

Abstract
The formation of amyloid fibrils, protofibrils and oligomers from the β-amyloid (Aβ) peptide represents a hallmark of Alzheimer’s disease. Aβ peptide-derived assemblies might be crucial for disease onset, but determining their atomic structures has proven to be a major challenge. Progress over the last five years has yielded substantial new data obtained with improved methodologies including electron cryo-microscopy and NMR. It is now possible to resolve the global fibril topology and the cross-β sheet organization within protofilaments, and to identify residues that are critical for stabilizing secondary structural elements and peptide conformations within specific assemblies. These data have significantly enhanced our understanding of the mechanism of Aβ aggregation and illuminated the possible relevance of specific conformers for neurodegenerative pathologies.
The structural diversity of β-amyloid aggregates
The β-amyloid (Aβ) peptide occurs naturally inside the human brain as a proteolytic fragment of the amyloid precursor protein [1–3]. The peptide possesses an amphiphilic structure with a hydrophilic N- and a hydrophobic C-terminus. The C-terminal end of the peptide is variable, producing peptides with lengths ranging at least from 37 to 42 residues [1,4]. The two most intensively studied Aβ alloforms are Aβ(1–40) and Aβ(1–42) [3], which consist of 40 and 42 residues, respectively. More than ten single-site sequence variants of this peptide have been described, most of which relate to familial forms of Alzheimer s disease (AD; termed FAD) [1]. Naturally occurring Aβ peptides can be chemically modified, for example oxidized side chains, truncated main chains and pyroglutamate modified N-termini have been described [1,5]. Aβ amyloid fibrils form the core of dense amyloid plaques within the brain parenchyma, one of the hallmarks of AD, or they accumulate at the walls of cerebral blood vessels, associated with cerebral amyloid angiopathy (CAA) [1]. Amyloid fibrils are the end products of a complex fibrillation pathway, and their formation is preceded by numerous on- or off-pathway intermediates. These intermediate structures can include Aβ dimers, oligomers, amyloid derived diffusible ligands, globulomers, paranuclei and protofibrils (not to be confused with protofilament, see below) [1,2,6,7]. However, the definition of these states strongly depends on the context of their preparation in different laboratories, and they often cannot be isolated or easily purified. Therefore, reliable structural information on Aβ amyloid fibrils and other Aβ-derived aggregated states is difficult to obtain, although knowledge about these states is crucial for understanding their biological properties and for the design of novel structure-specific ligands or inhibitors.
In this review, we summarize the progress made over the last five years towards understanding the structures of Aβ peptide aggregates. The different structural levels of Aβ amyloid fibrils are presented, including quaternary structure, protofilament organization and packing of β-sheets. Moreover, we discuss several topics of particular interest, including the structural polymorphism of amyloid fibrils, oligomers, and the comparison of Aβ(1–40) and Aβ(1–42) fibrils. Key methods of structural investigation are introduced to help the reader assess the published, and sometimes seemingly conflicting, structural models, which have been proposed to explain the new experimental data. The available structural data also shed light on the various conformational states adopted by Aβ and their possible role in human disease.
The cross-β structure of Aβ amyloid fibrils
Amyloid fibrils can be defined as fibrillar polypeptide aggregates with a cross-β structure [8]. Cross-β structures represent intermolecular polypeptide assemblies, where the β-sheet plane and the backbone hydrogen bonds that connect the β-strands are oriented parallel to the main fibril axis. It follows that the β-strands run perpendicular (“cross”) to this direction. The presence of a common cross-β structure in all amyloid fibrils was initially shown by X-ray diffraction measurements [9]. More recently, crystallographic studies of peptide microcrystals revealed so-called steric zippers [10,11] which are presumed to occur in many amyloid fibrils. Steric zipper units consist of a pair of two cross-β sheets with interdigitating side chains (Fig. 1a). They can be formed by several different short peptide chains (usually 4–7 amino acids), such as from Aβ residues 37–42 (Fig. 1a) or 35–40 [11]. It was suggested that steric zippers constitute, in the context of full-length polypeptide chains, the structural spine of amyloid fibrils. Indeed, the cross-β structure of Aβ fibrils was shown to be formed by discrete sequence segments at the peptide centre or C-terminus [20–24]. Most studies describe 2–4 β-strands, usually involving residues 16–20 and 31–36 (Fig. 1b). Differences exist concerning the exact residues forming the β-sheet segments, either reflecting real structural differences or experimental ambiguities (e.g. gaps within the structural assignment).
Figure 1. Structural hierarchy and β-sheet structure of Aβ amyloid fibrils.

Mature amyloid fibrils encompass one or several protofilaments. Their cores are formed by peptides adopting a cross-β structure. (a) Structural hierarchy of amyloid fibrils, exemplifying the cross-β structure by a steric zipper structure from Aβ residues 37–42 (PDB-code: 2ONV) [11]. Only the backbone is drawn; the upper three strands are shown in stick representation, highlighting the hydrogen bonds; the bottom three strands are displayed as a ribbon diagram. The cross-β structure forms the structural spine of an amyloid protofilament, the filamentous substructure of mature Aβ fibrils. (b) Possible β-sheet forming residues of Aβ(1–40) and Aβ(1–42), as suggested by chemical shift data (CS) [21,29,34], protection from hydrogen exchange (HX) [20,22,41,42], mutational perturbation without (M) [39] and with hydrogen exchange (M/HX) [22], mutagenic changes combined with structural order analysis (M/O) [23].
The amyloid fibrils formed from the full-length Aβ peptide display the typical characteristics of amyloid fibrils, including a high affinity to amyloid-specific dyes (Congo red and/or Thioflavin T) and fibril-specific conformation-sensitive antibodies [12,13]. Based on the shape of their amide I regions, their infrared spectroscopic properties also correspond to other amyloid fibrils [14–16] and additionally suggest a parallel orientation of the β-sheets in Aβ(1–40) and Aβ(1–42) amyloid fibrils [12,17,18]. This assembly of full-length Aβ fibrils differs from the antiparallel β-sheet characteristics seen in the fibrils formed, for instance, from the Aβ(11–25) peptide fragment, Aβ-derived variant peptides carrying the Iowa Asp23Arg substitution [19] and certain Aβ-derived steric zippers [11]. These examples illustrate the β-sheet variability of the fibrils arising from Aβ and its derived peptide fragments or variants.
The general topology and polymorphism of mature amyloid fibrils
Transmission electron microscopy (TEM) and atomic force microscopy experiments show that mature amyloid fibrils can have a length of more than one micrometer, whereas the lateral width of previously analyzed fibrils rarely exceeds 25 nm [15,20,25–27]. Mature Aβ amyloid fibrils comprise one or several protofilaments (not to be confused with protofibril) [4,7]. Analogous to other filamentous structures, such as microtubules, amyloid protofilaments represent the substructures of mature fibrils (Fig. 1a). Mature Aβ fibrils are often, but not always, twisted (Fig. 2a) and result in regular crossovers that are visible by TEM (Fig. 2a). All analyzed superhelical Aβ(1–40) and Aβ(1–42) fibrils have a left-handed twist [14,15,26,27], and they usually possess a polar structure [14,15,27]. Polarity refers in this case to the directionality of the fibril structure, similar to the polarity of actin filaments or microtubules. Most of the three-dimensional (3D) reconstructions of Aβ amyloid fibrils are limited to resolutions of 8 to 30 Å at which they exhibit two-fold fibril symmetry [15,25,27,28]; i.e. the fibril cross-section superimposes with itself after a 180° rotation. However, a minority of the obtained reconstructions do not comply with a two-fold symmetry [27]. Indeed, non-two-fold symmetrical fibrils had been assumed by some published models [29].
Figure 2. Polymorphism and structural deformations of Aβ amyloid fibrils.
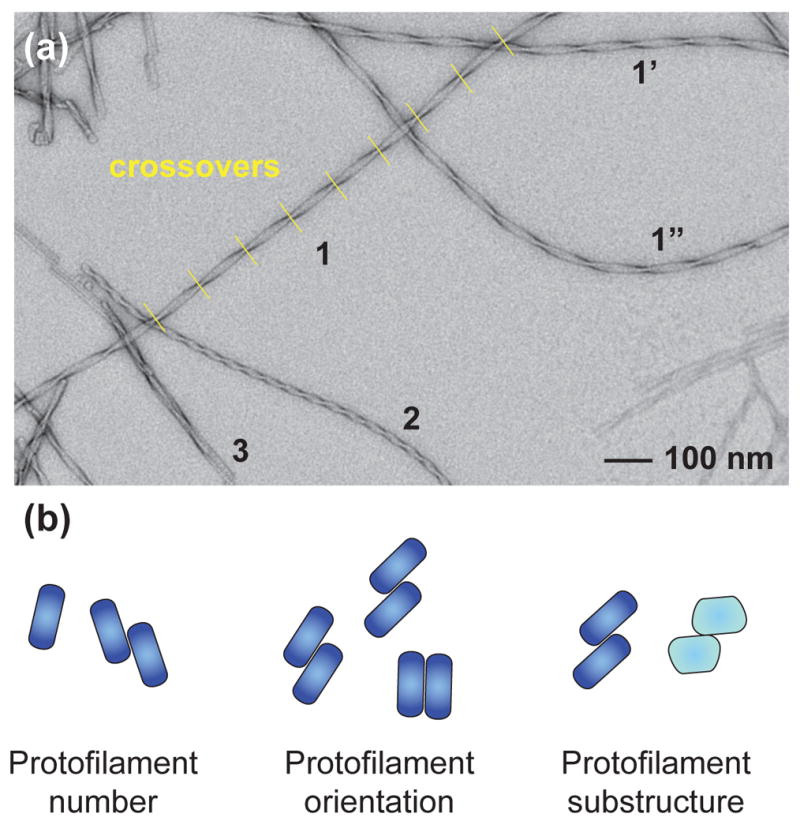
Typical Aβ fibril samples are affected by heterogeneity, which arises both from the intra-sample polymorphism of different fibril structures and from their deformations from ideal helical symmetry (bending & twist variability). (a) Negative stain TEM image of Aβ(1–40) amyloid fibrils illustrating the fibril variability. Fibrils 1, 2 and 3 show fibril polymorphism. Isomorphous fibrils 1, 1 and 1 differ in bending. The twist variability (crossover distance) of fibril 1 is highlighted in yellow. (b) Schematic representations of fibril cross-sections to illustrate three types of fibril polymorphism. Differences depend on the number, orientation or substructure of the underlying protofilaments.
An important structural feature emerging from these and many other studies is the immense structural polymorphism of amyloid fibrils. Structural polymorphism is defined here as the variability in peptide conformation or intra-fibrillar arrangement of different fibrils. Specific fibril morphologies can be identified, for instance, by TEM, by their characteristic widths or crossover distances (Fig. 2a). 3D reconstructions of polymorphic amyloid fibrils revealed three distinct types of polymorphisms, i.e. fibrils differing in (i) the number of protofilaments (Fig. 2b); (ii) the relative protofilament orientation (Fig. 2b); and (iii) the internal protofilament substructure (Fig. 2b) [30]. The latter polymorphism type can be further subdivided, as indicated by a recent analysis of different steric zippers, depending on differences in the participating sequence segments and packing modes; thus they were termed segmental, registration, combinatorial and packing polymorphisms [31].
Polymorphism is potentially relevant for human disease, as it could underlie the natural variability of some amyloid diseases, such as amyloid light chain (AL) amyloidosis, or the protein-encoded inheritance phenomena of prion strains [31]. It also constitutes an important biophysical difference between amyloid fibril formation and native protein folding reactions. Whereas the latter are usually characterized by a unique correspondence between amino acid sequence and folded state, amyloid fibril formation can lead, for the same polypeptide sequence, to many well-defined end states.
The term polymorphism frequently refers to an inter-sample variance that can arise from different incubation or solution conditions. For example, different fibril morphologies occur in the presence of salt, Zn2+ ions, or in conjunction with the use of different buffer systems [27,32,33]. In addition, it has been reported that agitation or quiescent incubation conditions can produce different fibril morphologies visible by TEM and encompassing different peptide folds, as judged by NMR [34]. Whereas these studies were self-consistent to the extent that the experiments in each study were carried out with comparable peptide aliquots and pre-treatments, cross-study comparisons are often problematic. Besides differences in the incubation conditions, variability is also caused by different peptide batches or sample pre-treatments. Numerous attempts have been reported trying to reduce these problems by application of disaggregation protocols [35,36], but the comparability of peptide samples remains a major problem in the analysis of fibril structures.
In addition to inter-sample polymorphism, the detailed examination of Aβ fibril samples with single particle techniques revealed substantial intra-sample polymorphism [27,33]. For example, a systematic analysis of Aβ(1–40) fibrils formed in 50 mM sodium borate, pH 9.0, revealed variations in the fibril width from 13 to 29 nm; by contrast, most fibrils present well-resolved crossover distances from 100 to 200 nm [33]. A dramatically different spectrum of morphologies was obtained when fibrils were grown in the presence of 0.5 M salt. The average width of fibrils was significantly decreased, with many fibrils being thinner than 13 nm or having no discernible crossover distances [33]. However, all these samples presented intra-sample polymorphism, indicating that changing the incubation conditions does not necessarily cause a switch between single fibril morphologies. Instead, it alters the distribution of morphologies and favors one polymorphic ensemble over another. Thus, intra-sample polymorphism constitutes a major obstacle to high-resolution structural techniques that cannot separate the signal coming from different morphologies.
Structural deformations report on the nanoscale flexibility properties of amyloid fibrils
Besides polymorphism, structural deformation is one further cause for the heterogeneity of amyloid samples. These deformations manifest themselves in different degrees of bending or twisting, evidenced by the variable crossover distances within the same fibril (Fig. 2a). Although these deformations create a further potential problem to structural analysis, they can be used to infer nanoscale mechanical properties of amyloid fibrils. For an Aβ(1–40) fibril morphology with an average crossover distance of 140 nm and a width of 19 nm, the values for the Young s modulus and shear modulus were found to be similar to most other protein filaments [37]. The considerable bending rigidity of amyloid fibrils is relevant when considering the pathogenic activity of rigid structures that can occur and impair normal function in naturally contractile or elastic tissues. For example, CAA is known to be associated with micro-hemorrhages caused by the deposition of Aβ fibrils within cerebral vessel walls [1].
Structural methods for studying amyloid fibrils
So far, atomic structures of full-length Aβ amyloid fibrils have not been determined. Suitable crystals of full-length Aβ amyloid fibrils for X-ray crystallography have not been obtained, and the large fibril size precludes conventional solution-state NMR techniques. The absence of atomic structures led to the rise of numerous structural models. Before entering a more detailed discussion of the available structural data on Aβ fibrils, it is important to clarify the difference between structure and model (Boxes 1 & 2).
Box 1. Use of the term “structure”.
The spatial coordinates of atoms and specification of their covalent and non-covalent interactions are collectively referred to as the structure of a protein or peptide if they are primarily determined based on experimental data obtained, for example, by X-ray crystallography, NMR or electron microscopy. The quality of the experimental data must allow the accurate placement of the major structural elements, such as amino acids and their side chains. In X-ray crystallography and electron microscopy, this accuracy is directly related to the spatial resolution of the experiment and usually needs to be better than 4 Å to allow reliable placement of amino acids. NMR experiments provide distance information and other data that constrain the arrangement of atoms, giving rise to a number of possible atomic coordinates. The variability of these coordinates is expressed as a root mean square deviation (RMSD) and values below 1.5 Å are usually sufficient for reliable amino acid placements. However, a structure can also be represented through a density map at lower resolution. For example, a cryo-EM map at 10 Å resolution can also be referred to as a structure (although it is often called reconstruction). In this case, the major structural elements could be the subunits of a multi-subunit complex. If a representation that is based more or less equally on experimental and other information, the distinction between structure and model becomes more difficult. However, if it is called a structure, spatial resolution information or RMSD values should always be provided in the initial publication.
Box 2. Use of the term “model”.
If the arrangement of atoms is primarily based on general reasoning, homology between molecules, general side chain geometry or chemical bond lengths obtained from a database, it is commonly called a model. A model can still be partially based on experimental data to guide model building. For example, the strong demand for detailed descriptors of amyloid structures often leads to atomic models in the literature that are based on some experimental data, but that encompass many details that have not been established experimentally (hence they are models). When comparing models, it is important to consider not only the structural detail shown in the model, but also the primary experimental data leading to it and the degree of extrapolation the model represents. An atomic model that includes the coordinates of side chains is, therefore, not necessarily more informative than a simpler model that includes only a backbone trace or, indeed, a model that outlines only the folding motif of a peptide. Models can also consist of more simplistic representations and symbols, for example cylinders and arrows to symbolize α-helical and β-sheet secondary structural elements, or a simple line to indicate the path of the peptide backbone.
Models can be partially based on structural constraints from biophysical measurements. Their critical assessment requires at least some knowledge about the structural constraints that can be derived from the employed biophysical techniques. Direct information about the secondary structure of fibrils and aggregates can be obtained by X-ray fiber diffraction, circular dichroism (CD) and infrared spectroscopy [4,14,16,38]. In addition, the two spectroscopy methods can be used to quantify the secondary structural content of a sample. Combined with isotope editing, infrared spectroscopy can even provide direct, residue-specific structural information [38].
Residue-specific information can also be obtained by site-specific mutagenesis coupled with kinetic or thermodynamic measurements [39,40], hydrogen exchange in conjunction with NMR or mass spectrometry [20,22,40–42], or electron paramagnetic resonance spectroscopy [23]. However, the structural data resulting from these techniques are more indirect, because the methods are, in contrast to CD or infrared spectroscopy, not able to distinguish β-sheet structure from other stable and ordered conformations. Although changes to these properties are usually interpreted with β-sheet formation, the more immediate readouts are the speed of aggregation, the aggregate stability [39,40], protection from hydrogen exchange [20,22,40–42], and the hydrophobicity and structural order at discrete sites after cysteine replacement and cysteine side chain modification [23].
More direct structural information can be generated by solid-state NMR and electron cryo-microscopy (cryo-EM). These two techniques have the potential to determine the structure of Aβ amyloid fibrils at atomic or near-atomic resolution. Solid-state NMR determines structural constraints such as chemical shift values, bond angles or specific interatomic distances. It allows the direct identification of the residues of Aβ participating in the β-sheet structure in fibrils (Fig. 1b). Such measurements revealed, for example, similar structural characteristics of in vitro grown amyloid fibrils and fibrils seeded with brain extracts [43]. The second technique, cryo-EM, directly visualizes the fibrils and allows the calculation of their 3D density (reconstruction). The observation of individual fibrils enables selection of specific fibril morphologies. Therefore, only data from a chosen morphology will be averaged in a 3D reconstruction. This possibility allows cryo-EM to avoid many of the problems of intra-sample polymorphism. Given a sufficient amount of data and accurate alignment, near-atomic resolution (3–4 Å) can be obtained [44,45]. In the case of Aβ amyloid fibrils, this technique was able to visualize the 3D structure of several fibril morphologies at up to 8 Å resolution, providing information about the global fibril architecture, protofilament substructure, and location of the cross-β structure within the symmetrical fibril helix [14, 15, 25, 27].
Protofilament structure of mature Aβ amyloid fibrils
The protofilament substructure of an Aβ fibril morphology was identified by cryo-EM [15,25]. The observed protofilaments show cross-sectional dimensions of 4 × 11 nm and a cross-sectional subdivision into a single central region of quasi two-fold symmetry (4 × 5 nm) and two peripheral regions (Fig. 3a–e). The central region is formed by two paired elongated density cores, corresponding to two cross-β sheets. Mass-per-length (MPL) measurements suggest that each protofilament contains ~2.5 peptides per cross-sectional layer. This protofilament structure was observed in two fibril reconstructions, one obtained with the Aβ(1–40) peptide [25], and the other with Aβ(1–42) [15]. Whereas the analyzed Aβ(1–40) fibril contains two such protofilaments, there was only one within the Aβ(1–42) fibril. The single-protofilament Aβ(1–42) fibril presents two equally shaped peripheral regions that are fully solvent-exposed and structurally disordered (Fig. 3d). By contrast, the two-protofilament Aβ(1–40) fibril contains an arch-shaped peripheral region at the protofilament–protofilament interface, whereas the other peripheral region is solvent-exposed and structurally disordered (Fig. 3a–c). This protofilament architecture is apparently conserved in different Aβ amyloid fibrils, and several other Aβ(1–40) fibril reconstructions are consistent with this protofilament substructure [27]. Nevertheless, some evidence points to additional types of Aβ protofilament substructures, as suggested by cryo-EM reconstructions [27, 28] (Fig. 3c) or MPL measurements of certain fibril morphologies [26,29,34].
Figure 3. Cryo-EM cross-sections of different Aβ fibril morphologies.

Cryo-EM reconstructions reveal significant similarities in the protofilament structure of two Aβ(1–40) and Aβ(1–42) fibril morphologies, suggesting homologous peptide assemblies. (a–c) Aβ(1–40) fibril cross-section filtered at 8 Å (b) and 15 Å (c) [25]. Structural interpretation of the cross-section (a). (d) Cross-section of the Aβ(1–42) fibril structure from pH 7.4 at 15 Å resolution [15]. (e) Structural interpretation of the cross-section. Structures in (b–d) present a similar subdivision into one central region (C, red), which possesses approximate two-fold symmetry (blue symbol), and two peripheral regions (P, green). (f) Aβ(1–42) fibril at pH ~2 [28].
Possible fold(s) and intermolecular arrangement of fibrillar Aβ peptide
Several models of the Aβ fold in amyloid fibrils have been proposed, sometimes combining structural constraints from different experimental techniques or from samples that were formed under different conditions. However, different conditions can produce fibrils with different peptide conformations (see above); thus, caution should be exercised when comparing the respective models. Most fibril models assume a U-shaped peptide fold (Fig. 4a,b). This fold has been termed a β-arc (or β-arch) and should not be confused with the terms “β-turn” or “β-hairpin” [46]. U-shaped peptide models were derived from molecular dynamics simulations, partially implementing structural constraints from solid-state NMR or other biophysical techniques. Occasionally, it was suggested that these models fit cryo-EM reconstructions [14,28]. However, the cryo-EM reconstructions used in these comparisons do not resolve the β-sheet leaflet structure, whereas the reconstruction of an Aβ(1–40) fibril at higher resolution (8 Å), in which the cross β-sheet structure is clearly resolved, does not comply with the previous U-shaped peptide models [25,37,47]. The 8 Å reconstruction presents cross-sectional dimensions that are significantly larger than predicted by the U-shaped models and therefore must visualize the peptide in a different structural arrangement (Fig. 4c–e).
Figure 4. Structural models of Aβ fibrils.

Previous structural models deviate significantly for the most highly resolved Aβ fibril structures, which were obtained by cryo-EM. Hence, the latter encompass a different peptide assembly. (a, b) Early structural models assuming a U-shaped peptide fold; side views and top views shown; only residues 9 to 40 modeled. (a) Three Aβ(1–40) molecules per cross-sectional layer [29]. (b) Two Aβ(1–40) molecules per cross-sectional layer [24]. (c–d) Cryo-EM structure of an Aβ(1–40) amyloid fibril (8 Å). (c,d) side views, (e) cross-section. Images in (c,e) are superimposed with a β-sheet model, which is derived from these cryo-EM data and highlights the peptides forming the cross-β regions in yellow or blue (the lines are not meant to imply continuous β-strands over their entire length; these regions might instead contain several shorter strands). Images in (a–c) and (e) are displayed at the same scale.
Based on MPL measurements as well as the shape, symmetry and subdivision of the protofilament cross-section of the 8 Å reconstruction, one protofilament cross-β repeat was proposed to comprise two oppositely directed Aβ molecules (Fig. 3a–e, Fig. 4e). These peptides are part of two separate, paired cross-β sheets. Therefore, the basic peptide arrangement resembles that of a steric zipper structure (Fig. 1a). Whereas the two peptides are structurally equivalent in the single-protofilament fibril (Fig. 3d–e), the cross-sectional density in the two-protofilament fibril implies that the two peptides differ in conformation (Fig. 3a–c). In addition, the peptide forming the protofilament–protofilament interface is arch-shaped (with dimensions different from the published β-arch), whereas the other peptide contains significant structural disorder [47]. The possibility that a specific Aβ(1–40) fibril morphology might encompass two different peptide folds was also raised by solid-state NMR [29]. Nevertheless, the current cryo-EM structures do not exclude the possibility that other Aβ fibril morphologies exist that possess an U-shaped peptide architecture and a protofilament substructure different from the one described here. All current models are consistent in assuming that a significant fraction of the peptides in the fibril have solvent exposed N-termini. This conjecture is consistent with findings that the peptide N-terminus can be flexible and constitutes an important epitope for AD immunotherapy [1–3].
Structural comparison of Aβ(1–40) and Aβ(1–42) fibrils
The Aβ(1–42) peptide is generally believed to be more pathogenic than the Aβ(1–40) peptide [1–3]. When expressed in Drosophila melanogaster, the Aβ(1–42) peptide is highly toxic and reduces the life-span of the affected animals, whereas Aβ(1–40)-transgenic flies do not present a discernible phenotype [48]. The significant chemical similarities of the two peptides (the first 40 residues are identical) suggest that their conformational properties are also largely similar. Yet, inevitable differences must exist, associated with the additional two C-terminal residues of Aβ(1–42). The clearest biophysical difference is the higher aggregation propensity of Aβ(1–42) [49]. Moreover, Aβ(1–40) can affect, in mixtures, the aggregation mechanism of Aβ(1–42), thereby preventing the formation of mature Aβ(1–42) fibrils [50] by stabilizing intermediate conformations [51].
The available cryo-EM fibril reconstructions from the two peptides show marked differences in protofilament packing. Published Aβ(1–42) fibrils possess either a single-protofilament arrangement or a two-protofilament assembly with a hollow core [15, 28]. All of the more than ten published Aβ(1–40) fibril reconstructions are differently structured [14, 15, 25,27,37]. By contrast, there is also evidence that the protofilaments of Aβ(1–42) and Aβ(1–40) fibrils are highly similar. For example, they can produce identical MPL values, cross-sectional areas and shapes, and their protofilament cross-sections show a similar division into one central and two peripheral regions [15]. These data suggest similar peptide folds in the two fibrils, which is consistent with the fact that the β-sheet structure is usually assigned to similar sequence segments of Aβ(1–40) or Aβ(1–42) peptides (Fig. 1b). In addition, infrared and NMR data indicate that both fibrils contain a parallel β-sheet structure [14, 15, 20, 24], and there is long-standing evidence that Aβ(1–42) fibrils are able to seed fibril formation from Aβ(1–40) peptides [49]. In addition to these similarities, there is evidence that fibrils formed by the two peptides differ, at least slightly, in the exact residues forming the secondary structural elements (Fig. 1b). Taken together, these data demonstrate numerous similarities, but also significant differences between specific samples prepared by the two peptides. Additional analyses will be required to establish whether these differences represent sample-specific variations or systematic differences between the two Aβ alloforms, and a disease-specific fibril guise cannot be currently identified.
Structure of Aβ(1–40) and Aβ(1–42) oligomers
In vitro Aβ fibrillation reactions involve a range of different on- or off-pathway intermediates, and it is thought that similar structures, such as oligomers or protofibrils, can become stabilized also during disease [2,6,7,52]. In fact, the onset of AD is thought to depend on the action of Aβ fibrillation intermediates [1–3], but there is uncertainty about the exact mechanism, and different possible pathways to disease have been proposed. For example, it was suggested that annular Aβ aggregates are toxic and kill the affected cells by perforating their membranes [7,53]. Other mechanisms involve Aβ-dependent excitotoxicity reactions, which require dendritic tau protein [54], or mitochondrial dysfunctions [18]. The available data suggest that although the Aβ peptide and its derived amyloid fibrils are typically located extracellularly, the formation of these deposits and their pathogenic activity arises from intracellular Aβ structures [55,56].
Although there are no atomic structures of Aβ amyloid fibrils, addressing the structures of their various precursors is even more challenging. Such aggregates can exhibit variable molecular weights and overall shapes, including spherical oligomers, curvilinear protofibrils and annular pores. Despite evidence for random coil-like conformations in some assemblies [57], many analyzed Aβ(1–40) and Aβ(1–42) oligomers display appreciable β-sheet content [12,17,58,59]. Chemical shift measurements by solid state NMR and Fourier transform infrared spectroscopy (FTIR) analyses consistently demonstrate that similar residues are involved in the formation of the β-sheet structure of Aβ(1–40) oligomers and fibrils, and that the two states display similar FTIR spectral characteristics [12,17,18,59]. Yet, FTIR also indicates that the β-sheet structure of Aβ(1–40) and Aβ(1–42) oligomers is considerably antiparallel, whereas full-length Aβ amyloid fibrils possess parallel β-sheet characteristics [12,17,18]. Moreover, NMR experiments suggest that the β-sheet packing distance and assembly could be different in Aβ(1–42) oligomers and Aβ(1–42) fibrils [58]. Other important structural differences between Aβ oligomers and fibrils are their higher diffusibility, hydrophobicity and ability to interact with membranes, which enable the oligomer-dependent pathogenicity mechanisms described above.
Concluding remarks
Recent improvements in techniques, in particular cryo-EM and NMR, have enabled the localization of the cross-β structure within mature Aβ fibrils and have identified some of the residues that stabilize fibrils and oligomers. The structures underlying the different aggregation states of the Aβ peptide are important for a mechanistic understanding of related diseases. For example, Aβ amyloid fibrils seem to underlie the pathogenicity in CAA, whereas oligomers or other premature Aβ aggregates are likely more relevant for AD [1–3]. To understand any one disease, it might be necessary to study entire populations if oligomers or fibrils that capture the natural polymorphic states of Aβ peptide. Continued technical developments raise hopes that more comprehensive structural information will become available in the next five to ten years that could potentially form the basis for further studies and developments, such as in disease treatment as well as in understanding the aggregation pathways that lead to the characteristic polymorphism seen in all amyloid fibrils.
Acknowledgments
We are very grateful to C. Sachse for contributing the image in Fig. 2b and to R. Tycko for providing the coordinate files of the Aβ structural models displayed in Fig. 4a/b. M.F. was supported by DFG (SFB 610), Alzheimer Forschungs-Initiative and the Exzellenznetzwerk Biowissenschaften (Sachsen-Anhalt). NG was supported by NIH Grant P01 GM-62580.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jakob-Roetne R, Jacobsen H. Alzheimer's disease: from pathology to therapeutic approaches. Angew Chem Int Ed Engl. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 2.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 3.Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis. 2007;4:13–27. doi: 10.1159/000100355. [DOI] [PubMed] [Google Scholar]
- 4.Makin OS, Serpell LC. Structures for amyloid fibrils. FEBS J. 2005;272:5950–5961. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 5.Schilling S, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 6.Broersen K, et al. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer's disease: oligomer size or conformation? Alzheimers Res Ther. 2010;2:12. doi: 10.1186/alzrt36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lashuel HA, Lansbury PT., Jr Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q Rev Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 8.Fandrich M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell Mol Life Sci. 2007;64:2066–2078. doi: 10.1007/s00018-007-7110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunde M, et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 10.Nelson R, et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 12.Habicht G, et al. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Abeta protofibrils. Proc Natl Acad Sci U S A. 2007;104:19232–19237. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Sachse C, et al. Quaternary structure of a mature amyloid fibril from Alzheimer's Abeta(1–40) peptide. J Mol Biol. 2006;362:347–354. doi: 10.1016/j.jmb.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt M, et al. Comparison of Alzheimer Abeta(1–40) and Abeta(1–42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zandomeneghi G, et al. FTIR reveals structural differences between native beta-sheet proteins and amyloid fibrils. Protein Sci. 2004;13:3314–3321. doi: 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerf E, et al. Antiparallel beta-sheet: a signature structure of the oligomeric amyloid beta-peptide. Biochem J. 2009;421:415–423. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 18.Eckert A, et al. Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J Mol Med. 2008;86:1255–1267. doi: 10.1007/s00109-008-0391-6. [DOI] [PubMed] [Google Scholar]
- 19.Tycko R, et al. Evidence for novel beta-sheet structures in Iowa mutant beta-amyloid fibrils. Biochemistry. 2009;48:6072–6084. doi: 10.1021/bi9002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luhrs T, et al. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petkova AT, et al. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kheterpal I, et al. Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange--mass spectrometry with on-line proteolytic fragmentation. J Mol Biol. 2006;361:785–795. doi: 10.1016/j.jmb.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 23.Torok M, et al. Structural and dynamic features of Alzheimer's Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 24.Petkova AT, et al. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sachse C, et al. Paired beta-sheet structure of an Abeta(1–40) amyloid fibril revealed by electron microscopy. Proc Natl Acad Sci U S A. 2008;105:7462–7466. doi: 10.1073/pnas.0712290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldsbury CS, et al. Studies on the in vitro assembly of a beta 1–40: implications for the search for a beta fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 27.Meinhardt J, et al. Abeta(1–40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J Mol Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang R, et al. Interprotofilament interactions between Alzheimer's Abeta1–42 peptides in amyloid fibrils revealed by cryoEM. Proc Natl Acad Sci U S A. 2009;106:4653–4658. doi: 10.1073/pnas.0901085106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paravastu AK, et al. Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fandrich M, et al. Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion. 2009;3:89–93. doi: 10.4161/pri.3.2.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiltzius JJ, et al. Molecular mechanisms for protein-encoded inheritance. Nat Struct Mol Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kodali R, et al. Abeta(1–40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klement K, et al. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer's abeta(1–40) amyloid fibrils. J Mol Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 34.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 35.Hellstrand E, et al. Amyloid beta-Protein Aggregation Produces Highly Reproducible Kinetic Data and Occurs by a Two-Phase Process. Acs Chemical Neuroscience. 2010;1:13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zagorski MG, et al. Methodological and chemical factors affecting amyloid beta peptide amyloidogenicity. Methods Enzymol. 1999;309:189–204. doi: 10.1016/s0076-6879(99)09015-1. [DOI] [PubMed] [Google Scholar]
- 37.Sachse C, et al. Nanoscale flexibility parameters of Alzheimer amyloid fibrils determined by electron cryo-microscopy. Angew Chem Int Ed Engl. 2010;49:1321–1323. doi: 10.1002/anie.200904781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim YS, et al. Two-dimensional infrared spectra of isotopically diluted amyloid fibrils from Abeta40. Proc Natl Acad Sci U S A. 2008;105:7720–7725. doi: 10.1073/pnas.0802993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto A, et al. Analysis of the secondary structure of beta-amyloid (Abeta42) fibrils by systematic proline replacement. J Biol Chem. 2004;279:52781–52788. doi: 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- 40.Williams AD, et al. Alanine scanning mutagenesis of Abeta(1–40) amyloid fibril stability. J Mol Biol. 2006;357:1283–1294. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 41.Olofsson A, et al. The solvent protection of alzheimer amyloid-beta-(1–42) fibrils as determined by solution NMR spectroscopy. J Biol Chem. 2006;281:477–483. doi: 10.1074/jbc.M508962200. [DOI] [PubMed] [Google Scholar]
- 42.Olofsson A, et al. Amide solvent protection analysis demonstrates that amyloid-beta(1–40) and amyloid-beta(1–42) form different fibrillar structures under identical conditions. Biochem J. 2007;404:63–70. doi: 10.1042/BJ20061561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paravastu AK, et al. Seeded growth of beta-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci U S A. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen JZ, et al. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc Natl Acad Sci U S A. 2009;106:10644–10648. doi: 10.1073/pnas.0904024106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachse C, et al. High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol. 2007;371:812–835. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kajava AV, et al. beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. Faseb Journal. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caspar DL. Inconvenient facts about pathological amyloid fibrils. Proc Natl Acad Sci U S A. 2009;106:20555–20556. doi: 10.1073/pnas.0910978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crowther DC, et al. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 49.Jarrett JT, et al. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 50.Jan A, et al. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem. 2008;283:28176–28189. doi: 10.1074/jbc.M803159200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuperstein I, et al. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ono K, et al. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jang H, et al. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer's Disease and Down syndrome. Proc Natl Acad Sci U S A. 2010;107:6538–6543. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 55.Hu X, et al. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedrich RP, et al. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A. 2010;107:1942–1947. doi: 10.1073/pnas.0904532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandberg A, et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc Natl Acad Sci U S A. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahmed M, et al. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chimon S, et al. Evidence of fibril-like beta-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's beta-amyloid. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]