

Introduction
The structure, function and components of the glomerular barrier have been a subject of debate among researchers for decades. Recent advances have identified many of the molecular components of the barrier, but their functional interactions and individual or collective contribution to the selective barrier remains elusive. The filtration barrier is comprised of the single glomerular capillary lined by the glomerular endothelial cells, the glomerular basement membrane and the specialized epithelial cells- podocytes that cover the basement membrane on the its side facing the urinary space (Figure 1). On the capillary side of the basement membrane smooth muscle like cells called mesangial cells occupy space between the capillary loops and provide structural support to the capillaries.
Figure 1.
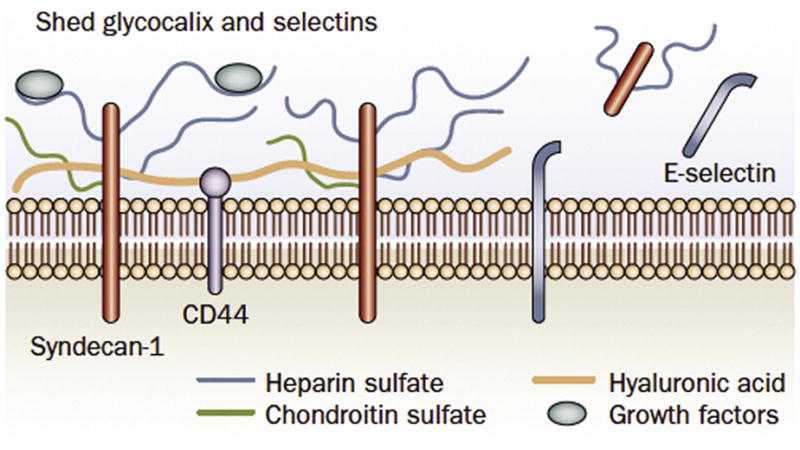
The glycocalyx contains anchoring proteoglycans (e.g. syndecan 1 and CD44) and connecting glycosoaminoglycans (e.g. heparan sulfate, chondroitin sulfate and hyaluronic acid). The negative charge of the proteins constitutes an important charge barrier against filtration of albumin. During endothelial activation, the glycocalyx is modified to allow leukocytes and platelets to interact with the endothelial surface. Glycocalyx components are then modified and released into the circulation. With prolonged activation and early apoptotic events, adhesion molecules such as E-selectin may also be shed. (Reprinted by permission from Nature review Nephrology).
The glomerular barrier is a complex biological sieve. It allows for high filtration rates of water, passage of small and mid-sized molecules, while completely restricting serum albumin and larger proteins. Based on a glomerular filtration rate of 100 ml/min, close to 180 liters of primary urine is produced every day at capillary pressures far exceeding pressures in any other capillary bed in the body. The majority of the filtrate is reclaimed by the tubules, raising the possibility of their role in protein reabsorption as well.
Perturbation of the components of the filtration barrier or molecular pathways can result in the clinical end points of proteinuria and progression to end-stage renal disease. There appears to be a relationship between development of effacement (spreading) of podocyte foot processes and proteinuria, though the reverse is not always true. This has resulted in a primary focus on podocytes as the key player in protein leak. Whether foot process effacement is a manifestation of podocyte injury resulting from leakage of normal or pathological protein or a yet unknown molecule; or it is the primary event which occurs prior to initiation of proteinuria remains to be seen. Despite the podocyte dominant view of the glomerular filter there is no diminishing the contribution of the other components of the filtration barrier as well as the role of hemodynamics, tubular re-absorption and diffusion gradient across the GBM. The role of mesangial cells as a primary cell responsible for development of proteinuria remains unclear. There is data to support the mesangial cells are influenced by ongoing proteinuria. They produce cytokines and inflammatory products in response to albuminuria or in response to glycation end products in diabetes. This leads to progressive scarring by deposition of matrix proteins. Bone marrow transplantation from db/db mice to normo-glycemic B6 mice resulted in development of albuminuria and glomerular lesion, presumably from transfer of mesangial cell progenitor (1).
The molecular mechanisms that lead to proteinuria are poorly understood; therefore, targeted therapies are lacking. However, a large body of information has emerged in this field, which has advanced our understanding of the molecular mechanisms that are in play during both development and maintenance of the filtration barrier. In this review we provide a broad overview of the different aspects of the filtration barrier and the pathogenesis of proteinuria.
Glomerular Endothelial Cells
The inside of the glomerular capillaries are covered with highly specialized endothelium. Secondary to signals from the podocytes and mesangium, which include VEGF, the endothelium acquires a highly fenestrated phenotype (2). The fenestrations cover up to 20% of the endothelial surface and facilitate high flux filtration of fluid and small solutes. In addition, the glomerular endothelial cells actively synthesize the glycocalyx and basement membrane. The glycocalyx is a 100–300 nm thick layer of membrane associated proteoglycans, glycosoaminoglycans, glycolipids and trapped plasma proteins (see figure 2.). The presence and potential importance of the glycocalyx surface has been overlooked due to the technical complexity to study its function-structure relationship. Only after specialized preservation techniques (e.g. high pressure freezing and the use of alcian blue) one can visualize, for example, the glycoso-aminoglycans. Using such techniques, Rosgard et al. were able to demonstrate that glomerular endothelial cells are not only covered with proteoglycans, but that in fact these proteoglycans formed a plug in the fenestrae that stretched out to the glomerular basement membrane (3). Electron microscopic studies suggest that these plugs are organized in a regular periodic way that may thus create pathways of different diameters for diffusion while the negative electrical charges of the glycosaminoglycan molecules may form an important charge barrier to prevent albumin filtration. The presence of charge selectivity has recently been debated, as measurements of glomerular sieving coefficients using intravital 2-photon microscopy suggest that such a charge barrier does not exist (4). Although the barrier against filtration of albumin most likely is not absolute, one has to realize that assessment of filtration sieving coefficients with these techniques is subject to multiple factors that influence these measurements of albumin filtration. Variations include the effect of tissue depth of measurements, detector sensitivity (the amount of laser excitation given), and the nature of the probes used (degree of polydispersion). Moreover, genetic strain, age and nutritional status may affect sieving coefficient as well (e.g. aged Fawn Hooded male rats have spontaneous proteinuria)(5). In support of an important role of the endothelial glycocalyx in prevention of albumin filtration, we recently observed that specific disruption of the endothelial glycosurface by the enzyme hyaluronidase resulted in albuminuria, while podocyte and tubular ultrastructures were unaltered (unpublished observations). Similar findings were reported by Gelberg et al. when they injected sialidase intraperitoneally in mice who developed foot process effacement and proteinuria due to loss of charge on the endothelial and podocyte cell surface (6).
Figure 2. Glomerular filtration barrier and tubules.

A. Nephrin/Neph1 tyrosine phosphorylation dependent recruitment of protein complexes involved in actin regulation. Fyn mediated tyrosine phosphorylation of Neph1 on Y637 and Y638 results in recruitment of adaptor protein Grb2. Similarly nephrin phosphorylation on its tyrosine residues Y1191, Y1208 and Y1232 recruits Nck, Crk (Y1198 and Y1225) and P85 subunit (Y1128, Y1153 and Y1154) of PI3 kinase. B. Schematic cross section of podocyte foot process, glomerular basement membrane and endothelial cells. Also illustrated are the important proteins (mutations or deletions) which have been identified to result in proteinuria in human diseases or mouse models. C. Schematic of the nephron with the proximal tubule illustrating tubular proteins cubulin and megalin. Abbreviations: TrpC6, transient receptor potential C6; nWasp: neural Wiskott Aldrich syndrome protein; Arp2/3: actin related protein 2 and 3; AT1 receptor: Angiotensin receptor 1; SSH1: slingshot 1; PPase: phosphatase.
Interestingly, there is probably a direct relationship between endothelial cell activation and loss of glycocalyx surface. Normally the endothelium maintains a quiescent phenotype where nitric oxide signaling dominates. Upon exposure to cardiovascular risk factors or inflammatory molecules the endothelial cell may switch to redox signaling and loose glycocalyx structure (7,8). As endothelial activation has also been implicated in the development of cardiovascular disease, this phenomenon may explain the strong epidemiological association of the occurrence of albuminuria with the development of cardiovascular risk (9).
In general in the microcirculation, endothelial cells always require signals such as angiopoietins and VEGF’s from their direct neighboring cells to maintain a stable and viable phenotype. At the same time, endothelial cells also control the phenotype of these adjacent cells by secreting factors such as PDGF, jagged 1 and CNP (10). Extrapolating these observations to the glomerulus, it is most likely that the endothelium and its function also are important in maintaining the glomerular architecture. For example, endothelial specific overexpression of the Receptor for Advance Glycation End-products results in glomerulosclerosis and albuminuria (11).
Glomerular Basement Membrane
GBM is mainly comprised of type IV collagen (collagen α3, α4, and α5 chains), proteoglycans and laminins. GBM was the primary focus of the initial investigations into the selectivity of the glomerular filtration barrier. It was considered to play a central role in filtration of macromolecules and function both as a size selective and charge selective barrier. While collagen IV and laminin provide the structural support to the capillary wall the proteoglycans perlecan and agrin and their negatively charged glycosaminoglycan side chains provide the GBM with its attribute of charge selectivity. One argument against its role as the primary barrier are illustrated by diseases like Alport’s syndrome and thin basement membrane disease, where mutation in the gene encoding the type IV collagen or the basement membrane thickness do not result in significant proteinuria. In mice, a mutation in α3 type IV collagen leads to elimination of all three chains as there are abnormalities in assembly of the tertiary structure (12). These mice show evidence of GBM splitting, thinning and basket weave appearance though the podocyte foot process and the endothelial layer remains intact. They develop proteinuria initially followed by foot process effacement, suggesting that effacement is a manifestation of podocyte injury (13).
Laminins are heterotrimeric proteins that self-assemble into a network in the basement membrane. Laminin 11 (α5, β2 and γ1 chains) is found in mature GBM and connect to collagen IV via nidogen and entactin. Human mutation in the Laminin β2 gene (Lamb2) results in Pierson’s syndrome which is a rare congenital nephrotic disorder with ocular abnormalities (14). Mice lacking the β2 chain (Lamb2−/−) develop proteinuria and die in the perinatal period (15). Interestingly, these mice had evidence of GBM disorganization that preceded proteinuria while podocytes and the slit diaphragm appeared normal initially (16). Tracer studies further confirmed increase in GBM permeability in the knock-out mice. These results suggest a role of GBM in the filtration barrier permselectivity. Alternatively, one may postulate that such severe disruptions of the basement membrane that occur in the lamb 2 −/− mice might also affect endothelial and podocyte function, thus indirectly promoting the development of albuminuria.
It has been hypothesized that the negatively charged heparan sulfates that are present in the glomerular basement membrane may also contribute to charge selectivity of filtration and prevent albumin leak. Seminal studies by Farquhar et. al demonstrated that on removal of glycosaminoglycans in the GBM by perfusion of bacterial glycosaminoglycan-degrading enzymes led to increased passage of labeled bovine serum albumin through the GBM (17). In contrast, removal of the anionic sites in the GBM using heparanase did not result in albuminuria (18). Also, mice that over-expressed the enzyme heparanase, displaying a fivefold reduction of anionic sites in the GBM did not develop albuminuria (19), questioning the importance of heparan sulfates in creating charge selectivity of filtration.
Podocytes
Podocytes are terminally differentiated epithelial cells, with similarities to neurons, which cover the GBM facing the urinary space. They have octopus like extensions from the cell body called primary processes which further branch to form secondary and tertiary processes and engulf the capillary loops. The junction between the tertiary foot processes is a specialized region termed the slit diaphragm and is thought to be a modified adherens junction. The slit diaphragm contains transmembrane proteins like Nephrin and Neph1 which are unique to the podocytes but also proteins like FAT1, P-cadherins and catenins which are typical for an adherens junction. Interestingly tight junction proteins like JAM-A, occludin and ZO-1 have also been demonstrated to be at the slit diaphragm using fractionation, immunofluorescence and immunoelectron microscopy (20). Unlike podocytes most other epithelial cells contain tight junctions and adherens junction as distinct separate protein complexes. The tight junction separates the apical and basolateral domains and help in defining and maintaining cell polarity. Adherens junction is located more basal to the tight junction and forms a tether for the actin cytoskeleton. During development the slit diaphragm originates as a typical tight junction which then migrates down as the foot process grow in length and persists as the interdigitation of the tertiary foot processes (21). In recent years, the discovery of the molecular basis of (dys)regulation of slit diaphragm structure and function, and its direct link to genetic forms of nephrotic syndrome, has put the podocyte in the center of thinking in regards to development of albuminuria. It is, however, important to discriminate albuminuria in the setting of genetic mutations of slit diaphragm proteins, from albuminuria that develops in the context of diabetes, hypertension and progressive renal disease. In the latter conditions, as discussed previously, endothelial activation and loss of charge selectivity of a highly fenestrated endothelium probably is the first event to occur. This consequently exposes the podocytes to the deleterious effects of albumin and other macromolecules. In metabolic diseases such as diabetes, albumin will have undergone chemical modifications such as glycation, nitration and binding of (modified) lipoproteins and free fatty acids (22). It is therefore likely that exposure to (modified) albumin may directly lead to alterations in podocyte function and rearrangement of slit diaphragm structure. Interestingly, podocytes can express scavenger receptors, such as the receptor for advanced glycation end products, megalin and CXCL16 (23). In this scenario, podocyte effacement could then be looked upon as a response to injury, and spreading and migration would serve to maintain coverage of the glomerulus by the podocytes.
Podocytes are also covered by glycocalyx, principally podocalyxin, a heavily glycosylated protein (24). In puromycin treated rats that develop proteinuria and foot process effacement show lower sialic content of podocalyxin (25) linking effacement to reduced negative surface charge of podocyte. Despite a large body of available literature the study of podocyte biology has been severely hamstringed by the lack of a good cell culture model that would recapitulate in-vivo podocyte phenotype. Based on the available data, podocytes have a remarkable structure function relationship that can be broadly examined by the signaling occurring at its intercellular junction (the slit diaphragm) and the relationship of the podocytes with the basement membrane (podocyte-GBM).
1. Slit Diaphragm: Regulation of Actin Cytoskeleton
Podocytes and specifically their intercellular junction termed the slit diaphragm have become the major focus of investigation in diseases presenting with proteinuria. This has been fueled by the discovery of single gene mutations in protein expressed at the slit diaphragm, that are responsible for the development of nephrotic syndrome in both human diseases and animal gene knock out models. The first such protein to be identified at the slit diaphragm was nephrin (NPHS1) which was found to be mutated in newborns suffering from the congenital nephrotic syndrome of the Finnish type (26). Nephrin belongs to the immunoglobulin superfamily whose members are involved in cell-cell adhesion. It is thought Nephrin binds across the junction to itself or a similar protein called Neph1 (27,28), forming a structure reminiscent of the zipper like structure described by Karnofsky and Rodewald in 1974. This cross junctional binding has been postulated to form a physical sieve that creates a size-selective pore in the slit diaphragm (29). In the absence of nephrin, there is failure to develop the slit diaphragm. Similarly, mutations in Podocin (NPHS2) a member of the stomatin family is also responsible for familial nephrotic syndrome (30). The lack of significant proteinuria in patients with Alports syndrome with mutations in GBM component type IV collagen when compared to the massive proteinuria of patients with mutant NPHS1 and NPHS2 provides evidence that the slit diaphragm plays a crucial role in forming the macromolecular glomerular filter. Nephrin-Neph1-Podocin is postulated to represent a junctional receptor. Based on available evidence it appears that this receptor complex plays a role in orchestrating the regulation of the actin cytoskeleton (31). Using either the Cre-lox system, classical knock down or transgenic animal models, numerous proteins have been deleted or over-expressed resulting in either normal development or foot process abnormalities and proteinuria (32–49) (see table 1 for a list of proteins/genes implicated in development of foot process and proteinuria). Interestingly many of these proteins are directly or indirectly involved in regulation of actin dynamics. A number of investigations demonstrate that the Nephrin-Neph1 receptor complex assembles a complex of proteins important in not only the nucleation of actin filament but also in regulating its morphology and elongation (50). Nephrin interacts with Nck in a tyrosine phosphorylation dependent manner resulting in nucleation of actin filaments forming actin pedestals (51–53). Furthermore, deletion of Nck1/2 conditionally in podocytes resulted in failure of foot process development, a phenotype similar to nephrin deletion (53) emphasizing the structure function relationship of these cells. Similarly, Neph1 recruits Grb2 and cooperates with Nephrin-Nck in actin polymerization (31). In a related model, phosphorylation of vaccinia viral protein A36R, by Src kinase, results in recruitment of Nck and associated actin polymerization machinery resulting in robust formation of actin filaments (54). Phosphorylation of a second A36R tyrosine residue leads recruits Grb2 and appears to cooperate with Nck to facilitate actin polymerization (55). The importance of the role of Src family tyrosine kinase Fyn in this process is highlighted by the observation that Fyn null mice have attenuated nephrin phosphorylation and abnormal foot process morphology (56,57).
Table 1.
List of genes and proteins deleted or over-expressed in glomerulus or the tubules resulting in proteinuria, foot process developmental abnormality or normal phenotype.
| Gene/Protein | Human/Animal | Disease/Phenotype | Response to Injury | Age of Onset | References |
|---|---|---|---|---|---|
| Podocytes | |||||
| NPHS1/Nephrin | Human/Mouse | FP Effacement/Proteinuria | NA | At birth | Kestila et. al (26) |
| NPHS2/Podocin | Human | FP Effacement/Proteinuria | NA | Late | Boute et. a (30) |
| PLC epsilon 1 | Human/Zebra Fish | Diffuse Mesangial Sclerosis (DMS) | NA | Early | Hinkes et. al (92) |
| Nck | Mouse | FP Formation Defect | NA | At birth | Jones et. al (53) |
| Fyn | Mouse | Subtle changes in FP | NA | Early | Yu et. al (56) |
| Fyn/Yes Combined | Mouse | FP effacement/Proteinuria | NA | Early | Stein et. al (32) |
| Kirrel/Neph1 | Mouse | FP Effacement/Proteinuria | NA | Early | Donoviel et. al (58) |
| TRPC6 | Human | FSGS | NA | NA | Winn et. al (65), Reiser et. al (66) |
| Combined CD2ap/Fyn | Mouse | FSGS | NA | Late | Huber et. al (47) |
| ACTN4/Alpha-Actinin-4 | Human/Mouse | FSGS | NA | Late | Kaplan et. al (64) |
| Notch1 Transgenic | Mouse | FSGS | NA | NA | Niranjan et. al (33) |
| NFAT Transgenic | Mouse | FSGS | NA | Early | Wang et. al (34) |
| uPAR | Mouse | NA | Protected | NA | Wei et. al (71) |
| Focal Adhesion Kinase | Mouse | NA | Protected | NA | Ma et. al (70) |
| COQ2/Coenzyme Q10 | Human | FSGS | NA | Late | Scalgia et. al (36) |
| HGF/C-met | Mouse | Normal | injury and proteinuria following adriamycin | Dai et. al (48) | |
| aPKC Lambda/iota | Mouse | FSGS/FP Effacement/Proteinuria | NA | Late | Huber et. al(61), Hirose et. al (62) |
| PDSS2/Prenly Diphosphate Synthase Subunit 2 | Human/Mouse | FP effacement/Proteinuria | NA | Late | Peng et. al (37) |
| Synaptopodin | Mouse | Normal | Slow Recovery | NA | Asanuma et al (40) |
| Cfl1/Cofilin 1 | Mouse/Zebra Fish | FP Effacement/Proteinuria | process morphology | Late | Garg et. al (50), Ashworth et. al. (60) |
| Sidekick Transgene | Mouse | FSGS | NA | Late | Kaufman et. al (49) |
| INF2 | Human | FSGS | NA | Late | Brown et. al (68) |
| ATG5 | Mouse | Glomerulosclerosis/Proteinuria | NA | Late | Hartleben et. al (90) |
| PI3Kc II | Mouse | DMS/FP effacement/Proteinuria | NA | Late | Harris et. al (91) |
| EP4 receptor Transgenic | Mouse | Proteinuria | 5/6 th Nephrectomy required | NA | Stitt-Cavanagh et. al (39) |
| AT1 Transgenic | Mouse | FP Effacement | NA | Early | Hoffman et. al (41) |
| Beta Catenin | Mouse | NA | Protected | NA | Dai et. al (42) |
| Podocalyxin Transgenic | Mouse | FP formation Defect | NA | NA | Economou et al. (43) |
| FAT1 | Mouse | FP Effacement/Proteinuria | NA | Perinatal Lethality | Ciani et. al (59) |
| GLEPP1 | Mouse | Broadening of FP/No Proteinuria | NA | NA | Wharram et. al (35) |
| VEGF transgenic | Mouse | Collapsing glomeruolopathy | NA | Early | Eremina et. al (44) |
| VEGF Heterozygous deletion | Mouse | Endotheliosis/Proteinuria | NA | Early | Eremina et. al (44) |
| Glomerular Basement Membrane | |||||
| Lamb2/Laminin β2 | Human/Mouse | DMS/FSGS (Pierson syndrome) | NA | Perinatal Lethality | Jarad et. al (15) |
| Beta 1 Integrin | Mouse | Proteinuria | NA | At Birth | Pozzi et. al (46) |
| Integrin alpha 3 subunit | Mouse | Disorganized GBM/FP formation defect/Proteinuria | NA | Early | Kang et. al (101) |
| Integrin Linked Kinase | Mouse | GBM alteration/FSGS | NA | Early | El Aouni et. al (102) |
| Tubules | |||||
| Cubulin | Mouse | Normal/Minimal Proteinuria | NA | NA | Amsellen et. al (109) |
| Megalin | Mouse | LMW proteinuria | NA | Early | Leheste et. al (45) |
| Transcription Factors | |||||
| WT1 | Human | Denys Drash and Frasier Syndrome | NA | Early | Yang et. al (88), Guo et. al (89) |
| PTIP | Mouse | Proteinuria | NA | Late | LeFevre et. al (87) |
| LMX1B/LIM homeobox transcription factor | Human | Nail Patella syndrome | NA | Early | Rohr et al. (80) |
| SMARCAL1 | Human | FSGS/Steroid Resistant NS | NA | Late | Boerkoel et al.(77) |
The accumulating evidence from the different mouse models indicates that actin dynamics is not only important in the initial development of the podocyte foot process and its intercellular junction but also plays a role during injury and repair. Deletion of NPHS1 (26), NPHS2 (30), Neph1 (58), or FAT1(59) each result in abnormal foot process development whereas deletion of other genes like Cfl1 (50,60), aPKC (61,62) and CD2AP (63) result in foot process abnormalities in mature podocytes following normal initial development. The latter is also observed in human diseases with actin polymerization related gene mutations like α-actinin 4 (64), TrpC6 (65–67) and INF2 (68). This suggests ongoing actin dynamics in mature healthy podocytes and supported by recent data using in-vivo imaging techniques showing that podocytes are constantly motile (69). Although these studies are limited by the resolution of the techniques used to image in-vivo podocytes and are still debatable, the existing paradigm of a stable quiescent podocyte foot process and its intercellular junction is being questioned.
The processes of podocyte injury and repair also involve dynamic actin polymerization and turnover. Effacement which originally was thought to be a passive process, is actually a very active actin reorganization mediated by a specific set of signaling intermediaries. It is not surprising that conditional deletion of some of these signaling proteins such as FAK (70), uPAR (71), Crk1/2 (presented as an abstract at Renal Week, 2009), Cdc42, or cathepsin (72) result in the absence of foot process effacement in podocyte injury models. Whether this affected the proteinuria as well is not known. The primary focus in these studies has been the preservation of the foot process morphology. Breakdown of the podocyte intercellular junction is associated with loss of junctional proteins like nephrin, zona occludens-1 (ZO-1) and P-cadherins (73,74). Numerous studies have tried to link this to an EMT like phenomenon (75–77). This would suggest a change in transcriptional regulation of these proteins resulting in junctional breakdown and detachment of podocytes. It is not surprising then that numerous transcriptional factors have been implicated in the development of foot processes and proteinuria (78–83). A role for transcription factor Pax2 in early renal development is well established (84,85). While healthy mature podocytes do not express Pax2, persistent expression of Pax2 in transgenic mice results in severe glomerulopathy (86). Deletion of Pax2 interacting protein (PTIP) conditionally in podocytes results in abnormal foot process development and proteinuria (87). Mutations in zinc finger transcription factor WT1 results in Denys-Drash and Frasier syndromes (88,89) where patients present with mesangial sclerosis (Denys-Drash) and focal segmental glomerulosclerosis (Frasier). Similarly, transcription factor LMX1B implicated in nail-patella syndrome is also required for normal podocyte development (See table 1 for other transcriptional factors implicated in glomerular development abnormalities). Targeted disruption of LMX1B results in developmental arrest of podocytes as they fail to form foot processes or a slit diaphragm (80). Besides the overwhelming data suggesting a predominant role of actin dynamics in podocyte structure and function, there is evidence that if there is podocyte specific perturbation of processes or molecules not directly related to actin dynamics, as in autophagy (90), PI3 kinase class II activity (91), or mutations in Phospholipase C ε1 (92) gene, can also result in proteinuria and foot process abnormalities. Even though there appears to be strong correlation between foot process effacement and proteinuria, there are conditions in which the relationship does not hold true, further suggesting that there are other components necessary for preventing protein leak (93,94).
The relationship between podocyte injury and progression to chronic kidney disease has been elegantly demonstrated by Wiggins et. al and other investigators. Rodents lack diphtheria toxoid receptor and are oblivious to the diphtheria toxin. Taking advantage of this attribute of rats they generated a transgenic rat which expresses human diphtheria toxin receptor (hDTR) on podocytes (95). When injected with diphtheria toxin following uptake there is selective injury to the podocytes within the glomerulus. Depending on the magnitude of the initial injury, determined by the dose or the frequency of the administered toxin, there is either recovery or progressive scarring of the renal glomerulus. They were further able to predict the progression of the disease (sclerosis) based on the ratio of Nephrin and Podocin mRNA levels from the podocytes shed in the urine (96,97).
2. Podocyte-Glomerular Basement membrane
Podocytes adhere to the GBM via adhesion protein complexes like dystroglycans and integrins. Disruption of the podocyte-GBM adhesion is required for movement of the podocytes such as in effacement. The dystroglycan gene knock-out results in embryonic lethality prior to glomerular development (98). Its role in basement membrane and podocyte structure and function can be only assessed by yet to be done podocyte specific knockout of the gene. The density of dystroglycans is decreased in minimal change disease (99). Whether this represents a primary event or a result of foot process effacement remains to be elucidated.
Podocytes also anchor to the GBM by an α3β1-integrin complex. Integrin hetrodimers cluster at the focal adhesions and are linked to the actin cytoskeleton. In podocytes integrins localize exclusively to the basal membrane. Using an antibody against glomerular integrin results in podocyte detachment (100) and α3-integrin knock-out mice develop congenital nephrotic syndrome (101). Ligation of integrin results in recruitment of its related kinase integrin linked kinase (ILK). Podocyte specific deletion of ILK results in foot process effacement, proteinuria and progressive glomerulosclerosis (102). ILK also appears to interact with nephrin and α-actinin-4 and this interaction is important for slit diaphragm integrity and podocyte morphology (102). Focal adhesion kinase (FAK) a cytoplasmic receptor tyrosine kinase is an important part of the integrin-matrix complex that links with the actin cytoskeleton via talin (103). Deletion of FAK (70) and uPAR (71) in podocytes resulted in a lack of both foot process effacement and proteinuria in injury models suggesting the importance of podocyte-GBM interaction and its regulation of podocyte cytoskeleton (70).
Tubular Reabsorption of Proteinuria
The tubular epithelial cells re-absorb most of the primary filtrate that comes through the glomerulus. Normally the final urine contains <30 mg/d of albumin. Assuming that the glomerular barrier is normally leaky to proteins and other molecules like vitamins then defects in their re-absorption by tubular cells could result in proteinuria. Several observations refute that this occurs, Maunsbach in the mid-1960’s noted no significant uptake of albumin from the tubular lumen to blood (104). Identification of apical membrane transport proteins like megalin by Kerjaschki and Farquhar (105) followed by cubulin (106–108) provided evidence of expression of proteins in the tubular epithelium which are capable of internalizing albumin, vitamin A, B12 and D. Amsellem et. al reported that cubulin, when conditionally knocked out in tubular epithelial cells resulted in six fold increase in albuminuria compared to basal levels of <30mg/day (109). Though this is not a very significant amount when compared to nephrotic levels proteinuria it would be interesting to study the magnitude of protein leak in a model of glomerular proteinuria with a similar conditional tubular cubulin knock out. This could result in significant amount of proteinuria in the absence of tubular reabsorption. These findings also raise an important teleological question: why do tubular cells express proteins involved in transporting albumin and other macromolecules if there is no passage of protein past the glomerular filter? It is possible that a mechanism to absorb protein leak could provide an evolutionary survival benefit. There is glomerular damage and proteinuria in response to transient infections and immunologic injury. In the absence of mechanisms to absorb these transient and small magnitude leaks the organism might succumb to the injury. It is when the tubular absorption is overwhelmed by either injury to the glomerular filter or tubules that a higher magnitude of proteinuria becomes evident. The role of tubular reabsorption in urine protein homeostasis is further reviewed by Nielson and Christensen (110). In recent, years there has been an extensive debate whether tubular albumin reabsorption and the subsequent lysosomal degradation, may in fact result in tubular injury (so called protein overload injury). Theilig and colleagues used an elegant cre-recombinase induced deletion of megalin in mice to address this question. They found a mosaic deletion of megalin and thus were able to observe the renal response both in the presence and absence of tubular albumin reabsorption in the same kidney (23). Upon induction of glomerular injury, the tubules that could reabsorb albumin demonstrated increased expression of pro-inflammatory cytokines and TGF-beta. However, the development of tubulointerstitial injury was entirely related to the extent of glomerular injury and not to tubular activation.
Conclusions
The development of albuminuria is probably a multistep process where initially, loss of the endothelial barrier function may play a role. Endothelial activation and subsequent shedding of the glycocalyx surface allows albumin to penetrate the sub-podocyte space. Podocytes may then take up albumin through scavenger receptors and display actin skeleton rearrangements and injury. Compensatory tubular reabsorption and the inflammatory responses that go with it, may then further contribute to the structural interstitial damage that has been associated with albuminuria. Though there has been a tremendous advances in understanding the role of various components of the filtration barrier in proteinuria there continues to be a lack of meaningful targeted therapies. The paucity of well defined injury models that can be extrapolated to human diseases limits the derived conclusions of many studies. Lack of an in vivo phenotype of podocytes in culture prevents us from answering some of the outstanding questions in this field in regards to the development of the processes and junctional dynamics. One big void in the podocyte development field has been the role of microtubules and its regulation in the development of the primary and secondary processes. The major impetus has been the regulation of actin by the slit diaphragm protein Nephrin/Neph1 which probably applies only to the tertiary processes. This is further supported by the SEM of Nephrin null mice which show abnormalities in the tertiary processes only (unpublished data). Investigators have convincingly demonstrated the relationship between foot process spreading and proteinuria. The recent observation that foot process spreading is an active dynamic process provides an important target for therapy. The resolution of proteinuria on reversal or inhibition of foot process spreading would prove the relationship further and provide impetus to generating therapies that could prevent the foot process spreading and hence proteinuria which would eventually impact disease progression. In the near future, improvement in available technology to develop model systems which can then be used to answer some of the difficult questions posed by the unique attributes of the filtration barrier, along with large scale expression profiling, and transgenic mouse lines will further expand our understanding of the glomerular function and pathology. Only then we can hope to develop novel target molecules for the therapy of proteinuric kidney diseases.
Acknowledgments
This work is supported by National Institutes of Health grants DK081403 (P.G.), and National Kidney Foundation Young Investigator Award (P.G.).
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibiliography
- 1.Zheng F, Cornacchia F, Schulman I, Banerjee A, Cheng QL, Potier M, Plati AR, Berho M, Elliot SJ, Li J, Fornoni A, Zang YJ, Zisman A, Striker LJ, Striker GE. Diabetes. 2004;53(9):2420–2427. doi: 10.2337/diabetes.53.9.2420. [DOI] [PubMed] [Google Scholar]
- 2.Eremina V, Quaggin SE. Current opinion in nephrology and hypertension. 2004;13(1):9–15. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Rostgaard J, Qvortrup K. Cells, tissues, organs. 2002;170(2–3):132–138. doi: 10.1159/000046186. [DOI] [PubMed] [Google Scholar]
- 4.Comper WD, Russo LM. Current opinion in nephrology and hypertension. 2009;18(4):336–342. doi: 10.1097/MNH.0b013e32832cb96a. [DOI] [PubMed] [Google Scholar]
- 5.Kreisberg JI, Karnovsky MJ. The American journal of pathology. 1978;92(3):637–652. [PMC free article] [PubMed] [Google Scholar]
- 6.Gelberg H, Healy L, Whiteley H, Miller LA, Vimr E. Laboratory investigation; a journal of technical methods and pathology. 1996;74(5):907–920. [PubMed] [Google Scholar]
- 7.Singh A, Friden V, Dasgupta I, Foster RR, Welsh GI, Tooke JE, Haraldsson B, Mathieson PW, Satchell SC. American journal of physiology. 300(1):F40–48. doi: 10.1152/ajprenal.00103.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ, Stroes ES. Diabetes. 2006;55(2):480–486. doi: 10.2337/diabetes.55.02.06.db05-1103. [DOI] [PubMed] [Google Scholar]
- 9.Grams M, Coresh J. Lancet. 376(9758):2046–2048. doi: 10.1016/S0140-6736(10)61498-5. [DOI] [PubMed] [Google Scholar]
- 10.Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L., Jr Histology and histopathology. 2009;24(7):909–969. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Takeuchi M, Watanabe T, Yamagishi S, Sakurai S, Takasawa S, Okamoto H, Yamamoto H. The Journal of clinical investigation. 2001;108(2):261–268. doi: 10.1172/JCI11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalluri R, Cosgrove D. The Journal of biological chemistry. 2000;275(17):12719–12724. doi: 10.1074/jbc.275.17.12719. [DOI] [PubMed] [Google Scholar]
- 13.Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, Samuelson GC. Genes & development. 1996;10(23):2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 14.Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dotsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A. Human molecular genetics. 2004;13(21):2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 15.Jarad G, Cunningham J, Shaw AS, Miner JH. The Journal of clinical investigation. 2006;116(8):2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. Nature genetics. 1995;10(4):400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 17.Kanwar YS, Linker A, Farquhar MG. The Journal of cell biology. 1980;86(2):688–693. doi: 10.1083/jcb.86.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wijnhoven TJ, Lensen JF, Wismans RG, Lefeber DJ, Rops AL, van der Vlag J, Berden JH, van den Heuvel LP, van Kuppevelt TH. J Am Soc Nephrol. 2007;18(12):3119–3127. doi: 10.1681/ASN.2007020198. [DOI] [PubMed] [Google Scholar]
- 19.van den Hoven MJ, Wijnhoven TJ, Li JP, Zcharia E, Dijkman HB, Wismans RG, Rops AL, Lensen JF, van den Heuvel LP, van Kuppevelt TH, Vlodavsky I, Berden JH, van der Vlag J. Kidney international. 2008;73(3):278–287. doi: 10.1038/sj.ki.5002706. [DOI] [PubMed] [Google Scholar]
- 20.Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. J Am Soc Nephrol. 2009;20(7):1491–1503. doi: 10.1681/ASN.2008101117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reeves W, Caulfield JP, Farquhar MG. Laboratory investigation; a journal of technical methods and pathology. 1978;39(2):90–100. [PubMed] [Google Scholar]
- 22.Torres-Rasgado E, Fouret G, Carbonneau MA, Leger CL. Free radical research. 2007;41(3):367–375. doi: 10.1080/10715760601064706. [DOI] [PubMed] [Google Scholar]
- 23.Theilig F, Kriz W, Jerichow T, Schrade P, Hahnel B, Willnow T, Le Hir M, Bachmann S. J Am Soc Nephrol. 2007;18(6):1824–1834. doi: 10.1681/ASN.2006111266. [DOI] [PubMed] [Google Scholar]
- 24.Kerjaschki D, Sharkey DJ, Farquhar MG. The Journal of cell biology. 1984;98(4):1591–1596. doi: 10.1083/jcb.98.4.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerjaschki D, Vernillo AT, Farquhar MG. The American journal of pathology. 1985;118(3):343–349. [PMC free article] [PubMed] [Google Scholar]
- 26.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Molecular cell. 1998;1(4):575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 27.Khoshnoodi J, Sigmundsson K, Ofverstedt LG, Skoglund U, Obrink B, Wartiovaara J, Tryggvason K. The American journal of pathology. 2003;163(6):2337–2346. doi: 10.1016/S0002-9440(10)63590-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB. The Journal of biological chemistry. 2003;278(21):19266–19271. doi: 10.1074/jbc.M301279200. [DOI] [PubMed] [Google Scholar]
- 29.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K. The Journal of clinical investigation. 2004;114(10):1475–1483. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. Nature genetics. 2000;24(4):349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 31.Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Molecular and cellular biology. 2007;27(24):8698–8712. doi: 10.1128/MCB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein PL, Vogel H, Soriano P. Genes & development. 1994;8(17):1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 33.Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K. Nature medicine. 2008;14(3):290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F. J Am Soc Nephrol. 21(10):1657–1666. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wharram BL, Goyal M, Gillespie PJ, Wiggins JE, Kershaw DB, Holzman LB, Dysko RC, Saunders TL, Samuelson LC, Wiggins RC. The Journal of clinical investigation. 2000;106(10):1281–1290. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scaglia F, Vogel H, Hawkins EP, Vladutiu GD, Liu LL, Wong LJ. American journal of medical genetics. 2003;123A(2):172–178. doi: 10.1002/ajmg.a.20315. [DOI] [PubMed] [Google Scholar]
- 37.Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, Yudkoff M, Hancock WW, Meade R, Saiki R, Lunceford AL, Clarke CF, Gasser DL. PLoS genetics. 2008;4(4):e1000061. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kazama I, Mahoney Z, Miner JH, Graf D, Economides AN, Kreidberg JA. J Am Soc Nephrol. 2008;19(11):2181–2191. doi: 10.1681/ASN.2007111212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stitt-Cavanagh EM, Faour WH, Takami K, Carter A, Vanderhyden B, Guan Y, Schneider A, Breyer MD, Kennedy CR. J Am Soc Nephrol. 21(10):1678–1690. doi: 10.1681/ASN.2009121234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P. The Journal of clinical investigation. 2005;115(5):1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann S, Podlich D, Hahnel B, Kriz W, Gretz N. J Am Soc Nephrol. 2004;15(6):1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 42.Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y. J Am Soc Nephrol. 2009;20(9):1997–2008. doi: 10.1681/ASN.2009010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Economou CG, Kitsiou PV, Tzinia AK, Panagopoulou E, Marinos E, Kershaw DB, Kerjaschki D, Tsilibary EC. Journal of cell science. 2004;117(Pt 15):3281–3294. doi: 10.1242/jcs.01163. [DOI] [PubMed] [Google Scholar]
- 44.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. The Journal of clinical investigation. 2003;111(5):707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, Aucouturier P, Moskaug JO, Otto A, Christensen EI, Willnow TE. The American journal of pathology. 1999;155(4):1361–1370. doi: 10.1016/S0002-9440(10)65238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pozzi A, Jarad G, Moeckel GW, Coffa S, Zhang X, Gewin L, Eremina V, Hudson BG, Borza DB, Harris RC, Holzman LB, Phillips CL, Fassler R, Quaggin SE, Miner JH, Zent R. Developmental biology. 2008;316(2):288–301. doi: 10.1016/j.ydbio.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber TB, Kwoh C, Wu H, Asanuma K, Godel M, Hartleben B, Blumer KJ, Miner JH, Mundel P, Shaw AS. The Journal of clinical investigation. 2006;116(5):1337–1345. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai C, Saleem MA, Holzman LB, Mathieson P, Liu Y. Kidney international. 77(11):962–973. doi: 10.1038/ki.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaufman L, Potla U, Coleman S, Dikiy S, Hata Y, Kurihara H, He JC, D’Agati VD, Klotman PE. The Journal of biological chemistry. 285(33):25677–25685. doi: 10.1074/jbc.M110.133959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garg P, Verma R, Cook L, Soofi A, Venkatareddy M, George B, Mizuno K, Gurniak C, Witke W, Holzman LB. The Journal of biological chemistry. 285(29):22676–22688. doi: 10.1074/jbc.M110.122929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tryggvason K, Pikkarainen T, Patrakka J. Cell. 2006;125(2):221–224. doi: 10.1016/j.cell.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. The Journal of clinical investigation. 2006;116(5):1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, Huang H, Larose L, Li SS, Takano T, Quaggin SE, Pawson T. Nature. 2006;440(7085):818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 54.Frischknecht F, Moreau V, Rottger S, Gonfloni S, Reckmann I, Superti-Furga G, Way M. Nature. 1999;401(6756):926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- 55.Scaplehorn N, Holmstrom A, Moreau V, Frischknecht F, Reckmann I, Way M. Curr Biol. 2002;12(9):740–745. doi: 10.1016/s0960-9822(02)00812-6. [DOI] [PubMed] [Google Scholar]
- 56.Yu CC, Yen TS, Lowell CA, DeFranco AL. Curr Biol. 2001;11(1):34–38. doi: 10.1016/s0960-9822(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 57.Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, Wiggins RC, Killen P, Holzman LB. The Journal of biological chemistry. 2003;278(23):20716–20723. doi: 10.1074/jbc.M301689200. [DOI] [PubMed] [Google Scholar]
- 58.Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, Mathur BN, Turner CA, Geske R, Montgomery CA, Starbuck M, Brandt M, Gupta A, Ramirez-Solis R, Zambrowicz BP, Powell DR. Molecular and cellular biology. 2001;21(14):4829–4836. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciani L, Patel A, Allen ND, ffrench-Constant C. Molecular and cellular biology. 2003;23(10):3575–3582. doi: 10.1128/MCB.23.10.3575-3582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashworth S, Teng B, Kaufeld J, Miller E, Tossidou I, Englert C, Bollig F, Staggs L, Roberts IS, Park JK, Haller H, Schiffer M. PloS one. 5(9):e12626. doi: 10.1371/journal.pone.0012626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, Walz G, Haller H, Schiffer M. J Am Soc Nephrol. 2009;20(4):798–806. doi: 10.1681/ASN.2008080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirose T, Satoh D, Kurihara H, Kusaka C, Hirose H, Akimoto K, Matsusaka T, Ichikawa I, Noda T, Ohno S. PloS one. 2009;4(1):e4194. doi: 10.1371/journal.pone.0004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS. Science (New York, NY. 2003;300(5623):1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 64.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR. Nature genetics. 2000;24(3):251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 65.Winn MP, Daskalakis N, Spurney RF, Middleton JP. J Am Soc Nephrol. 2006;17(2):378–387. doi: 10.1681/ASN.2005090962. [DOI] [PubMed] [Google Scholar]
- 66.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. Nature genetics. 2005;37(7):739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. Science (New York, NY. 2005;308(5729):1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 68.Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Nature genetics. 42(1):72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peti-Peterdi J, Sipos A. J Am Soc Nephrol. 21(11):1835–1841. doi: 10.1681/ASN.2010040378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma H, Togawa A, Soda K, Zhang J, Lee S, Ma M, Yu Z, Ardito T, Czyzyk J, Diggs L, Joly D, Hatakeyama S, Kawahara E, Holzman L, Guan JL, Ishibe S. J Am Soc Nephrol. 21(7):1145–1156. doi: 10.1681/ASN.2009090991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J. Nature medicine. 2008;14(1):55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 72.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. Nature medicine. 2008;14(9):931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee DB, Huang E, Ward HJ. American journal of physiology. 2006;290(1):F20–34. doi: 10.1152/ajprenal.00052.2005. [DOI] [PubMed] [Google Scholar]
- 74.Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. The American journal of pathology. 2008;172(2):299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strutz FM. Kidney international. 2009;75(5):475–481. doi: 10.1038/ki.2008.425. [DOI] [PubMed] [Google Scholar]
- 76.Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS. J Am Soc Nephrol. 2009;20(3):593–603. doi: 10.1681/ASN.2008040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, Andre JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Frund S, Illies F, Joseph M, Kaitila I, Lama G, Loirat C, McLeod DR, Milford DV, Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J, Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW. Nature genetics. 2002;30(2):215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 78.Srichai MB, Konieczkowski M, Padiyar A, Konieczkowski DJ, Mukherjee A, Hayden PS, Kamat S, El-Meanawy MA, Khan S, Mundel P, Lee SB, Bruggeman LA, Schelling JR, Sedor JR. The Journal of biological chemistry. 2004;279(14):14398–14408. doi: 10.1074/jbc.M314155200. [DOI] [PubMed] [Google Scholar]
- 79.Quaggin SE. Microscopy research and technique. 2002;57(4):208–211. doi: 10.1002/jemt.10076. [DOI] [PubMed] [Google Scholar]
- 80.Rohr C, Prestel J, Heidet L, Hosser H, Kriz W, Johnson RL, Antignac C, Witzgall R. The Journal of clinical investigation. 2002;109(8):1073–1082. doi: 10.1172/JCI13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miner JH, Morello R, Andrews KL, Li C, Antignac C, Shaw AS, Lee B. The Journal of clinical investigation. 2002;109(8):1065–1072. doi: 10.1172/JCI13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chugh SS. Transl Res. 2007;149(5):237–242. doi: 10.1016/j.trsl.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suleiman H, Heudobler D, Raschta AS, Zhao Y, Zhao Q, Hertting I, Vitzthum H, Moeller MJ, Holzman LB, Rachel R, Johnson R, Westphal H, Rascle A, Witzgall R. Developmental biology. 2007;304(2):701–712. doi: 10.1016/j.ydbio.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 84.Dressler GR, Woolf AS. The International journal of developmental biology. 1999;43(5):463–468. [PubMed] [Google Scholar]
- 85.Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT, Williams-Simons L, Westphal H. Nature. 1993;362(6415):65–67. doi: 10.1038/362065a0. [DOI] [PubMed] [Google Scholar]
- 86.Ohtaka A, Ootaka T, Sato H, Soma J, Sato T, Saito T, Ito S. Am J Kidney Dis. 2002;39(3):475–485. doi: 10.1053/ajkd.2002.31391. [DOI] [PubMed] [Google Scholar]
- 87.Lefevre GM, Patel SR, Kim D, Tessarollo L, Dressler GR. PLoS genetics. 6(10):e1001142. doi: 10.1371/journal.pgen.1001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Y, Jeanpierre C, Dressler GR, Lacoste M, Niaudet P, Gubler MC. The American journal of pathology. 1999;154(1):181–192. doi: 10.1016/S0002-9440(10)65264-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, Hammes A, Hastie ND, Schedl A. Human molecular genetics. 2002;11(6):651–659. doi: 10.1093/hmg/11.6.651. [DOI] [PubMed] [Google Scholar]
- 90.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstadt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB. The Journal of clinical investigation. 120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harris DP, Vogel P, Wims M, Moberg K, Humphries J, Jhaver KG, Dacosta CM, Shadoan MK, Xu N, Hansen GM, Balakrishnan S, Domin J, Powell DR, Oravecz T. Molecular and cellular biology. doi: 10.1128/MCB.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Muller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’Toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nurnberg P, Hildebrandt F. Nature genetics. 2006;38(12):1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 93.van den Berg JG, van den Bergh Weerman MA, Assmann KJ, Weening JJ, Florquin S. Kidney international. 2004;66(5):1901–1906. doi: 10.1111/j.1523-1755.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- 94.Kalluri R. J Am Soc Nephrol. 2006;17(9):2383–2389. doi: 10.1681/ASN.2006060628. [DOI] [PubMed] [Google Scholar]
- 95.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. J Am Soc Nephrol. 2005;16(10):2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 96.Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, Kuick RD, Wiggins RC. J Am Soc Nephrol. 2005;16(10):2953–2966. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- 97.Wiggins RC. Kidney international. 2007;71(12):1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 98.Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Human molecular genetics. 1997;6(6):831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- 99.Regele HM, Fillipovic E, Langer B, Poczewki H, Kraxberger I, Bittner RE, Kerjaschki D. J Am Soc Nephrol. 2000;11(3):403–412. doi: 10.1681/ASN.V113403. [DOI] [PubMed] [Google Scholar]
- 100.Adler S, Chen X. The American journal of physiology. 1992;262(5 Pt 2):F770–776. doi: 10.1152/ajprenal.1992.262.5.F770. [DOI] [PubMed] [Google Scholar]
- 101.Kang YS, Li Y, Dai C, Kiss LP, Wu C, Liu Y. Kidney international. 78(4):363–373. doi: 10.1038/ki.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.El-Aouni C, Herbach N, Blattner SM, Henger A, Rastaldi MP, Jarad G, Miner JH, Moeller MJ, St-Arnaud R, Dedhar S, Holzman LB, Wanke R, Kretzler M. J Am Soc Nephrol. 2006;17(5):1334–1344. doi: 10.1681/ASN.2005090921. [DOI] [PubMed] [Google Scholar]
- 103.Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. The Journal of biological chemistry. 1995;270(28):16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- 104.Maunsbach AB. Journal of ultrastructure research. 1966;15(3):197–241. doi: 10.1016/s0022-5320(66)80108-9. [DOI] [PubMed] [Google Scholar]
- 105.Kerjaschki D, Exner M, Ullrich R, Susani M, Curtiss LK, Witztum JL, Farquhar MG, Orlando RA. The Journal of clinical investigation. 1997;100(9):2303–2309. doi: 10.1172/JCI119768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ramanujam KS, Seetharam S, Ramasamy M, Seetharam B. Biochimica et biophysica acta. 1990;1030(1):157–164. doi: 10.1016/0005-2736(90)90251-i. [DOI] [PubMed] [Google Scholar]
- 107.Hammad SM, Barth JL, Knaak C, Argraves WS. The Journal of biological chemistry. 2000;275(16):12003–12008. doi: 10.1074/jbc.275.16.12003. [DOI] [PubMed] [Google Scholar]
- 108.Birn H, Fyfe JC, Jacobsen C, Mounier F, Verroust PJ, Orskov H, Willnow TE, Moestrup SK, Christensen EI. The Journal of clinical investigation. 2000;105(10):1353–1361. doi: 10.1172/JCI8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amsellem S, Gburek J, Hamard G, Nielsen R, Willnow TE, Devuyst O, Nexo E, Verroust PJ, Christensen EI, Kozyraki R. J Am Soc Nephrol. 21(11):1859–1867. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nielsen R, Christensen EI. Pediatric nephrology (Berlin, Germany) 25(5):813–822. doi: 10.1007/s00467-009-1381-9. [DOI] [PubMed] [Google Scholar]