

The crystal structure of an extremely thermostable multicopper oxidase from a hyperthermophile was determined.
Keywords: hyperthermophiles, Pyrobaculum aerophilum, multicopper oxidases, archaea
Abstract
The crystal structure of an extremely thermostable multicopper oxidase (McoP) from the hyperthermophilic archaeon Pyrobaculum aerophilum was determined at a resolution of 2.0 Å. The overall fold was comprised of three cupredoxin-like domains and the main-chain coordinates of the enzyme were similar to those of multicopper oxidases from Escherichia coli (CueO) and Bacillus subtilis (CotA). However, there were clear topological differences around domain 3 between McoP and the other two enzymes: a methionine-rich helix in CueO and a protruding helix in CotA were not present in McoP. Instead, a large loop (PL-1) covered the T1 copper centre of McoP and a short α-helix in domain 3 extended near the N-terminal end of PL-1. In addition, the sizes of several surface loops in McoP were markedly smaller than the corresponding loops in CueO and CotA. Structural comparison revealed that the presence of extensive hydrophobic interactions and a smaller cavity volume are likely to be the main factors contributing to the hyperthermostability of McoP.
1. Introduction
Multicopper oxidases (MCOs) are a large family of enzymes that couple the four-electron reduction of oxygen to water with the oxidation of a broad range of organic and/or inorganic substrates (Claus, 2004 ▶; Solomon et al., 1996 ▶). MCOs often contain four Cu atoms, which are classified as type 1 (T1), type 2 (T2) or type 3 (T3). The mononuclear T1 centre of an MCO mediates the intramolecular transfer of one electron from a substrate to a trinuclear T2–T3 centre formed by a T2 Cu coordinated with a T3 Cu pair. The T2–T3 centre subsequently reduces molecular oxygen to water. Because of their wide reaction capabilities, as well as their broad substrate specificity, these enzymes possess a great deal of biotechnological potential, for example in dye bleaching (Claus et al., 2002 ▶) and in the production of polymers (Hüttermann et al., 2001 ▶) and of biosensors (Peter & Wollenberger, 1997 ▶).
Recently, several MCOs have been detected in various thermophiles and hyperthermophiles (Fernandes et al., 2007 ▶, 2010 ▶; Miyazaki, 2005 ▶). Biochemical characterization of these enzymes showed that they are highly stable and have great potential for industrial application. Among them was an MCO from the hyperthermophilic archaeon Pyrobaculum aerophilum (McoP), which reportedly exhibits extreme thermostability and a half-life for inactivation of ∼6 h at 353 K (Fernandes et al., 2010 ▶). Using comparative modelling techniques, a structural model of the P. aerophilum enzyme was derived (Fernandes et al., 2010 ▶). The model revealed McoP to have the same overall fold as two other MCOs assembled from three cupredoxin domains: CueO from Escherichia coli and CotA from Bacillus subtilis. On the other hand, the structural features underlying the thermostability of McoP were not reported. In the present study, we examined the same protein from P. aerophilum and succeeded in determining its crystal structure. This is the first crystal structure of an MCO from a hyperthermophile. Through comparison with CueO and CotA, we also evaluated the structural features responsible for the high thermostability of McoP.
2. Materials and methods
2.1. Overexpression and purification of recombinant protein
We initially carried out PCR using the following pair of oligonucleotide primers to amplify a PAE1888 gene fragment (without the first 87 nucleotides): 5′-CATATGACTGGTGAAGTCAAGAGGCCTG-3′, which contains a unique NdeI restriction site overlapping the 5′ initial codon, and 5′-CTTGGATCCTTATTTAACTGCTATGTTT-3′, which contains a unique BamHI restriction site proximal to the 3′-end of the termination codon. Chromosomal DNA prepared from P. aerophilum using a genomic DNA isolation kit for bacteria (Nexttec GmbH Biotechnologie, Leverkusen, Germany) served as the template. The amplified 1.4 kb fragment was digested with NdeI and BamHI and ligated into the expression vector pET-11a linearized with NdeI and BamHI, yielding pET1888. E. coli strain BL21 (DE3) Codon Plus RIL (Stratagene) was then transformed with pET1888, after which the transformants were cultivated at 310 K in 0.5 l of a medium consisting of 6 g tryptone, 12 g yeast extract, 2.5 ml glycerol, 6.25 g K2HPO4, 1.9 g KH2PO4 and 50 µg ml−1 ampicillin until the optical density at 600 nm reached 0.6. Expression was then induced by adding 0.1 mM isopropyl β-d-1-thiogalactopyranoside to the medium and cultivation was continued for an additional 21 h at 293 K. The cells were then harvested by centrifugation, suspended in 10 mM Tris–HCl pH 7.2 and lysed by sonication. After centrifugation (15 000g for 20 min) of the lysate, CuSO4 was added to the supernatant to a concentration of 1 mM and the solution was incubated for 16 h at 277 K.
To isolate McoP, the enzyme solution was heated to 353 K for 10 min and denatured protein was removed by centrifugation (15 000g for 10 min). The resultant supernatant was loaded onto a DEAE-Toyopearl 650M column (Tosoh, Japan) equilibrated with 10 mM Tris–HCl pH 7.2 and the column was washed with the same buffer. The enzyme was eluted without absorption. The active fractions in the flowthrough were pooled and loaded onto a HiPrep 16/10 SP XL column (GE Healthcare) equilibrated with 10 mM Tris–HCl pH 7.2. After washing the column with the same buffer, the active fractions in the flowthrough were collected, concentrated by ultrafiltration and subjected to gel filtration on a Superdex 200 26/60 column (GE Healthcare) equilibrated with 10 mM Tris–HCl pH 7.2. The entire procedure was carried out at room temperature (∼298 K). The enzyme activity was determined at 323 K in 100 mM sodium acetate buffer pH 3 using 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) as a substrate (Miyazaki, 2005 ▶).
For the expression of selenomethionyl McoP, E. coli BL21 (DE3) Codon Plus RIL cells were transformed with pET1888 in a modified M9 medium containing selenomethionine (Doublié, 1997 ▶). The overexpression and purification of selenomethionyl McoP were performed using the same procedures as used for the native enzyme.
2.2. Crystallization and structure determination
The crystallization of native and selenomethionyl McoP was performed using the sitting-drop vapour-diffusion method. Drops (1 µl) of protein solution (1.4 mg ml−1) were mixed with an equal volume of 0.2 M ammonium acetate, 0.1 M trisodium citrate dihydrate pH 5.6 and 30%(v/v) PEG 4000 and equilibrated against 0.1 ml reservoir solution. The crystals grew in 6 d at 293 K. Selenium multiple-wavelength anomalous dispersion data and native diffraction data were collected using an ADSC CCD detector system on the BL5A beamline at the Photon Factory, Tsukuba, Japan. All measurements were carried out on crystals cryoprotected with Paratone-N (Hampton Research) and cooled to 100 K in a stream of nitrogen gas. The data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). SOLVE (Terwilliger & Berendzen, 1999 ▶) was used to refine the parameters of the Se atoms and to calculate the phases. The initial phase was improved by solvent flattering followed by autotracing using RESOLVE (Terwilliger, 1999 ▶). The model was built using Coot (Emsley & Cowtan, 2004 ▶) and XtalView (McRee, 1999 ▶). Refinement of the model structure was first carried out for the selenomethionyl protein diffraction data and continued for the native diffraction data using REFMAC5 (Murshudov et al., 2011 ▶) and CNS (Brünger et al., 1998 ▶). After several cycles of inspection of the 2F o − F c and F o − F c density maps, the model was rebuilt. A Cu—O—Cu bridge (T3 centre), two Cu atoms (T1 and T2 centres), one acetate molecule and 218 water molecules were included in the model. The N-terminal region (amino-acid residues 30–39) was disordered and was not visible in the electron-density map. The model geometry was analyzed using RAMPAGE (Lovell et al., 2003 ▶) and all of the main-chain atoms except His106 fell within the favourable region of the Ramachandran plot. Moreover, it was confirmed on the basis of the electron-density maps that the conformation of His106 was correct. The R-factor and R free values for the final model were 20.2% and 22.4%, respectively (Table 1 ▶).
Table 1. Data-collection and refinement statistics.
(a).
Data collection. Values in parentheses are for the highest resolution data shell in each data set.
| SeMet | ||||
|---|---|---|---|---|
| Native | Peak | Edge | Remote | |
| Space group | I41 | I41 | ||
| Unit-cell parameters () | a = 103.9, b = 103.9, c = 122.1 | a = 103.4, b = 103.4, c = 121.5 | ||
| Wavelength | 1.0 | 0.9791 | 0.9794 | 0.9640 |
| Maximum resolution () | 2.0 (2.12.0) | 1.9 (2.01.9) | 1.9 (2.01.9) | 1.9 (2.01.9) |
| Total No. of reflections | 661499 | 698907 | 703103 | 705288 |
| No. of unique reflections | 44415 | 92917 | 93005 | 93214 |
| Multiplicity | 14.9 (12.9) | 7.5 (5.9) | 7.6 (5.9) | 7.6 (5.9) |
| Completeness (%) | 99.9 (99.7) | 99.7 (97.9) | 99.2 (93.0) | 99.4 (94.9) |
| R merge † (%) | 5.2 (18.7) | 4.6 (14.6) | 4.5 (17.0) | 4.5 (21.4) |
| I/(I) | 20.4 (15.1) | 26.0 (16.3) | 23.1 (14.0) | 21.2 (10.9) |
(b).
Refinement. The refinement statistics for CotA and CueO are also indicated. R.m.s.d., root-mean-square deviation.
| McoP | CotA (1gsk) | CueO (1kv7) | |
|---|---|---|---|
| Resolution range () | 26.32.0 | 87.71.7 | 261.4 |
| R/R free ‡ (%) | 20.2/22.4 (21.7/24.2) | 17.7/19.8 | 18.5/22.1 |
| No. of protein atoms | 3425 | 4044 | 3561 |
| No. of water molecules | 218 | 480 | 507 |
| R.m.s.d. | |||
| Bond lengths () | 0.036 | 0.019 | 0.010 |
| Bond angles () | 1.8 | 1.8 | 0.026 |
| Average B factor (2) | 32.3 | 25.9 | 20.3 |
| Ramachandran statistics (%) | |||
| Favoured | 96.3 | 96.6 | 97.2 |
| Allowed | 3.4 | 3.4 | 2.8 |
| Outliers | 0.2 | 0 | 0 |
R
merge = 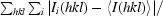
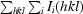 , where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl, I(hkl) is the mean value and the summation is over all measurements.
, where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl, I(hkl) is the mean value and the summation is over all measurements.
R free was calculated with randomly selected reflections (5%).
Hydrogen bonds were identified using the program CCP4mg (Potterton et al., 2002 ▶). Ion pairs (with a cutoff distance of 4.0 Å) and hydrophobic interactions were identified using the WHAT IF web server (Rodriguez et al., 1998 ▶). The solvent-accessible surface area (ASA; determined using a radius of 1.4 Å for the probe solvent molecule) was calculated using AREAIMOL in the CCP4 program suite (Winn et al., 2011 ▶). The volumes of the cavities within the structure were analyzed using Swiss-PDBViewer (Guex & Peitsch, 1997 ▶; http://www.expasy.org/spdbv/). A drawing quality of 6 (grid size of 0.47 Å) was used for the calculation.
3. Results and discussion
3.1. Overall structure, copper centres and structural homologues
The structure of McoP was determined at a resolution of 2.0 Å. The asymmetric unit contained one protein molecule, which gave a crystal volume per enzyme mass (V M) of 3.3 Å3 Da−1 and a solvent content of 62.7%. The overall fold contained three cupredoxin-like domains (Figs. 1 ▶ and 2 ▶): domain 1 (residues 40–155) was comprised of three short helices and ten strands organized into a β-barrel form, domain 2 (residues 160–321) was comprised of one short helix and a β-barrel composed of 12 strands, and domain 3 (residues 340–477) was comprised of four short helices and a β-barrel composed of ten strands and also contained the mononuclear copper centre (T1). The trinuclear copper centre (T2 and T3) was located at the interface between domains 1 and 3 (Fig. 1 ▶). In addition, one acetate molecule was bound to the side chain of Arg410 in helix α5.
Figure 1.

Overall structure of McoP. Domains 1, 2 and 3 are shown in blue, green and red, respectively. PL-1 and PL-2 are labelled in green and red, respectively. The linker peptides connecting the three domains are shown in yellow. Cu and O atoms in the T3 site are represented as orange and red balls, respectively. Cu atoms in the T1 and T2 sites are represented as magenta and cyan balls, respectively.
Figure 2.

Structure-based amino-acid sequence alignment of McoP, CueO and CotA. Asterisks indicate conserved residues. The residues comprising PL-1 and PL-2 in McoP are shown in red and those comprising EL-1–3 in CueO and BL-1–7 in CotA are shown in blue. The secondary-structural assignments for McoP are shown above the alignment. Domains 1 (blue), 2 (green) and 3 (red) are represented as solid bars over the secondary structure.
The copper-binding sites in McoP had the geometry typical of these sites in other MCOs. The T1 copper centre was coordinated by two histidine residues (His391 and His465), one cysteine residue (Cys460) and one methionine residue (Met470). The trinuclear copper centre was coordinated by eight histidine residues arranged as four His-X-His motifs. The two Cu atoms in the T3 site were coordinated by six of these histidine residues (His94, His132, His461, His134, His396 and His459); the remaining two (His92 and His394) were involved in coordinating the T2 Cu atom.
We next used the DALI server (Holm & Sander, 1998 ▶) to find proteins similar to our model McoP structure. The proteins with the greatest structural similarity to McoP were CueO from E. coli (r.m.s.d. of 2.0–2.1 Å), the SufI cell-division protein from E. coli (r.m.s.d. of 1.9–2.0 Å) and the CotA protein from B. subtilis (r.m.s.d. of 2.0 Å). Among these, SufI is structurally related to the multicopper oxidase superfamily but lacks metal cofactors (Tarry et al., 2009 ▶). Thus, the structure of McoP is closer to those of CueO (PDB entry 1kv7; Roberts et al., 2002 ▶) and CotA (PDB entry 1gsk; Enguita et al., 2003 ▶).
3.2. Structural comparison with CueO and CotA
As previously predicted from comparative modelling (Fernandes et al., 2010 ▶), the basic domain organization of McoP was similar to that of CueO and CotA. However, there were clear topological differences around domain 3 between McoP and the other two enzymes: (i) a methionine-rich helix and loop (EL-3; residues 356–379 of CueO) lying over the T1 copper centre in CueO was not observed in McoP (Fig. 3 ▶ a), (ii) a protruding section (BL-7; residues 436–453 of CotA) formed by a loop and a short α-helix lying over the substrate-binding site in CotA was also absent from McoP (Fig. 3 ▶ b), (iii) the T1 copper centre in McoP was covered by a large loop (PL-1; residues 289–307) protruding from domain 2 (Figs. 2 ▶ and 3 ▶) and (iv) a loop containing a short α-helix (α5) (PL-2; residues 408–420) in domain 3 extended near the N-terminal end of PL-1 in McoP. The presence of helix α5 increased the interactions between domains 2 and 3, as described below. Based on the comparative modelling method, the residues contributing to the occlusion of the T1 site in McoP have been predicted to be Trp355 (which corresponds to Asn408 in CueO and Leu386 in CotA), Met389 (which is structurally equivalent to Met441 of CueO), Met297 (which is in a similar position to Met303 of CueO) and Glu296 (which is in a similar position to Gln302 of CueO) (Fernandes et al., 2010 ▶). In the crystal structure of McoP, however, both Met297 and Glu296 are located within PL-1. Thus, the residues which correspond to Met297 and Glu296 are absent in CueO. On the other hand, we found a marked reduction in the size of several surface loops in McoP compared with the corresponding loops in CueO and CotA. The residues absent from the loop regions of McoP correspond to residues 42–47 (EL-1) and 54–66 (EL-2) of CueO (Figs. 2 ▶ and 3 ▶ a) and to residues 18–27 (BL-1), 35–48 (BL-2), 77–99 (BL-3), 127–136 (BL-4), 210–226 (BL-5), 378–383 (BL-6) and 436–453 (BL-7) of CotA (Figs. 2 ▶ and 3 ▶ b).
Figure 3.

Comparison of the structure of McoP with those of CueO and CotA. (a) The structure of CueO (yellow) is superimposed on that of McoP (green). PL-1 and PL-2 in McoP are shown in red and EL-1–3 in CueO are shown in blue. (b) The structure of CotA (yellow) is superimposed on that of McoP (green). PL-1 and PL-2 in McoP are shown in red and BL-1–7 in CotA are shown in blue. Cu and O atoms are represented as orange and red balls, respectively.
3.3. Structural features underlying thermostability
McoP has been reported to be a highly thermostable enzyme which shows a half-life for inactivation of ∼6 h at 353 K (Fernandes et al., 2010 ▶). In contrast, CotA and CueO exhibit much lower thermostability and their half-lives for inactivation are 2 h and 10–15 min, respectively, at the same temperature (Kim et al., 2001 ▶; Martins et al., 2002 ▶). Structural studies of hyperthermophilic proteins have suggested that greater numbers of ion pairs and hydrophobic interactions are mainly responsible for their high thermostability (Bhuiya et al., 2005 ▶; Hennig et al., 1995 ▶; Yip et al., 1995 ▶). We therefore determined the numbers of these interactions within the structure of McoP. We identified a total of 70 ion pairs (cutoff distance 4.0 Å) in McoP, which is somewhat fewer than in CueO (72) and CotA (79). In contrast, there were a total of 910 hydrophobic interactions in McoP, which was significantly more than in CueO (892) or CotA (814). In McoP we observed a large hydrophobic cluster formed by ten residues (Leu230, Phe265, Phe290, Pro390, Pro409, Val412, Leu415, Leu423, Leu434 and Trp436) at the interface between domains 2 and 3. Seven of these residues (Pro390, Pro409, Val412, Leu415, Leu423, Leu434 and Trp436) are derived from domain 3 and the remainder (Leu230, Phe265, and Phe290) are from domain 2. It is noteworthy that Pro409, Val412 and Leu415, which are located within α5 in domain 3, are situated near the N-terminal end of PL-1 in domain 2. This means that the presence of α5 leads to the formation of additional hydrophobic interactions and strengthens the interdomain interaction within McoP.
It has also been shown that smaller solvent-accessible surface areas (ASAs), smaller overall volumes and smaller cavity volumes all make large contributions to protein thermostability (Chan et al., 1995 ▶; Dalhus et al., 2002 ▶). The calculated ASA for McoP (16 264 Å2) was smaller than those for CueO (16 823 Å2) and CotA (19 885 Å2) and the overall volume of McoP (55 106 Å3) was also smaller than those of CueO and CotA (57 766 and 64 165 Å3, respectively). This may reflect the smaller size of the surface loops in McoP, as judged from a comparison with the corresponding loops in CueO and CotA (Fig. 2 ▶). However, CueO and CotA have 40–65 more residues than McoP. Moreover, the final models of CueO and CotA lack 26 and 11 residues, respectively, because of poor electron density. Therefore, direct comparison of ASA and overall volume among these enzymes may not be appropriate for estimation of the structural features responsible for thermostability. On the other hand, five cavities (>50 Å3) were found in McoP as well as in CotA, while seven cavities were found in CueO. The total volume of these cavities in McoP (291 Å3) was markedly smaller than in CotA (365 Å3) or CueO (760 Å3). Taken together, these observations suggest that the strength of its hydrophobic interactions and its compactness, as well as a reduction in the flexibility of its loop regions, all contribute to the greater overall stability of McoP.
Supplementary Material
PDB reference: multicopper oxidase, 3aw5
Acknowledgments
Data collection was performed on BL5A at the Photon Factory. We thank Drs Matsugaki, Igarashi and Wakatsuki for their kind assistance with the data collection. This work was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, the Thermostable Enzyme Laboratory Research Fund and the Novozymes Japan Research Fund 2010.
References
- Bhuiya, M. W., Sakuraba, H., Ohshima, T., Imagawa, T., Katunuma, N. & Tsuge, H. (2005). J. Mol. Biol. 345, 325–337. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chan, M. K., Mukund, S., Kletzin, A., Adams, M. W. & Rees, D. C. (1995). Science, 267, 1463–1469. [DOI] [PubMed]
- Claus, H. (2004). Micron, 35, 93–96. [DOI] [PubMed]
- Claus, H., Faber, G. & König, H. (2002). Appl. Microbiol. Biotechnol. 59, 672–678. [DOI] [PubMed]
- Dalhus, B., Saarinen, M., Sauer, U. H., Eklund, P., Johansson, K., Karlsson, A., Ramaswamy, S., Bjørk, A., Synstad, B., Naterstad, K., Sirevåg, R. & Eklund, H. (2002). J. Mol. Biol. 318, 707–721. [DOI] [PubMed]
- Doublié, S. (1997). Methods Enzymol. 276, 523–530. [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Enguita, F. J., Martins, L. O., Henriques, A. O. & Carrondo, M. A. (2003). J. Biol. Chem. 278, 19416–19425. [DOI] [PubMed]
- Fernandes, A. T., Damas, J. M., Todorovic, S., Huber, R., Baratto, M. C., Pogni, R., Soares, C. M. & Martins, L. O. (2010). FEBS J. 277, 3176–3189. [DOI] [PubMed]
- Fernandes, A. T., Soares, C. M., Pereira, M. M., Huber, R., Grass, G. & Martins, L. O. (2007). FEBS J. 274, 2683–2694. [DOI] [PubMed]
- Guex, N. & Peitsch, M. C. (1997). Electrophoresis, 18, 2714–2723. [DOI] [PubMed]
- Hennig, M., Darimont, B., Sterner, R., Kirschner, K. & Jansonius, J. N. (1995). Structure, 3, 1295–1306. [DOI] [PubMed]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res. 26, 316–319. [DOI] [PMC free article] [PubMed]
- Hüttermann, A., Mai, C. & Kharazipour, A. (2001). Appl. Microbiol. Biotechnol. 55, 387–394. [DOI] [PubMed]
- Kim, C., Lorenz, W. W., Hoopes, J. T. & Dean, J. F. (2001). J. Bacteriol. 183, 4866–4875. [DOI] [PMC free article] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Martins, L. O., Soares, C. M., Pereira, M. M., Teixeira, M., Costa, T., Jones, G. H. & Henriques, A. O. (2002). J. Biol. Chem. 277, 18849–18859. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Miyazaki, K. (2005). Extremophiles, 9, 415–425. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Peter, M. G. & Wollenberger, U. (1997). EXS, 80, 63–82. [DOI] [PubMed]
- Potterton, E., McNicholas, S., Krissinel, E., Cowtan, K. & Noble, M. (2002). Acta Cryst. D58, 1955–1957. [DOI] [PubMed]
- Roberts, S. A., Weichsel, A., Grass, G., Thakali, K., Hazzard, J. T., Tollin, G., Rensing, C. & Montfort, W. R. (2002). Proc. Natl Acad. Sci. USA, 99, 2766–2771. [DOI] [PMC free article] [PubMed]
- Rodriguez, R., Chinea, G., Lopez, N., Pons, T. & Vriend, G. (1998). Bioinformatics, 14, 523–528. [DOI] [PubMed]
- Solomon, E. I., Sundaram, U. M. & Machonkin, T. E. (1996). Chem. Rev. 96, 2563–2606. [DOI] [PubMed]
- Tarry, M., Arends, S. J., Roversi, P., Piette, E., Sargent, F., Berks, B. C., Weiss, D. S. & Lea, S. M. (2009). J. Mol. Biol. 386, 504–519. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (1999). Acta Cryst. D55, 1863–1871. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yip, K. S., Stillman, T. J., Britton, K. L., Artymiuk, P. J., Baker, P. J., Sedelnikova, S. E., Engel, P. C., Pasquo, A., Chiaraluce, R. & Consalvi, V. (1995). Structure, 3, 1147–1158. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: multicopper oxidase, 3aw5