

Abstract
The chicken embryo is a classical animal model for studying normal embryonic and fetal development and for xenotransplantation experiments to study the behavior of cells in a standardized in vivo environment. The main advantages of the chicken embryo include low cost, high accessibility, ease of surgical manipulation and lack of a fully developed immune system. Xenotransplantation into chicken embryos can provide valuable information about cell proliferation, differentiation and behavior, the responses of cells to signals in defined embryonic tissue niches, and tumorigenic potential. Transplanting cells into chicken embryos can also be a step towards transplantation experiments in other animal models. Recently the chicken embryo has been used to evaluate the neurogenic potential of human stem and progenitor cells following implantation into neural anlage1-6. In this video we document the entire procedure for transplanting human stem cells into the developing central nervous system of the chicken embryo. The procedure starts with incubation of fertilized eggs until embryos of the desired age have developed. The eggshell is then opened, and the embryo contrasted by injecting dye between the embryo and the yolk. Small lesions are made in the neural tube using microsurgery, creating a regenerative site for cell deposition that promotes subsequent integration into the host tissue. We demonstrate injections of human stem cells into such lesions made in the part of the neural tube that forms the hindbrain and the spinal cord, and into the lumen of the part of the neural tube that forms the brain. Systemic injections into extraembryonic veins and arteries are also demonstrated as an alternative way to deliver cells to vascularized tissues including the central nervous system. Finally we show how to remove the embryo from the egg after several days of further development and how to dissect the spinal cord free for subsequent physiological, histological or biochemical analyses.
Keywords: Cellular Biology, Issue 41, chicken embryo, xenotransplantation, transplantation, human stem cells, differentiation, regeneration, spinal cord, cell therapy
Protocol
1. Isolation and culture of human stem cells and preparation for injection into embryos
Human stem cells used as examples in this protocol (brain, adipose, bone marrow) are isolated in different ways from the different tissues. For example, adult neural stem cells are isolated from tissue pieces collected from the ventricular wall of the brain during endoscopic neurosurgery, which are then dissociated to individual cells that are cultured in vitro. The neural stem cells spontaneously form neurospheres that float in the culture medium while other cell types adhere to the bottom of the dish7. Adipose-derived mesenchymal stem cells are obtained from liposuction material that is enzymatically digested and strained to remove fibrous tissue and then centrifuged to remove mature adipocytes. The resulting cells are resuspended in culture medium supplemented with serum to promote the proliferation of stem cells over other cell types8 Hematopoietic stem and progenitor cells can be isolated from bone marrow aspirates by positive selection using specific antibodies conjugated to magnetic particles (Myltenyi Biotec, Germany or Dynal-Invitrogen, USA) and magnetic cell separation, and/or using antibodies conjugated to fluorophores and fluorescence activated cell sorting (FACS)9.
To facilitate later identification, label the stem cells with a fluorescent marker prior to implantation into chicken embryos. Stem cells can be labeled with fluorescent lipophilic dyes such as DiI (Invitrogen, USA) or PKH26 (Sigma, USA) using supplier-recommended protocols up to shortly before implantation, or, for a permanent label with essentially no danger of contaminating host cells, they can be transduced with the gene for a fluorescent protein like Green Fluorescent Protein, using lentiviral vectors, while still in culture10.
Protocols for the culture and propagation of human stem cells vary. Detailed protocols can be found in the literature. Briefly, sphere-forming stem cells are typically cultured in flasks, whereas adherent stem cells are typically cultured in petri dishes (which facilitate trituration when splitting, see below), in appropriate medium. For splitting or harvesting, pellet sphere-forming cells by 5 min centrifugation at 1200rpm and resuspend in pre-warmed trypsin/EDTA solution for no more than 5 min at 37 degrees Celsius. For adherent cells add the trypsin/EDTA solution to the petri dish, and triturate at the end of 5 minutes. Terminate the trypsinization by adding Ca2+-containing medium, sometimes complemented with albumin. To remove the trypsin/EDTA, centrifuge the cells (5 min at 1200rpm), remove the supernatant, resuspend in cold injection vehicle (typically medium or phosphate-buffered saline), and centrifuge again (5 min at 1200rpm). Remove most of this supernatant and gently resuspend the cells to create a highly concentrated cell suspension.
Keep the suspension of human stem cells in a small, capped vial or microfuge tube on ice for up to several hours prior to injection. Survival in this state may be cell type-dependent. The injection vehicle must be optimized according to the properties of the cells, including their adhesivity (e.g. a low-Ca2+ vehicle may help prevent clumping) and resistance to hypoxia and pH changes.
2. Egg incubation and preparation
Store fertilized eggs in a cooling chamber (for example: Termaks, Bergen, Norway) at 14-15°C prior to incubation. At this temperature development is arrested until incubation begins. Fertilized eggs stored for more than about 10 days or at lower temperatures have a lower rate of subsequent development and survival.
To reinitiate development, place the eggs in a humidified, forced draft incubator (for example: Binder, New York, USA) at 38-39°C, and incubate until the desired stage of development is reached (for staging, see ref. 11).
Before opening the egg, create an air pocket above the embryo by inserting an 18 gauge syringe needle through the egg shell (not too deeply to avoid piercing the yolk) and evacuating 4 ml of albumin (Figure A). This creates a pressure drop that within several minutes is equalized by the entry of air through the porous shell. As the air enters it collects at the highest point within the shell, thus separating the embryo from the inner surface of the shell.
Seal the hole created by the syringe needle with conventional plastic tape.
3. Pulling glass micropipettes
Pull glass micropipettes from borosilicate glass (for example, 1.2mm OD x 0.94 mm ID, Harvard Apparatus, USA) on a conventional electrode puller (for example, Model P-2000, Sutter Instruments, USA). Adjust puller settings to obtain micropipettes that have high input resistance when used as microelectrodes. Tips should be approximately 1-1.5 cm long. The shafts can be marked at 1 mm intervals for calculating volumes ejected.
Break the micropipette tips to a workable size by bending them with a forceps or pushing them against a solid surface while viewing under a microscope. The break should be as even as possible around the perimeter of the glass and the resultant inner diameter at the tip should accommodate the size of the cells to be used without getting clogged. Typically a 20-30 micron inner diameter works well.
4. Preparation of Fast Green dye solution (contrasting reagent)
Prepare a 0.1 % (w/v) solution of Fast Green dye (Sigma-Aldrich, USA) by dissolving in chicken physiological saline (137 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM Na-phosphate, 5 mM HEPES, 11 mM glucose, pH 7.4).
Filter the Fast Green solution through a 0.22 μm Millex GP filter (Millipore Co., Bedford, USA).
5. Opening the eggs and contrasting the embryo
Place two pieces of tape on the top of the egg, covering the dimensions of the air pocket. Keeping the egg oriented so that the taped area (and thus the air pocket) is uppermost, cut a small window within the borders of the taped area using a sturdy dissection scissors. The tape adds support and helps prevent eggshell fragments from falling onto the embryo. Air bubbles associated with the air pocket can be popped using the back end of a plastic pipette tip or other blunt probe.
Draw some Fast Green solution into a 1 ml syringe with a 25 gauge needle. Remove any air bubbles in the syringe and the needle. Bend the needle to a 45-degree angle, penetrate at a point outside the embryonic plate (identified by the ring of blood vessels that circumscribe it; piercing within the embryonic plate increases mortality) and carefully guide the needlepoint under the embryo. Inject the dye until desired contrast is achieved. The embryo, which is normally transparent, should now stand out as a whitish ghost against the dark green color (Figure B). Many investigators use India Ink (diluted to 10% v/v) instead of Fast Green. In this case it is important to use India Ink that is non-toxic, such as Pelikan Fount India Ink.
6. Making hindbrain and spinal cord neural tube lesions
Produce sharpened tungsten needles by flame-etching short (2-3 cm) pieces of 100 μm diameter tungsten rod (catalog number 719000, A&R Systems, Seattle, Washington), with an acetylene/oxygen torch. This is done by inserting the end of the rod very briefly into the torch flame until a spark is seen, which represents the explosive erosion of the outer layers of tungsten, leaving behind a sharp tip.
Using a sharpened tungsten needle, slice through and deflect the vitelline membrane covering the area for microsurgery.
Using a second sharpened tungsten needle (the tip of the first needle is often damaged after slicing through the vitelline membrane and not fit for subsequent microsurgery), cut carefully through the epithelium overlying the neural tube and deflect it, then cut around and under the piece of the neural tube to be removed, using short, quick upward strokes (Figure C). Making the transverse cuts first and then the longitudinal cuts is usually most successful. Do not cut too deep or you risk penetrating underlying blood vessels, which typically is fatal.
Gently flip or drag the resected tissue piece away from the lesion site using the tungsten needle. If this proves difficult, it can also be removed by mouth suction using a glass micropipette attached to flexible tubing.
7. Injection of human stem cells
a. Injection into a lesion of the neural tube
Draw a small amount of cells into the tip of a glass micropipette by mouth suction using a plastic tube. The working concentration of the cell suspension must be optimized for the cell type to be injected. Mount the micropipette on a micromanipulator and connect it to a microinjector pump (for example: PV830 Pneumatic PicoPump, WPI, Washington, USA). Under visual control using a dissection microscope, guide the micropipette tip into the lesion and carefully expel the cells by increasing the air pressure (either by pulses of constant pressure or by gradually ramping up the pressure) (Figure C). When the desired amount of cells is deposited within the lesion site, carefully withdraw the micropipette.
b. Injection into the lumen of the neural tube
In some cases it may be desirable to inject cells into the developing brain ventricles, for example if the cells are invasive enough to gain access to the neural tissue in the absence of lesions. Injection of cells into the lumen of any brain region requires using a developmental stage at which the lumen provides a large enough receptacle and is easily accessed; HH stages 12 to 18 are favorable in this regard. A lesion is not necessary. Simply pierce the wall of the neural tube with the glass micropipette prior to injection of cells. The key elements for the success of this method are the sharpness of the micropipette and the abruptness of the penetration; too slow a movement and the micropipette tip may only dimple the neural tube wall without penetrating.
c. Systemic injection
Systemic injections can be made via the extra-embryonic blood vessels (starting from HH stage 15, when these vessels are well developed). This is not uncommonly accompanied by some bleeding and since there are many small arteries and fewer larger veins, intra-arterial injections are generally safer when it comes to the embryo's survival. On the other hand, intravenous injections provide quicker access of the cells to the heart and thus probably a more even distribution of the cells. A successful injection requires a sharp micropipette with a diameter smaller than the vessel targeted. Approach the vessel along its axis at a small angle from the horizontal (about 30 degrees). Push the micropipette against the vessel until the vessel begins to occlude. Then push further in a firm, abrupt movement to penetrate. Retract slowly until blood is seen to draw by capillary action into the tip of the micropipette. Injection of cells should then be performed using constant pressure, after which the micropipette is retracted with a quick movement.
8. Sealing the egg
After the injection, let the egg stand still for a few minutes to allow the injected cells to settle and adhere. To avoid dehydration (which can occur quickly and can prove fatal), lay a piece of moistened tissue or filter paper over the window during this rest period.
After a few minutes and the cells have settled, dry the shell around the window and seal the window with conventional plastic tape. Make sure this seal is tight and that no albumin can leak through the opening or between the shell and the tape (this often proves fatal during subsequent incubation).
Replace the eggs to the forced-draft incubator for further development. Observation can be made at intervals by removing the tape over the window.
9. Embryo dissection
Once the embryo has reached the desired stage, crack open the egg into a bowl of ice-cold physiological saline or phosphate-buffered saline (the tape covering the window should be cut first to allow the two halves of the eggshell to separate). The ice-cold saline serves to anesthetize the embryo by hypothermia. The heart should quickly slow and stop within a couple of minutes.
Separate the embryo from the extraembryonic membranes and stage it using ref. 11.
Decapitate the embryo, transfer it to a dissection dish that has a silicone rubber floor and contains oxygenated physiological saline supplemented with glucose (137 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM Na-phosphate, 5 mM HEPES, 11 mM glucose, pH 7.4) and pin it fast ventral side up. Remove the abdominal and thoracic viscera. Using an angled scissors, make longitudinal incisions along each ventrolateral aspect of the vertebral column. Particularly at later stages (after day 6 of development), the initial incision is best made in the lumbosacral region where the space between the vertebrae and the spinal cord is greatest. Remove the ventral aspect of the vertebral column to reveal the spinal cord. Make sure that the physiological saline remains oxygenated by bubbling at approximately 10-minute intervals with pure medical oxygen.
Carefully cut the nascent dorsal and ventral roots on either side, transect the spinal cord at the desired levels, and gently lift it out of the vertebral canal. The spinal cord will survive for many hours in this state and can be subjected to anatomical12 and physiological13,14 experiments.
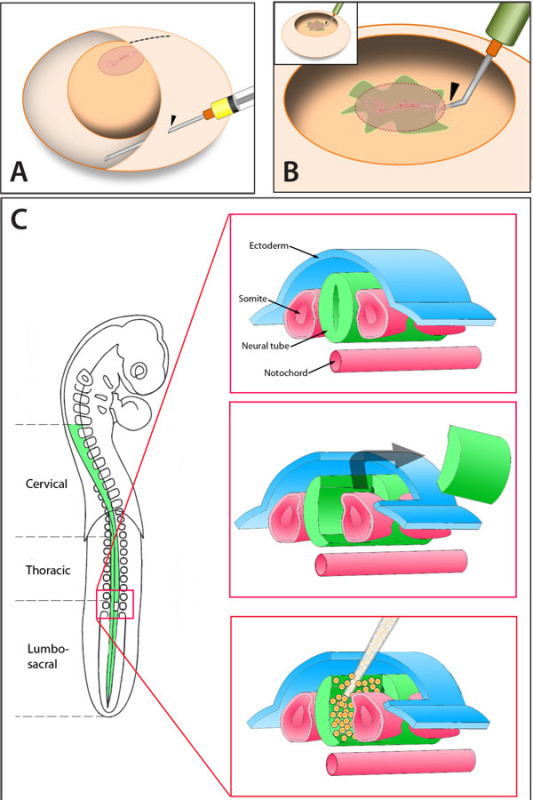 Figure 1. A) The appropriate approach for evacuation of albumin. Note the position of the embryo and the placement of the needle. B) Contrasting of the embryo. Note the penetration point of the needle outside the embryonic disc. C) Schematic representation of the procedure for unilateral spinal neural tube lesion followed by cell transplantation.
Figure 1. A) The appropriate approach for evacuation of albumin. Note the position of the embryo and the placement of the needle. B) Contrasting of the embryo. Note the penetration point of the needle outside the embryonic disc. C) Schematic representation of the procedure for unilateral spinal neural tube lesion followed by cell transplantation.
Discussion
The chicken embryo is a convenient and versatile animal model for xenotransplantation experiments. The lack of a functional immune system during embryonic and fetal development permits the survival and development of cells from any species that tolerates the incubation temperature. Chicken embryos have been used to test the regenerative potential of various types of stem cells (reviewed in ref. 15). Mammalian stem cells can integrate into chicken embryo tissues including the central nervous system, where they can give rise to neurons whose axonal projections, synaptic connectivity and electrophysiological properties can be assessed in the context of functional neural circuits1,2.
Because the surgical procedures are relatively easy, and injections are made under visual instead of stereotaxic control, the number of embryos can easily be increased to accommodate different experimental conditions within a single experiment. Typically cells can be injected into 20-30 embryos within a 4-hour experiment. The post-injection mortality of the embryos is the principal limiting factor. Precautions must be taken to avoid hemorrhaging and dehydration. Be aware that there are certain critical developmental stages at which the natural mortality rate increases. Supplementing the embryo with a few mL of warm, sterilized chicken ringer following injection and the day before critical developmental stages, as well as ensuring that the window in the egg is well-sealed and oriented to avoid leakage, improves survival. If leakage through the tape seal should occur, remove the tape, dry the surfaces and re-tape. Some investigators use wax and glass or plastic coverslips to seal the window. The mortality should not be substantially different in injected versus sham-operated embryos.
Although we have focused here on the injection of human stem cells into the anlage of the brain and spinal cord, stem cells can also be injected into the anlage of other organs (see for example ref. 4). Each organ poses its own challenges. For example, injection into the heart can be challenging because of the high mechanical resistance to pipette penetration and because of movements, which on the other hand can be slowed by cooling the embryos to room temperature prior to injection. Organs that lack a lumen may not accommodate the injection of sufficient volumes and may therefore require surgical lesion to create a receptacle. In our experience, post-lesion tissue regeneration is an advantage because it promotes the integration of the human stem cells into the tissue. Cells can also be injected into embryonic mesenchyme such as the dispersing somitic mesenchyme or the migrating neural crest as a way to achieve incorporation into mesenchymal derivatives, including neural crest-derived peripheral ganglia16,17.
Disclosures
No conflicts of interest declared.
Acknowledgments
The work was supported by grants from Helse og Rehabilitering (JLB), The Norwegian Research Council (GH and JCG) and the University of Oslo (NK and JCG).
References
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Sigurjonsson OE, Perreault MC, Egeland T, Glover JC. Adult human hematopoietic stem cells produce neurons efficiently in the regenerating chicken embryo spinal. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5227–5232. doi: 10.1073/pnas.0501029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochampally RR, Neville BT, Schwarz EJ, Li MM, Prockop DJ. Rat adult stem cells (marrow stromal cells) engraft and differentiate in chick embryos without evidence of cell fusion. Proc. Natl. Acad. Sci. U.S.A. 2004;101:9282–9285. doi: 10.1073/pnas.0401558101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RS. Transplantation of human embryonic stem cells to the chick embryo. Methods Mol. Biol. 2006;331:137–151. doi: 10.1385/1-59745-046-4:137. [DOI] [PubMed] [Google Scholar]
- Lee H, Shamy GA, Elkabetz Y, Schofield CM, Harrsion NL, Panagiotakos G, Socci ND, Tabar V, Studer L. Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells. 2007;25:1931–1939. doi: 10.1634/stemcells.2007-0097. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Peljto M, Nedelec S. Xenotransplantation of embryonic stem cell-derived motor neurons into the developing chick spinal cord. Methods Mol. Biol. 2009;482:171–183. doi: 10.1007/978-1-59745-060-7_11. [DOI] [PubMed] [Google Scholar]
- Westerlund U, Svensson M, Moe MC, Varghese M, Gustavsson B, Wallstedt L, Berg-Johnsen J, Langmoen IA. Endoscopically harvested stem cells: a putative method in future autotransplantation. Neurosurgery. 2005;57:779–784. [PubMed] [Google Scholar]
- Boquest AC, Shahdadfar A, Brinchmann JE, Collas P. Isolation of stromal stem cells from human adipose tissue. Methods Mol. Biol. 2006;325:35–46. doi: 10.1385/1-59745-005-7:35. [DOI] [PubMed] [Google Scholar]
- Smeland EB, Funderud S, Kvalheim G, Gaudernack G, Rasmussen AM, Rusten L, Wang MY, Tindle RW, Blomhoff HK, Egeland T. Isolation and characterization of human hematopoietic progenitor cells: an effective method for positive selection of CD34+ cells. Leukemia. 1992;6:845–852. [PubMed] [Google Scholar]
- Zhang XY, La Russa VF, Bao L, Kolls J, Schwarzenberger P, Reiser J. Lentiviral vectors for sustained transgene expression in human bone marrow-derived stromal cells. Mol. Ther. 2002;5:555–565. doi: 10.1006/mthe.2002.0585. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J. Morph. 1951;88(1):44–92. [PubMed] [Google Scholar]
- Glover JC. Retrograde and anterograde axonal tracing with fluorescent dextrans in the embryonic nervous system. Neuroscience Protocols. 1995;30:1–13. [Google Scholar]
- O'Donovan MJ, Bonnot A, Mentis GZ, Arai Y, Chub N, Shneider NA, Wenner P. Imaging the spatiotemporal organization of neural activity in the developing spinal. Dev. Neurobiol. 2008;68:788–803. doi: 10.1002/dneu.20620. [DOI] [PubMed] [Google Scholar]
- Mendelson B, Frank E. Specific monosynaptic sensory-motor connections form in the absence of patterned neural activity and motoneuronal cell death. J. Neurosci. 1991;11:1390–1403. doi: 10.1523/JNEUROSCI.11-05-01390.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JC, Boulland JL, Halasi G, Kasumacic N. Chimeric Animal Models in Human Stem Cell Biology. ILAR Journal. 2009;51:62–73. doi: 10.1093/ilar.51.1.62. [DOI] [PubMed] [Google Scholar]
- Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, Studer L. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- Jiang X, Gwye Y, McKeown SJ, Bronner-Fraser M, Lutzko C, Lawlor ER. Isolation and characterization of neural crest cells derived from in vitro differentiated human embryonic stem cells. Stem Cells Dev. 2008;18:1059–1070. doi: 10.1089/scd.2008.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]