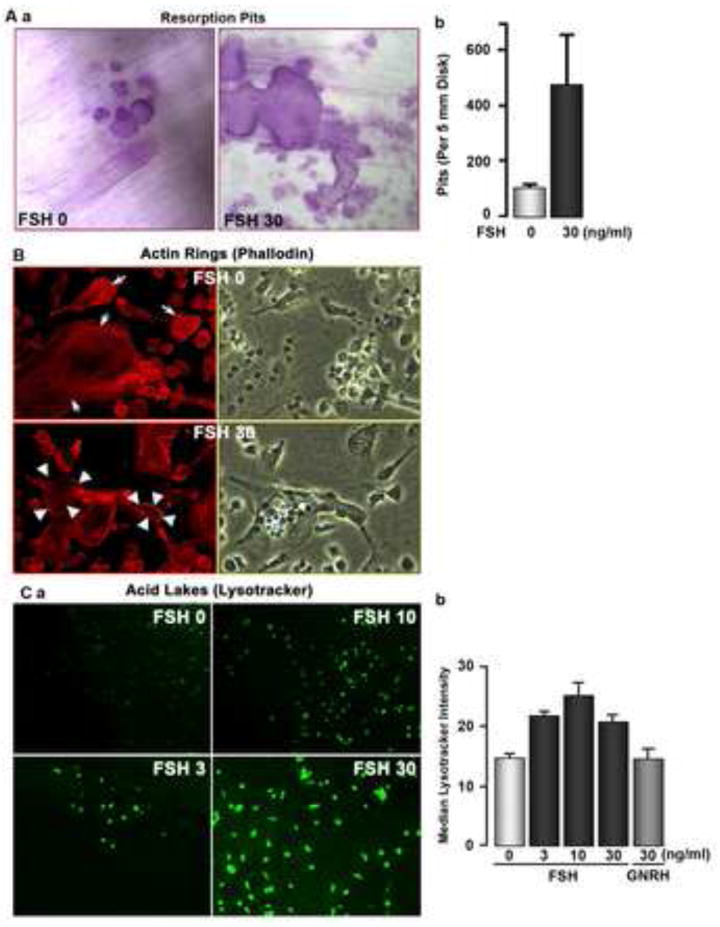
Response of human peripheral blood mononuclear cell-derived osteoclasts to FSH. (A) Resorption of devitalized dentine substrate (Aa) as photomicrographs and expressed as pits per slice (Ab); this response was variable over several experiments and on average an increase of ∼30-50% was seen, although striking increases in activity, as in this case, were not unusual. Photomicrographs are 300 μm2. (B) Osteoclasts on tissue culture plastic with phalloidin-Cy5 to label actin (red fluorescence, left frames) and in phase (right frames), without (top frames) or with (bottom frames) human FSH (30 ng/ml) added. Note that without added FSH, actin attachments are punctate (associated with podosomes), while after FSH, a continuous actin ring is present, similar to the tight attachment on bone. Fields are 200 μm across. One of triplicate cell cultures showing similar changes is illustrated. (C) Lysotracker labeling of human monocyte cultures on bone 7 days after addition of M-CSF (10 ng/ml) and RANKL (40 ng/ml) and indicated concentrations of human FSH in ng/ml. Note in the photomicrographs in (a) the trend toward increased labeling of acid compartments with exposure to FSH. Fields are 580 nm across. In a second experiment, labeling was quantified as fluorescence intensity for 20-100 cells of each culture type (b). There was considerable variability, with overlap between individual cells with each type of treatment, but the average increase in acid secretion with FSH addition is clear. An additional control, GnRH addition, is shown (right bar).
Finally, we studied the effect of FSH on the fate of RAW264.7 cells and osteoclasts by examining cell number and annexin V staining after treatment with a pro-apoptotic agent, camptothesin. There was a doubling of cell numbers with FSH within 24 hours in the presence of camptothesin, although not as dramatic as with M-CSF (Figure 4A). This was in agreement with parallel flow cytometry studies, where we found a concentration-dependent decrease in annexin-positive cells (Figure 4B). To differentiate dead cells from cells undergoing early apoptosis, cells were double-labeled with annexin V and propidium iodide, a nuclear stain that labels dead cells. There was a striking decline in annexin V-high, propidium iodide-low cells, concordant with a pro-survival action of FSH (Figure 4C). Overall, therefore, our studies provide more persuasive evidence for the stimulation of osteoclastogenesis and resorptive activity, and the inhibition of apoptosis by FSH.
Figure 4.
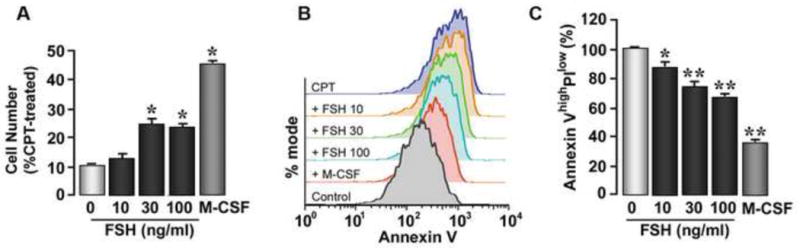
The pro-survival actions of FSH on RAW264.7 cells and primary murine bone marrow cells. Cell number (A) and annexin V staining (B,C) was assessed after treatment with a pro-apoptotic agent, camptothesin (CPT). Positive control: M-CSF. Propidium iodine labeling was used to differentiate dead cells from cells undergoing early apoptosis; flow cytometry allowed the quantitation of annexin V-high, propidium iodide-low cells (C). Statistics by Student's t-test, *p < 0.05, **p < 0.01, (A) 3 experiments pooled; (C) n = 4 wells/treatment.
Discussion
The question we ask is whether FSH has a role in hypogonadal, and in humans, peri- and postmenopausal bone loss, which has hitherto been attributed solely to declining estrogen levels. And, if so, what is the mechanism through which these effects are exerted? In our earlier study, we showed that FSH is potently osteoclastogenic and pro-resorptive in vitro and that the absence of FSH or its receptor protects against hypogonadal bone loss [2]. While the in vitro effects of FSH have since been replicated in other laboratories [7, 8], it was shown subsequently that part of the osteoprotection in chronic genetic FSHR deficiency arises from the accompanying hyperandrogenemia due to preserved luteal function [3]. However, the clinical paradox still remains: the fundamental observation that women lose bone dramatically and most rapidly during late perimenopausal and early post-menopausal years [4, 9, 10]. This precipitous bone loss [11, 12] cannot possibly be explained by hypoestrogenemia, particularly because, during this time, estrogen levels are relatively normal, while early follicular FSH levels have risen to over 5-fold of premenopausal values [13]. Indeed, a change in serum FSH correlates strongly with a change in bone mineral density [4] and women that fall within the highest tertile of serum FSH levels have highest bone resorption markers [14, 15]. Although correlative, these clinical observations come not only from SWAN (Study of Women's Health across the Nations) [4, 14, 15], but also from a Chinese cohort [9], and a younger cohort of American women aged between 20 and 50 years [10]. Most impressive, however, is that the AA rs6166 FSHR genotype is associated with low femoral neck and total body bone mineral density, independently of circulating estrogen [11]. We, and more recently others, have shown that human osteoclasts bear FSHRs [2, 10]. We also show that FSH stimulates the formation of human osteoclasts, as well as their resorptive function. These in vitro and human studies together provide a compelling rationale for further investigations on a role for FSH in the pathophysiology of post-menopausal bone loss.
Demonstrating the attenuation of ovariectomy-induced bone loss by inhibiting the rising FSH will constitute ultimate proof for causality. Our current studies nonetheless provide further affirmative evidence for its mechanism of action in both human osteoclasts and RAW264.7 cells. Namely, FSH has both direct and indirect actions to increase resorption. Using RAW264.7 cells, we show that FSH stimulates osteoclastogenesis, proven previously by us [2] and others [7, 8] using murine and human osteoclasts. It does this primarily by activating Erk phosphorylation via a Gi2α protein [2], which we demonstrate here as classically pertussis toxin-sensitive. It also enhances osteoclast activation, acid secretion and overall resorptive activity. Additionally, we show, for the first time, that FSH stimulates osteoclast survival, even in the presence of a potent pro-apoptotic stimulus, camptothesin. Together, we find that the exposure of osteoclast precursors to FSH results in more osteoclasts being formed, each with enhanced function and reduced propensity to undergo apoptosis.
In addition to the direct effects of FSH on osteoclast formation, the hormone also stimulates the production of several osteoclastogenic cytokines, namely TNFα, IL-1β and IL-6 [6, 10]. The accompanying manuscript demonstrating clear effects of FSH on TNFα production from human osteoclasts [17] confirms previous mouse studies [2]. We know that a key function of TNFα is to expand the osteoclast precursor pool [6], thus amplifying overall osteoclast formation. Nonetheless, whether the elevated peripheral blood mononuclear cell TNFα reported during human menopause [18] contributes to the increased osteoclastogenesis remains to be determined. A recent report however shows striking correlations between FSH and TNFα, IL-1β and IL-6 in serum as well as, between monocytic FSHR expression and serum cytokine levels [10]. FSH also stimulated cytokine production from monocyte precursors, confirming our results with both murine and human cells [6, 17]. However, serum cytokine levels did not relate to bone mineral density, whereas serum FSH correlated strongly [10].
We have established previously that, in contrast to FSH, thyroid stimulating hormone (TSH) negatively regulates TNFα production through an effect on gene transcription [19]. Elevated TNFα levels in bone marrow supernatants and macrophages from the TSHR-/- mice [20], and the rescue of their enhanced osteoclastogenesis in compound mutants also lacking TNFα [21] together attest to a role for TNFα in synergizing the bone loss of experimental TSH signaling deficiency. However, whether TNFα and other inflammatory cytokines play a role in human osteoporosis has yet to be determined. That said, TNFα blockade is unlikely to become a putative target for either hypogonadal or thyrotoxic osteoporosis, as infliximab administration to patients with Crohn disease, where the osteoporosis is overtly TNFα-driven, fails to prevent bone loss [22]. In contrast, a recent study shows that exogenous FSH enhances periodontitis-induced alveolar osteolysis, as direct evidence for synergism between FSH and locally-released cytokines [23]. Finally, and perhaps most intriguingly, Pacifici and colleagues show that FSH up-regulates the CD40-ligand on antigen presenting cells, in essence attributing the T-cell-mediated, hypogonadal bone loss, in part, to high FSH levels [24]. In all, evidence for a role of FSH in bone loss continues to mount.
Acknowledgments
This work was supported by the National Institute on Aging (AG23176 to M.Z.). M.Z., L.S. and H.C.B. also acknowledge the National Institutes of Health for grant support (DK70526, DK80490, AR053976, AR055208, and AR053566. H.C.B. and J.I. also acknowledges the Department of Veterans Affairs and the American Federation for Aging Research, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albright F, Smith PH, Richardson AM. Post menopausal osteoporosis. JAMA. 1941;1167:2465–2474. [Google Scholar]
- 2.Sun L, Peng Y, Sharrow AC, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 3.Gao J, Tiwari-Pandey R, Samadfam R, et al. Altered ovarian function affects skeletal homeostasis independent of the action of follicle stimulating hormone. Endocrinology. 2007;148:2613–2621. doi: 10.1210/en.2006-1404. [DOI] [PubMed] [Google Scholar]
- 4.Sowers MR, Jannausch M, McConnell D, et al. Hormone predictors of bone mineral density changes during the menopausal transition. J Clin Endocrinol Metabol. 2006;91:1261–1267. doi: 10.1210/jc.2005-1836. [DOI] [PubMed] [Google Scholar]
- 5.Devleta B, Adem B, Seneda S. Hypergonadotropic amenorrhea and bone density: new approach to an old problem. J Bone Min Res. 2004;22:360–364. doi: 10.1007/s00774-004-0495-1. [DOI] [PubMed] [Google Scholar]
- 6.Iqbal J, Sun L, Kumar TR, et al. Follicle stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci. 2006;103:14925–14930. doi: 10.1073/pnas.0606805103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Torcheria J, Yao W, et al. Bone microenvironment specific roles of ITAM adapter signaling during bone remodeling induced by acute estrogen deficiency. PlosOne. 2007;2:e586. doi: 10.1371/journal.pone.0000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicks KM, Akel NS, Suva LJ, Gaddy D. Inhibin directly targets suppression of isolated human osteoclast precursor development and activity. J Bone Miner Res. 2008;23:S150. [Google Scholar]
- 9.Wu XY, Wu XP, Xie H, et al. Age-related chanhes in biochemical markers of bone turnover and gonadotropins levels and their relationship among Chinese adult women. Osteoporosis Int. 2010;21:275–285. doi: 10.1007/s00198-009-0943-9. [DOI] [PubMed] [Google Scholar]
- 10.Cannon JG, Cortez-Cooper M, Meaders E, et al. Follicle stimulating hormone, interleukin-1 and bone density in adult women. Am J Physiol. 2009 Dec 8; doi: 10.1152/ajpregu.00728.2009. E-pub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Recker R, Lappe J, Davies KM, Heaney R. Bone remodeling increases substantially in the years after menopause and remains increased in older osteoporosis patients. J Bone Miner Res. 2004;19:1628–1633. doi: 10.1359/JBMR.040710. [DOI] [PubMed] [Google Scholar]
- 12.Akhter MP, Lappe JM, Davies KM, Recker RR. Transmenopausal changes in the trabecular bone structure. Bone. 2007;41:111–116. doi: 10.1016/j.bone.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 13.Randolph JF, Sowers M, Bondarenko IV, et al. Change in estradiol and follicle-stimulating hormone across the early menopausal transition: effects of ethinicity and age. J Clin Endocrinol Metabol. 2004;89:1555–1561. doi: 10.1210/jc.2003-031183. [DOI] [PubMed] [Google Scholar]
- 14.Sowers MR, Greendale GA, Bondarenko J, et al. Endogenous hormones and bone turnover markers in pre- and perimenopausal women: SWAN. Osteoporos Int. 2003;14:191–197. doi: 10.1007/s00198-002-1329-4. [DOI] [PubMed] [Google Scholar]
- 15.Xu ZR, Wang AH, Wu XP, et al. Relationship of age-related concentrations of serum FSH and LH with bone mineral density, prevalence of osteoporosis in native Chinese women. Clin Chim Acta. 2009;400:8–13. doi: 10.1016/j.cca.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 16.Rendina D, Gianfrancesco F, Di Fillipino G, et al. FSHR gene polymorphisms influence bone mineral density and bone turnover in post-menopausal women. Eur J Endocrinol. doi: 10.1530/EJE-10-0043. In the press. [DOI] [PubMed] [Google Scholar]
- 17.Robinson LJ, Tourkova I, Wang Y, et al. FSH-Receptor isoforms and FSH-dependent gene transcription in human monocytes and osteoclasts. doi: 10.1016/j.bbrc.2010.02.112. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacifici R. Estrogen, cytokines and pathogenesis of post-menopausal osteoporosis. J Bone Miner Res. 1996;11:1043–1051. doi: 10.1002/jbmr.5650110802. [DOI] [PubMed] [Google Scholar]
- 19.Hase H, Ando T, Eldeiry L, et al. TNFalpha mediates the skeletal effects of thyroid-stimulating hormone. Proc Natl Acad Sci. 2006;103:12849–12854. doi: 10.1073/pnas.0600427103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151–162. doi: 10.1016/s0092-8674(03)00771-2. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Vukicevic S, Baliram R, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci. 2008;105:4289–4294. doi: 10.1073/pnas.0712395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pazianas M, Rhim AD, Weinberg AM, et al. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn's disease. Ann NY Acad Sci. 2006;1068:543–556. doi: 10.1196/annals.1346.055. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Cheng Y, Fan M, et al. FSH aggravates periodontitis-related bone loss in ovariectomized rats. J Dent Res. 2010 February 5; doi: 10.1177/0022034509358822. E-pub. [DOI] [PubMed] [Google Scholar]
- 24.Pacifici R. Immune system, FSH and regulation of bone mass. Proceeding of the 1st Annual Meeting of the Società Italiana dell'Osteoporosi del Metabolismo Minerale e delle Malattie dello Scheletro, Torino, Clinical Cases in Mineral and Bone Metabolism; 2009. S32 (available at 24 http://siommms09.congress-online.it/siommms09/index.php/pacifici.html) [DOI] [PubMed] [Google Scholar]
- 25.Adebanjo OA, Anandathreethavarada HK, Koval AP, et al. A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol. 1999;7:409–414. doi: 10.1038/15640. [DOI] [PubMed] [Google Scholar]
- 26.Yaroslavskiy BB, Li Y, Ferguson DJ, et al. Autocrine and paracrine nitric oxide regulate attachment of human osteoclasts. J Cell Biochem. 2004;91:962–972. doi: 10.1002/jcb.20009. [DOI] [PubMed] [Google Scholar]