

Abstract
Experimental and clinical studies often require highly purified cell populations. FACS is a technique of choice to purify cell populations of known phenotype. Other bulk methods of purification include panning, complement depletion and magnetic bead separation. However, FACS has several advantages over other available methods. FACS is the preferred method when very high purity of the desired population is required, when the target cell population expresses a very low level of the identifying marker or when cell populations require separation based on differential marker density. In addition, FACS is the only available purification technique to isolate cells based on internal staining or intracellular protein expression, such as a genetically modified fluorescent protein marker. FACS allows the purification of individual cells based on size, granularity and fluorescence. In order to purify cells of interest, they are first stained with fluorescently-tagged monoclonal antibodies (mAb), which recognize specific surface markers on the desired cell population (1). Negative selection of unstained cells is also possible. FACS purification requires a flow cytometer with sorting capacity and the appropriate software. For FACS, cells in suspension are passed as a stream in droplets with each containing a single cell in front of a laser. The fluorescence detection system detects cells of interest based on predetermined fluorescent parameters of the cells. The instrument applies a charge to the droplet containing a cell of interest and an electrostatic deflection system facilitates collection of the charged droplets into appropriate collection tubes (2). The success of staining and thereby sorting depends largely on the selection of the identifying markers and the choice of mAb. Sorting parameters can be adjusted depending on the requirement of purity and yield. Although FACS requires specialized equipment and personnel training, it is the method of choice for isolation of highly purified cell populations.
Keywords: Immunology, Issue 41, cell sorting, monoclonal antibodies, compensation, antibody titration, FACS
Protocol
- Before starting the process, the following items need to be to collected and prepared:
- Ice
- 15 ml conical tubes for staining cells and 12 x 75 mm flow tubes for staining single color controls.
- Staining buffer: phosphate buffered saline (PBS) + 3% fetal calf serum (FCS)
- Pipettes
- Fc block (if required) and staining antibodies. Antibodies need to be titrated for optimal staining.
- Compensation beads
- Suspension buffer: Hank's Balanced Salt Solution (HBSS) + 25 mM HEPES + 3% FCS
- Trypan blue and hemocytometer
- Cell strainer (40 μm Nylon)
- Collection tubes: Two types of collection tubes can be used a) 12 x 75 mm polystyrene tubes (containing 300 μl FCS and 25 mM HEPES) or b) 15 ml conical tubes (containing 1 ml FCS + 25 mM HEPES). Any rich medium with high serum concentration can be used for the collection of sorted cells.
Generate a single cell suspension of the starting cell population.
Optional: Enrich for the desired cell population by a bulk purification method such as complement depletion or magnetic sorting. The main advantage of the enrichment step is that it reduces the sort time.
Wash cells once with staining buffer.
Discard the supernatant and resuspend the cells in staining buffer at a concentration up to 50 x 106 for efficient staining.
Optional: For cells expressing high levels of FcR, block the receptors using an appropriate blocking method. A method of choice is the use of a mAb that binds to FcγR on ice for 10-15 min. These antibodies are commercially available.
Add the appropriate mAb (at a predetermined concentration) to stain the desired cell population and incubate for 20-30 min on ice in the dark, followed by two washes with staining buffer. Optional: Staining with a vital dye, such as propidium iodide, can be included to discern dead cells (5). If multi-color staining is to be used, single color controls are required. We use BD CompBeads to prepare single color controls using the manufacturer's protocol.
If not using directly conjugated antibodies, repeat step 7 using the appropriate secondary antibody or Streptavidin conjugate.
After washing, resuspend the cells in culture medium and determine the cell concentration using a vital dye such as Trypan blue.
Adjust the cell concentration to 15-20 x 106/ml. For cell populations that form clusters, which can clog the instrument during sorting, filter the cells through a strainer.
- Set up and optimize the cell sorter. The process of setting up a flow cytometer is varied depending on the manufacture and needs to be performed by appropriately trained personnel.The general recommendations are as follows:
- Select the appropriate nozzle depending on the cell type to be sorted.
- For sterile sorts, sterilize the instrument.
- Perform instrument quality control with beads to verify lasers are functioning, and it is sorting accurately.
- Install the required collection device and set up the side streams.
- Look at the instrument's laser and detector set up to determine the fluorescent labels that can be used (our BD FACS Aria has three lasers and can detect up to nine colors).
- Once it is determined what fluorescent labels will be used on the cell sorter, compensation can be performed (as explained below).
- Perform compensation using the negative control sample and the single positive controls. Compensation is necessary to remove the spectrum overlap between two detectors. It is pertinent to note that compensation is not fool proof and is adversely affected by how brightly certain markers stain, and by the fluorescence of cells that have low autofluorescence which can lead to poor resolution between dim and negative populations. For best results, the experimental design should include using bright fluorochromes with markers that have low expression or that don't have a known staining pattern. Markers that provide good separation between cell populations and are highly expressed should be used with dimer fluorochromes such as ones excited by red or violet lasers.
- Compensation can be performed with compensation beads, which are polystyrene microparticles that have been coupled to an antibody specific for the Kappa light chain of Ig from mouse, rat, or rat/hamster. The beads are easy to stain, have a robust signal and provide an easy way of preparing single stained controls that have the same fluorophore as the experimental samples.
- Set up a template that includes a bivariate plot to display forward scatter (FSC) and side scatter (SSC), and one histogram for each fluorochrome that will be used.
- Run the negative control tube and adjust the forward scatter (FSC) and side scatter (SSC) to place the population of interest on scale.
- Adjust the fluorescence PMT settings to place the negative population or autofluorescence in the far left hand portion of the histogram.
- Record the negative control tube.
- Run the single positive control tubes and record the data for each tube.
- Draw an interval gate around the positive portion of the data on the histogram, and another interval gate on the negative portion of the histogram.
- Manual compensation is done by adjusting the median (median is a better estimate of central tendency than the mean on a logarithmic scale) of the positives until it is equal to the median of the negatives. This must be done for each of the fluorochrome labels used in the experiment.
- To adjust the median, adjust the spectral overlap values either higher or lower until the medians for each fluorescent parameter match as closely as possible.
- Compensation can also be done automatically with analysis software. Automatic compensation uses the recorded data from compensation controls to calculate an algebraic matrix for all the fluorophores used in the experiment. These values are used to subtract out the contributions of the non-primary colors that are bleeding into a primary color's detector.
- Record the experimental sample to be sorted and use gating tools and subsetting methods to define the populations of interest.
- Set up the experimental template with a dot plot that displays FSC vs. SSC and use a gating tool, polygon, rectangle, interval or quadrant to gate the population of interest. The scatter plot displays the physical properties of the cell which is distinct to the cell type.
- The best way to determine the fluorescence gating strategy is to use fluorescence minus one (FMO) controls. In a FMO control tube all reagents that are used in the experiment are included except for the one of interest. The FMO helps discriminate between dimly stained populations and broad negative populations.
- Use the data recorded from FMO tubes to determine where to place gates within the experimental tube.
Once gates have been determined, the gates can be selected for sorting into external collection tubes. Up to four gates (or populations) can be sorted at one time depending on the instrumentation available.
Run the experimental sample tube at 4 °C, turn on deflection plates, and sort the sample. 5 x 105 -1.5 x 106 cells can be sorted into a 12 x 75 mm tube and 1.5 x 106 - 4.5 x 106 cells can be sorted into a 15 ml conical.
Once the required number of cells has been obtained, manually stop sorting.
Perform a post-sort analysis to determine the purity of the sorted cell populations.
Representative results
We have shown here results for mouse splenic B and CD4 T cell sorting (Figure 1). Splenocytes were stained with PE Texas Red-conjugated anti-mouse B220, FITC-conjugated anti-mouse TCRβ and Alexafluor-700-conjugated anti-mouse CD4 to identify B cells (B220+TCRβ-) and CD4 T cells (CD4+TCRβ+). The sorting was performed with BD FACS Aria instrument. After sorting the B cell purity was >97% and the CD4 T cell purity was >98%.
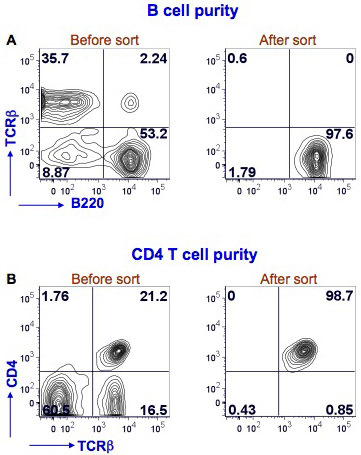 Figure 1. Purity of splenic B cells and CD4 T cells after FACS. Mouse splenocytes were stained with anti-mouse B220, TCRβ and CD4 antibodies. FACS for B cells was performed using a BD FACS Aria by gating on B220+TCRβ- cells (Fig. 1A, left panel, lower right quadrant). A post sort analysis was performed to determine the purity of the B cells (Fig. 1A, right panel). B220 staining is shown on the x-axis and TCRβ staining on the y-axis. FACS for CD4 T cells was performed using the same flow cytometer by gating on TCRβ+CD4+ cells (Fig. 1B, left panel, upper right quadrant). A post sort analysis was performed to determine the purity of the CD4 T cells (Fig. 1B, right panel). TCRβ staining is shown on the x-axis and CD4 staining on the y-axis. The percent positive cells in each quadrant is shown.
Figure 1. Purity of splenic B cells and CD4 T cells after FACS. Mouse splenocytes were stained with anti-mouse B220, TCRβ and CD4 antibodies. FACS for B cells was performed using a BD FACS Aria by gating on B220+TCRβ- cells (Fig. 1A, left panel, lower right quadrant). A post sort analysis was performed to determine the purity of the B cells (Fig. 1A, right panel). B220 staining is shown on the x-axis and TCRβ staining on the y-axis. FACS for CD4 T cells was performed using the same flow cytometer by gating on TCRβ+CD4+ cells (Fig. 1B, left panel, upper right quadrant). A post sort analysis was performed to determine the purity of the CD4 T cells (Fig. 1B, right panel). TCRβ staining is shown on the x-axis and CD4 staining on the y-axis. The percent positive cells in each quadrant is shown.
Discussion
FACS is a highly sophisticated technique for purifying cell populations of interest, in which a very high purity (95-100%) of the sorted population can be obtained. Therefore, this technique is particularly important for experiments, where high purity is an essential requirement (e.g. microarray analysis). FACS is particularly advantageous over other available methods of purification when a cell population needs to be purified based on a weekly expressed surface marker or when two or more populations need to be purified which have different levels of expression of the same surface marker. For example, the purification of marginal zone B cells (B220+CD21hiCD23int/low) and follicular B cells (B220+CD21int/lowCD23hi) based on expression levels of CD21 and CD23. Another growing application of FACS is single cell sorting allowing the analysis of individual cells (3, 4). FACS is also commonly used to sort cells expressing fluorescent proteins that are genetically expressed, such as green fluorescent protein tagged proteins (5). If a sterile sort is performed the cells can be cultured. However, cell viability and yield can be compromised during sorting. Viability can be improved by using a cell-dependent optimal temperature for the sorting and by processing collection tubes immediately after they reach capacity. Recovery can be increased by using polypropylene collection tubes, collecting sorted cells in serum rich media and inverting collection tubes intermittently, maintaining collection tubes at an optimal temperature and centrifuging sorted cells for ~10 min (http://cyto.mednet.ucla.edu/Protocol.htm).
The development of ultra high-speed sorters has extended the possibility of the application of flow sorting in clinical settings. The potential clinical applications of FACS include purification of blood stem cells from human blood for therapeutic purposes, applications in cancer therapy, amniocentesis replacement, sorting human sperm and early disease detections (6, 7).
For multicolor staining, antibody selection is a critical step and fluorochromes are chosen based on the detection system for each particular instrument. There are a number of companies that sell cell sorters and the laser configuration varies according to user needs. Thus one cannot simply use the exact same antibodies used in published studies without confirming compatibility. For optimal antibody selection, one should consider the expression level of the protein being examined. In general, the brightest available fluorochrome-tagged antibodies should be used to stain dimly expressed surface markers, while dim fluorochromes can be used for the staining of highly expressed surface markers (8). In addition, antibodies should be selected in such a way that overlap among their emission spectra is minimal. For multicolor staining, it is extremely difficult to choose fluorochrome that have no spectral overlap. In this situation, compensation is performed.
Compensation is necessary to mathematically eliminate the fluorescence overlap between the emissions spectrum of the various fluorochromes that is measureable by the detectors (8-10). In other words, the overlap fluorescence from one fluorochrome is subtracted from the emissions spectrum of another fluorochrome. The compensation calculation requires an unstained control and single positive controls for each fluorochrome used for a multicolor staining (9, 10). Compensation can be conducted using cells or beads. The beads are easy to stain, have a robust signal and provide an easy way of preparing single stained controls that have the same fluorophores as the experimental samples. Compensation beads are particularly advantageous when the cell surface protein of interest is expressed at a low level and when the cell population of interest is limiting in the initial starting cell population.
Once the antibodies have been chosen, their activity should be optimized by performing a titration analysis. This is particularly important if the FACS machine has highly powerful lasers. The use of suboptimal concentrations of staining antibodies can result in poor separation of the desired cell population from the total cell population. The use of high antibody concentrations increases the chance for antigen non-specific staining. Therefore antibody titration will allow the selection of the antibody concentration that gives the maximum brightness of the positive population and minimal background staining (8).
FACS is now a standard technique for the purification of subpopulations of cells. It can be used to separate any cell type in which a single cell suspension can be generated and antibodies are available to identify the desired cell population. FACS is the method of choice when highly pure cell populations are required.
Acknowledgments
We would like to thank Dr. Bill Cashdollar for his support in the use of the Flow Cytometry Core.
References
- Herzenberg LA, De Rosa SC. Monoclonal antibodies and the FACS: complementary tools for immunobiology and medicine. Immunol Today. 2000;21:383–390. doi: 10.1016/s0167-5699(00)01678-9. [DOI] [PubMed] [Google Scholar]
- Givan AL. Flow cytometry first principles. New York: Wiley-Liss; 2001. pp. 273–273. [Google Scholar]
- Hewitt Z, Forsyth NR, Waterfall M, Wojtacha D, Thomson AJ, McWhir J. Fluorescence-activated single cell sorting of human embryonic stem cells. Cloning Stem Cells. 2006;8:225–234. doi: 10.1089/clo.2006.8.225. [DOI] [PubMed] [Google Scholar]
- Walker A, Parkhill J. Single-cell genomics. Nat Rev Microbiol. 2008;6:176–177. doi: 10.1038/nrmicro1862. [DOI] [PubMed] [Google Scholar]
- Sorensen TU, Gram GJ, Nielsen SD, Hansen JE. Safe sorting of GFP-transduced live cells for subsequent culture using a modified FACS vantage. Cytometry. 1999;37:284–290. [PubMed] [Google Scholar]
- Diamond RA. In: In living color: Protocols in flow cytometry and cell sorting. DeMaggio S, editor. Berlin: Springer; 2000. pp. 800–800. [Google Scholar]
- Karabinus DS. Flow cytometric sorting of human sperm: MicroSort clinical trial update. Theriogenology. 2009;71:74–79. doi: 10.1016/j.theriogenology.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA, Tung J, Moore WA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7:681–685. doi: 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA. Modern flow cytometry: a practical approach. Clin Lab Med. 2007;27:453–468. doi: 10.1016/j.cll.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]