

The design, expression and purification of receptor protein tyrosine phosphatase γ and two mutants and their crystallization in three different crystal forms are described.
Keywords: protein tyrosine phosphatases, hydrolases, inhibitor complexes
Abstract
Protein tyrosine phosphatase γ is a membrane-bound receptor and is designated RPTPγ. RPTPγ and two mutants, RPTPγ(V948I, S970T) and RPTPγ(C858S, S970T), were recombinantly expressed and purified for X-ray crystallographic studies. The purified enzymes were crystallized using the hanging-drop vapor-diffusion method. Crystallographic data were obtained from several different crystal forms in the absence and the presence of inhibitor. In this paper, a description is given of how three different crystal forms were obtained that were used with various ligands. An orthorhombic crystal form and a trigonal crystal form were obtained both with and without ligand, and a monoclinic crystal form was only obtained in the presence of a particularly elaborated inhibitor.
1. Introduction
Protein tyrosine phosphatases (PTPs) represent a large family of enzymes (Almo et al., 2007 ▶; Barr et al., 2009 ▶). They play a very important role in cellular signaling within and between cells. PTPs work antagonistically with protein tyrosine kinases to regulate signal transduction in cells. Protein tyrosine kinases phosphorylate tyrosine residues on a substrate protein and PTPs dephosphorylate tyrosine residues on a substrate protein. Since the phosphorylation status of a protein can modulate its function, PTKs and PTPs work together to regulate protein function in response to a variety of signals, including hormones, mitogens and oncogenes (Zhang et al., 2002 ▶). The rationale for receptor PTPγ (RPTPγ) as a target for drug design has been reviewed in the introduction to Appiah et al. (2011 ▶). This work was undertaken to aid a structure-based drug-design program.
2. Experimental procedures
2.1. Construct design
A schematic of the RPTPγ protein is shown in Fig. 1 ▶ (see, for example, Barnea et al., 1993 ▶). The intracellular portion extends from residues 761 to 1445 and contains two PTP-like domains: 1 and 2. Only domain 1 is catalytically active. For high-throughput screening (HTS), a construct was designed that was a 78 kDa protein consisting of 671 amino acids. It included domains 1 and 2 of RPTPγ, 16 non-native amino acids at the N-terminus and 37 amino acids of the full-length protein N-terminal to domain 1 (Appiah et al., 2011 ▶).
Figure 1.

Schematic diagram of the RPTPγ sequence. The hydrophobic signal peptide (residues 1–19) and the transmembrane domain (residues 737–760) are indicated by filled rectangles. The extracellular portion of RPTPγ includes a carbonic anhydrase homology domain (CAH) and a fibronectin III homology domain (FN). The intracellular portion of RPTPγ includes a protein tyrosine phosphatase catalytic domain (D1) and a protein tyrosine phosphatase-like domain (D2) in which the catalytic cysteine is replaced by an aspartate. Domains D1 and D2 as shown in the diagram include the α1′ and α2′ helices as defined by Barford et al. (1994 ▶), since PTPμ (PDB entry 1rpm) on which the construct design for PTPγ was based had sequences in these regions that were helical.
We pursued construct design because the protein used for HTS was suboptimal for structural studies. Protein recovery was poor during the buffer exchange which was necessary for direct binding experiments (either NMR or thermal shift assay; Sheriff et al., 2011 ▶) of HTS compounds. In addition, no crystals of this protein were obtained in broad screening trials.
The RPTPγ(825–1128) construct was designed based on a homology model using the human receptor protein tyrosine phosphatase μ structure (PDB entry 1rpm) as a template (Hoffmann et al., 1997 ▶), which begins at residue 827 of RPTPγ. Over 30 constructs were designed and made. The constructs incorporated various N- and C-termini based on the homology model and proteolytic digestion studies. Soluble expression was observed for constructs consisting of domain 1 or domain 2 but not both domains. The construct consisting of residues 825–1128 produced soluble protein and was further pursued for structural studies. Domain 1 (825–1128) produced approximately 20 mg isolated protein per litre of culture.
2.2. Cloning of RPTPγ, RPTPγ(V948I, S970T) and RPTPγ(C858S, S970T)
Two mutants were pursued to make a domain 1 construct whose sequence more closely resembled the sequence of the construct used for HTS and for enzymatic assay, which had V948I and S970T point mutations. A sample of plasmid DNA containing the RPTPγ gene (RefSeq NM_002841.3) encoding the cytoplasmic portion (amino acids 790–1445) of the RPTPγ protein was obtained from Yi Hu of Lexicon Pharmaceuticals Inc. PCR primers BamHI-487-M825-FP (5′-CCAGTGGATCCATGGAAGCCATTCCTGTCAAAC-3′) and KpnI-487-S1128-RP (5′-CCAGTGGTACCTCAAGATACTTCAGTCTCCTTTCCAAGA-3′), 5′-phosphorylated mutagenesis primers BCOL-52 (5′-P-AAACACTGGAATCATTATCATGATTACGAACC-3′) and BCOL-51 (5′-P-GCCAACAGAGAACACTGAGGAATATGGAA-3′), and mutagenesis primers BCOL-58 (5′-GAGGAAGTCCAGCGCTCTACTGCTGATATGAAC-3′) and BCOL-59 (5′-GTTCATATCAGCAGTAGAGCGCTGGACTTCCTC-3′) were obtained from Operon Biotechnologies Inc. (Huntsville, Alabama, USA). The coding sequence for RPTPγ amino acids 825–1128 was amplified from this plasmid template by PCR with Platinum PFX DNA polymerase (Invitrogen, Carlsbad, California, USA) with primers BamHI-487-M825-FP and KpnI-487-S1128-RP. The PCR product was cloned into a modified version of the commercially available pET28 vector (Novagen, Madison, Wisconsin, USA) between BamHI and KpnI restriction sites to give His-Tb-RPTPγ(825–1128)-pET28. The resulting expression vector expresses RPTPγ(825–1128) fused with an N-terminal hexahistidine tag and thrombin cleavage site, which was confirmed by sequencing analysis.
The V948I and S970T mutations were generated in His-Tb-RPTPγ(825–1128)-pET28 using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, Cedar Creek, Texas, USA) with primers BCOL-51 and BCOL-52. The final construct, His-Tb-RPTPγ(825–1128)-V948I-S970T-pET28, was confirmed by sequencing analysis. During the generation of His-Tb-RPTPγ(825–1128)-V948I-S970T-pET28 a secondary clone was obtained which was confirmed to be His-Tb-RPTPγ(825–1128)-S970T-pET28 by sequencing analysis. This clone was further mutagenized using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) with primers BCOL-58 and BCOL-59. The final construct, His-Tb-RPTPγ(825–1128)-C858S-S970T-pET28, was confirmed by sequencing analysis. The sequences are shown in Fig. 2 ▶.
Figure 2.

Amino-acid sequences of the (a) His-Tb-RPTPγ, (b) His-Tb-RPTPγ(825–1128)/(V948I, S970T) and (c) His-Tb-RPTPγ(825–1128)/(C858I, S970T) constructs used to produce protein for crystallization studies. The hexa-His tag and the thrombin cleavage site are underlined; the amino acids from residues 825 to 1128 of RPTPγ are not underlined. The first residue following thrombin cleavage is marked with a down arrow (↓). Residues mutated in the V948I, S970T and C858S, S970T constructs are denoted in red bold letters.
2.3. Expression and purification of RPTPγ
Transformed Escherichia coli Rosetta (DE3) cells (Novagen) were propagated in minimal medium (15N-labeled) overnight at 310 K. The culture was induced at OD600 = 1.0 with 0.25 mM IPTG. After induction, the temperature of the culture was lowered to 293 K and the cells were harvested after 30 h. All lysis, clarification and purification steps were performed at 277 K unless otherwise specified. The harvested cells from 4 l culture medium were suspended in 200 ml 25 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM dithiothreitol (DTT), 5 mM ethylenediamine tetraacetic acid (EDTA), 0.5 M urea and 1 ml protease-inhibitor cocktail (Sigma, St Louis, Missouri, USA). The cells were sonicated and the homogenate was clarified by centrifugation at 26 000g for 10 min (Sorval SS34 rotor). The supernatant was saturated with ammonium sulfate to 80% final concentration in an ice bath for 40 min while stirring. The precipitated protein was collected by centrifugation at 11 900g for 30 min (Sorval SS34 rotor), dissolved in 100 ml 25 mM Tris–HCl pH 8.0, 500 mM NaCl, 2 mM DTT, 1 ml protease-inhibitor cocktail (Sigma) and dialyzed overnight at 277 K against 2 l 25 mM Tris–HCl pH 8.0, 100 mM NaCl, 2 mM DTT, 5% glycerol, 1 mM PMSF. The protein solution after dialysis was applied onto a 15 ml Ni-charged affinity column (His-Ni-Select, affinity gel, Sigma, St Louis, Missouri, USA). The column was washed with two volumes of dialysis buffer, washed with 100 ml 1 M NaCl, 25 mM Tris–HCl pH 8.0, 2 mM DTT and washed with two volumes of 25 mM Tris–HCl pH 8.0, 2 mM DTT. The protein was eluted with 0.25 M imidazole. To remove the His tag, the protein (3 mg ml−1) was cleaved with human thrombin (10 units mg−1) at room temperature for 1 h. The protein was concentrated using a 30 000 Da molecular-weight cutoff membrane (Millipore Corporation, Bedford, Massachusetts, USA) and diluted twice with 25 mM Tris–HCl pH 7.5. The cleaved protein was passed through 5 ml Q Sepharose resin (GE Healthcare, Piscataway, New Jersey, USA). The flowthrough was applied onto 5 ml SP Sepharose resin (GE Healthcare). The SP Sepharose was washed with (two volumes of) low-salt buffer (100 mM NaCl, 25 mM Tris–HCl pH 7.5, 2 mM EDTA, 5 mM DTT) and the protein was eluted with 0.8 M NaCl, 25 mM Tris–HCl pH 7.5, 2 mM EDTA, 5 mM DTT. The eluted protein was passed through a 1 ml syringe containing HiTrap Benzamidine FF (GE Healthcare) and concentrated to 12 mg ml−1 using a filtering device with a 30 000 Da molecular-weight cutoff membrane (Millipore Corporation) and exchanged into the final buffer: 25 mM Tris pH 7.5, 200 mM NaCl, 2 mM EDTA, 5 mM DTT. Typical yields were 40 mg per litre of culture. The protein was used immediately for crystallization trials or stored at 193 K with 10% glycerol. Protein concentrations were determined using guanidine–HCl and an extinction coefficient of 43 720 M −1 cm−1. The protein purity was ≥95%. The identity of the purified protein was confirmed by mass spectrometry, with an observed mass of 36 097 Da for 15N-labeled protein compared with a theoretical mass of 36 101 Da.
2.4. Expression and purification of RPTPγ(V948I, S970T)
The RPTPγ(V948I, S970T) plasmid was transformed using chemically competent E. coli BL21 (DE3) cells (Novagen). Transformants were picked, inoculated into starter cultures and expanded into large-scale cultures for expression in defined M9–glucose medium with casamino acids (Difco, BD Diagnostics, Franklin Lakes, New Jersey, USA) and trace minerals using a procedure described previously (Sack et al., 2008 ▶). Briefly, the culture was induced at OD600 = 1.0 with 0.25 mM IPTG. After induction, the temperature of the culture was lowered to 293 K and the cells were harvested after 30 h. Cells were harvested by centrifugation (5900g for 30 min), resuspended in phosphate-buffered saline (20 ml per litre of culture), pelleted (3000g for 30 min) and stored at 193 K. All lysis, clarification and purification steps were performed at 277 K unless otherwise specified. The frozen cells from 0.5 l culture (6 g paste) were suspended in 100 ml buffer A (25 mM Tris–HCl pH 7.5, 500 mM NaCl, 5% glycerol, 10 mM imidazole, 2 mM dithiothreitol) plus 0.5 mM tris(2-carboxyethyl)phosphine (TCEP; Pierce, Rockford, Illinois, USA) and two tablets of Complete EDTA-free protease-inhibitor cocktail (Roche Applied Science, Mannheim, Germany). The cells were lysed by homogenization using one pass through a microfluidizer (Rannie, Albertslund, Denmark) at 69 MPa. The homogenate was clarified by sedimentation at 100 000g for 45 min (Beckman 45Ti rotor, Beckman Coulter, Brea, California, USA). The resulting supernatant was applied onto a Nickel Sepharose 6 Fast Flow (GE Healthcare) column (2.5 × 2.0 cm) equilibrated with buffer A. The column was washed to baseline absorbance at 280 nm using buffer A and was eluted with a linear gradient of 10–350 mM imidazole in buffer A. Fractions were pooled (40 ml) from the gradient between 150 and 250 mM imidazole and dialyzed overnight against 2 l 25 mM Tris–HCl pH 7.5, 0.4 M NaCl, 5% glycerol, 2 mM EDTA, 2 mM DTT. The dialyzed sample was diluted with an equal volume of buffer B (25 mM Tris–HCl pH 7.5, 5% glycerol, 2 mM EDTA, 2 mM DTT) and loaded onto an SP-Sepharose Fast Flow (GE Healthcare) column (2.5 × 5.0 cm) equilibrated with buffer B containing 0.2 M NaCl. The column was washed to baseline absorbance at 280 nm with buffer B containing 0.2 M NaCl and eluted with a linear gradient of 0.2–0.8 M NaCl in buffer B. The protein eluted in the gradient between 0.40 and 0.48 M NaCl in a volume of 26 ml. The protein was concentrated to a volume of 10 ml and a protein concentration of 1.5 mg ml−1 using an Amicon Ultra 15 ml concentration unit with 10 000 Da cutoff (Millipore). The His tag was cleaved with 10 units mg−1 human α-thrombin (Enzyme Research Laboratories, South Bend, Indiana, USA) for 2 h at room temperature. To remove the thrombin, the reaction mixture was applied onto a Benzamidine Sepharose 6B (GE Healthcare) column (1.5 × 1.0 cm) equilibrated with buffer B containing 0.45 M NaCl. The column-flowthrough fraction (17 ml) was concentrated to 3 ml and applied onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with 25 mM Tris–HCl pH 7.5, 200 mM NaCl, 2 mM EDTA, 5 mM DTT. The final purified protein eluted as a single major peak centered at 90 ml. This peak was collected in a volume of 7.5 ml and the protein concentration was determined to be 1.0 mg ml−1 from its absorbance at 280 nm (theoretical extinction coefficient at 280 nm = 43 360 M −1 cm−1). The final yield of purified RPTPγ(V948I, S970T) was 15 mg per litre of culture. The protein was concentrated and used immediately for crystallization trials or stored at 193 K. The protein purity was ≥95% according to Coomassie Brilliant Blue-stained SDS–PAGE (Fig. 3 ▶). The identity of the purified protein was confirmed by mass spectrometry, with an observed mass of 35 681 Da compared with a theoretical mass of 35 681 Da.
Figure 3.

SDS–PAGE analysis of the purification of RPTPγ(825–1128)/(V948I, S970T) from E. coli. Samples were electrophoresed on a 4–20% Tris–glycine gradient gel and stained with Coomassie Blue. Lane 1, molecular-mass markers (kDa); lane 2, crude lysate (10 µg); lane 3, purified His-Tb-RPTPγ(825–1128)/(V948I, S970T) following nickel-affinity chromatography (5 µg); lane 4, purified His-Tb-RPTPγ(825–1128)/(V948I, S970T) following SP-Sepharose chromatography (5 µg); lanes 5, 6 and 7, purified RPTPγ(825–1128)/(V948I, S970T) following thrombin cleavage, Benzamidine Sepharose and size-exclusion chromatography (2, 4 and 8 µg, respectively).
2.5. Expression and purification of RPTPγ(C858S, S970T)
RPTPγ(C858S, S970T) was purified using the same procedure as described for the purification of RPTPγ(V948I, S970T). A HiLoad 26/60 Superdex 200 column was used and the final purified protein eluted as a single major peak centered at 225 ml. The main peak was collected in a volume of 20 ml and a protein concentration of 1.4 mg ml−1 was determined from its absorbance at 280 nm (theoretical extinction coefficient at 280 nm = 43 330 M −1 cm−1). The final yield of purified RPTPγ(C858S, S970T) was 28 mg per litre of culture. The identity of the purified protein was confirmed by mass spectrometry, with an observed mass of 35 651 Da compared with a theoretical mass of 35 649 Da.
2.6. Crystallization
2.6.1. Protein manipulation and crystallization of an orthorhombic crystal form of RPTPγ
Crystallization trials of RPTPγ domain 1 (residues 825–1128) used the hanging-drop vapor-diffusion method. The domain 1 protein stock solution consisted of 10.2 mg ml−1 protein (0.283 mM based on the calculated molecular weight of 36 101 Da) in 200 mM NaCl, 1 mM EDTA, 5 mM DTT buffered with 50 mM Tris–HCl pH 7.5. Initial crystallization screens were prepared using a Screenmaker 96+8 (Innovadyne, Santa Rosa, California, USA) in Neuro Probe (Gaithersburg, Maryland, USA) hanging-drop trays using Hampton Research (Aliso Viejo, California, USA) crystallization screening solutions: Crystal Screen, Crystal Screen 2, Natrix, MembFac, Index and SaltRX. Initial crystals were observed and conditions were successfully optimized for harvesting and data collection.
The optimized conditions consisted of a reservoir solution consisting of 15–30%(w/v) PEG 3350, 200 mM ammonium sulfate, 0.1 M Bis-Tris pH 6.5. Drops were formed from 1 µl protein solution and 1 µl reservoir solution (total initial volume of 2 µl), mixed and placed at 277 K to equilibrate. Crystals appeared within one week. Single crystals were removed and prepared for data collection at 100 K using a cryosolution that consisted of 25%(w/v) sucrose added to the reservoir solution. Diffraction from these crystals yielded unit-cell parameters a = 74.9, b = 79.5, c = 123.9 Å, α = β = γ = 90°. The symmetry was consistent with point group 222 and the systematic absences suggested that the space group was P212121. From these unit-cell parameters and point group, two molecules per asymmetric unit were expected to yield a Matthews coefficient (Matthews, 1968 ▶) of 2.6 Å3 Da−1 and a solvent fraction of 53%.
Cocrystallization with compound 1 (Table 1 ▶; Fig. 4 ▶) used the same conditions described above in addition to 1.42 mM compound 1, but yielded crystals with somewhat different unit-cell parameters: a = 74.9, b = 82.3, c = 126.8 Å, α = β = γ = 90°.
Table 1. IUPAC names of compounds 1, S1, 2, 4, 5, 7, 10 and S2 of Sheriff et al. (2011 ▶).
Structures are shown in Fig. 3 ▶.
| Designation | IUPAC name |
|---|---|
| Compound 1 | 3-(3,4-Dichlorobenzylthio)thiophene-2-carboxylic acid |
| Compound S1 | 3-(3-Bromo-4-chlorobenzylthio)thiophene-2-carboxylic acid |
| Compound 2 | 3-(3,4-Dichlorobenzylthio)-N-(methylsulfonyl)thiophene-2-carboxamide |
| Compound 4 | N-(3-Aminophenylsulfonyl)-3-(3,4-dichlorobenzylthio)thiophene-2-carboxamide |
| Compound 5 | 5-{N-[3-(3,4-Dichlorobenzylthio)thiophene-2-carbonyl]sulfamoyl}-2-methoxybenzoic acid |
| Compound 7 | 2-(3,4-Dichlorobenzylthio)benzoic acid |
| Compound 10 | 2-(3,4-Dichlorobenzylthio)-4-(4-hydroxybut-1-ynyl)benzoic acid |
| Compound S2 | 2-(3,4-Dichlorobenzylthio)-4-[(3-{2-[2-(methylamino)ethylamino]acetamido}phenyl)ethynyl]benzoic acid |
Figure 4.

Two-dimensional structures of compounds 1, S1, 2, 4, 5, 7, 10 and S2 of Sheriff et al. (2011 ▶). The compounds are named in Table 1 ▶.
Apo crystals were also soaked with compound 1 for 3 d. The soaking solution consisted of 20%(w/v) PEG 3350, 5%(v/v) saturated ammonium sulfate, 0.1 M Bis-Tris pH 6.5, 3 mM compound 1. This yielded very similar unit-cell parameters to those from cocrystallization: a = 74.9, b = 82.3, c = 127.0 Å, α = β = γ = 90° (Fig. 5 ▶).
Figure 5.

Orthorhombic crystal form of RPTPγ. The crystals shown have approximate dimensions of 85 × 35 × 35 µm.
Apo crystals used for soaking in sodium orthovanadate grew from a reservoir condition consisting of 25%(w/v) PEG 3350, 0.2 M magnesium chloride, 0.1 M HEPES pH 7.5. All other crystallization protocols were as described above. Apo crystals were soaked with sodium orthovanadate for 6 h. The soaking solution consisted of 25%(w/v) PEG 3350, 0.2 M magnesium chloride, 0.1 M HEPES pH 7.5, 10 mM sodium orthovanadate. This yielded similar unit-cell parameters to those from apo crystals: a = 74.9, b = 77.7, c = 123.3 Å, α = β = γ = 90°.
2.6.2. Protein manipulation and crystallization of a trigonal crystal form of RPTPγ and RPTPγ(C858S, S970T)
A second crystal form of RPTPγ domain 1 (residues 825–1128) was prepared by the hanging-drop vapor-diffusion method on siliconized cover slips in 24-well Linbro plates. The domain 1 protein stock solution consisted of 10.2 mg ml−1 protein (0.286 mM based on the calculated molecular weight of 35 649 Da) in 200 mM NaCl, 1 mM EDTA, 5 mM DTT buffered with 50 mM Tris–HCl pH 7.5. The 1 ml reservoir solution consisted of 4.0 M ammonium acetate, 100 mM Bis-Tris propane pH 7.0. Drops were formed from 1 µl protein solution and 1 µl reservoir solution, mixed and placed at 277 K to equilibrate. Crystals appeared within 3 d and continued to grow for an additional 5–10 d.
Apo crystals were placed in a soaking solution consisting of 4.0 M ammonium acetate, 100 mM bis-tris propane pH 7.0 and 3 mM ligand (compounds S1, 2, 4, 5, 7 and 10; Table 1 ▶; Fig. 4 ▶) for 3 d or 10 mM sodium orthovanadate for 6 h. Single crystals were removed and placed into a drop of reservoir solution. The stabilizing reservoir solution was serially replaced with cryosolution consisting of 25%(w/v) sucrose added to reservoir solution. Approximately 20% of the total volume was added, mixed and removed. This was repeated until the drop contained 25%(w/v) sucrose. Crystals were gently transferred into a large drop of Paratone-N. Excess aqueous solution was removed, the crystals were flash-cooled directly in the cryo-stream and data were collected at 100 K. The diffraction patterns from the crystals gave unit-cell parameters a = b ≃ 75.5, c ≃ 152 Å, α = β = 90, γ = 120°. The symmetry was consistent with point group 321 and systematic absences were consistent with one of the enantiomeric pair of space groups P3121 or P3221. From these unit-cell parameters and point group, one molecule per asymmetric unit was expected to yield a Matthews coefficient (Matthews, 1968 ▶) of 3.5 Å3 Da−1 and a solvent fraction of 65% (Fig. 6 ▶).
Figure 6.

Trigonal crystal form of RPTPγ. The crystal shown has approximate dimensions of 100 × 50 × 150 µm.
The crystal-growth and soaking procedures for the RPTPγ(C858S, S970T) domain 1 (residues 825–1128) double mutant were identical to those described above for RPTPγ.
2.6.3. Protein manipulation and crystallization of a monoclinic crystal form of RPTPγ(V948I, S970T)
Crystallization trials of RPTPγ(V948I, S970T) domain 1 (residues 825–1128) were prepared using the hanging-drop vapor-diffusion method. The domain 1 protein stock solution consisted of 6.0 mg ml−1 protein (0.168 mM based on the calculated molecular weight of 35 681 Da) in 200 mM NaCl, 2 mM EDTA, 5 mM DTT buffered with 25 mM Tris–HCl pH 7.5. The protein was complexed with 0.84 mM compound S2 (Table 1 ▶; Fig. 4 ▶) from a 50 mM stock solution dissolved in 100% DMSO. The protein–ligand complex was incubated on ice for 2 h and was clarified by centrifugation. Initial crystallization screens were prepared using a Screenmaker 96+8 (Innovadyne) in Neuro Probe hanging-drop trays using Hampton Research crystallization screening solutions: Crystal Screen, Crystal Screen 2, Natrix, MembFac, Index and SaltRX. Initial crystals were observed and conditions were successfully optimized for harvesting and data collection.
The optimized conditions consisted of a reservoir solution consisting of 15–20%(w/v) PEG 20 000 and 100 mM MES pH 6.7. Drops were formed from 1 µl protein solution and 1 µl reservoir solution (total initial volume of 2 µl), mixed and placed at 277 K to equilibrate. Crystals appeared within one week. Single crystals were removed and serially transferred into a final cryosolution consisting of 25%(w/v) sucrose, transferred to Paratone-N and flash-cooled directly in the cryostream as for the trigonal crystal form (see §2.6.2). Diffraction from these crystals yielded unit-cell parameters a = 59.6, b = 74.6, c = 81.9 Å, α = γ = 90, β = 98.3°. The symmetry was consistent with point group 2 and the systematic absences suggested that the space group was P21. From these unit-cell parameters and point group, two molecules per asymmetric unit were expected to yield a Matthews coefficient (Matthews, 1968 ▶) of 2.5 Å3 Da−1 and a solvent fraction of 51% (Fig. 7 ▶).
Figure 7.

Monoclinic crystal form of RPTPγ(V948I, S970T): small and large thin plates. The large prominent crystal pictured has dimensions of 200 × 50 × 7.5 µm.
2.7. Data collection and processing
The source, detector, frame width, collection time per frame, number of frames and crystal-to-detector distance for 13 data sets are reported in Tables 2 ▶, 3 ▶ and 4 ▶. All data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶) with the resolutions and statistics shown in Tables 2 ▶, 3 ▶ and 4 ▶.
Table 2. Data-collection and processing statistics (orthorhombic crystal form).
Values in parentheses are for the highest resolution shell.
| Compound | Apo | Vanadate | 1 (soak) | 1 (cocrystal) |
|---|---|---|---|---|
| Source | † | ‡ | ‡ | † |
| Wavelength | 1.54178 | 1.0 | 1.0 | 1.54178 |
| Detector | Rigaku Saturn92 | ADSC Q210 | ADSC Q210 | Rigaku Saturn92 |
| Crystal-to-detector distance (mm) | 50 | 200 | 200 | 55 |
| Rotation range/image (°) | 0.5 | 0.5 | 0.5 | 0.5 |
| Total rotation range (°) | 180 | 180 | 180 | 180 |
| Exposure time per image (s) | 30 | 10 | 10 | 60 |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters (Å, °) | a = 74.9, b = 79.5, c = 123.9 | a = 74.9, b = 77.7, c = 123.7 | a = 74.9, b = 82.3, c = 126.8 | a = 74.9, b = 82.5, c = 127.0 |
| Mosaicity range (°) | 0.6–0.7 | 0.8–1.7 | 0.1–0.3 | 0.5–0.8 |
| Resolution range (Å) | 50–2.1 (2.18–2.10) | 50–2.1 (2.18–2.10) | 50–2.1 (2.18–2.10) | 50–2.5 (2.59–2.50) |
| Total No. of measured reflections§ | 282477 (≥18843) | 254533 (≥21305) | 336283 (≥29754) | 186734 (≥16916) |
| Unique reflections | 43827 (4260) | 39702 (3912) | 46575 (4546) | 27695 (2755) |
| Multiplicity | 6.4 (4.7) | 6.4 (5.9) | 7.2 (7.3) | 6.7 (6.6) |
| Completeness (%) | 99.8 (99.0) | 94.2 (94.8) | 99.9 (100) | 99.4 (100) |
| Rmerge¶ (%) | 10.4 (46.3) | 13.6 (40.3) | 8.7 (57.8) | 9.9 (5.95) |
| Mean I/σ(I) | 18.8 (3.2) | 11.6 (4.0) | 21.5 (3.7) | 18.4 (3.3) |
| Overall B from Wilson plot (Å2) | 28 | 42 | 33 | 56 |
Rigaku FR-E running at 45 kV and 45 mA and equipped with Rigaku MicroMax confocal optics.
Beamline 17-ID (IMCA-CAT) at the Advanced Photon Source at Argonne National Laboratory, Argonne, Illinois, USA as configured in 2005–2006.
If any reflections are measured more than four times then one cannot extract from HKL-2000 output the total number of measured reflections in a shell but merely a lower limit, hence the use of the ≥ sign.
R
merge = 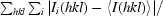
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Table 3. Data-collection and processing statistics (trigonal crystal form).
Values in parentheses are for the highest resolution shell.
| Compound | Apo | Vanadate | S1 | 2 | 4 | 5 | 7 | 10 |
|---|---|---|---|---|---|---|---|---|
| Source | † | ‡ | † | † | † | † | † | † |
| Wavelength | 1.54178 | 1.0 | 1.54178 | 1.54178 | 1.54178 | 1.54178 | 1.54178 | 1.54178 |
| Detector | Rigaku R-AXIS IV++ | ADSC Q210 | Rigaku R-AXIS IV++ | Rigaku R-AXIS IV++ | Rigaku R-AXIS IV++ | Rigaku R-AXIS IV++ | Rigaku R-AXIS IV++ | Rigaku R-AXIS IV++ |
| Crystal-to-detector distance (mm) | 180 | 170 | 150 | 175 | 175 | 175 | 120 | 180 |
| Rotation range/image (°) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Total rotation range (°) | 180 | 180 | 120 | 180 | 180 | 180 | 120 | 180 |
| Exposure time per image (min) | 4 | 5 s | 3 | 3 | 3 | 8 | 4 | 4 |
| Space group | P3221 | P3221 | P3221 | P3221 | P3221 | P3221 | P3221 | P3221 |
| Unit-cell parameters (Å, °) | a = b = 75.2, c = 152.3 | a = b = 75.4, c = 151.0 | a = b = 75.5, c = 152.1 | a = b = 75.5, c = 152.9 | a = b = 75.4, c = 153.8 | a = b = 75.7, c = 152.2 | a = b = 75.5, c = 152.6 | a = b = 75.5, c = 151.8 |
| Mosaicity range (°) | 1.3–2.0 | 0.5–0.6 | 0.4 | 0.8–1.4 | 0.7–1.1 | 0.7–0.9 | 1.0–1.1 | 0.4–0.9 |
| Resolution range (Å) | 50–2.41 (2.5–2.41) | 50–1.8 (1.86–1.80) | 50–2.05 (2.12–2.05) | 50–2.40 (2.49–2.4) | 50–2.26 (2.34–2.26) | 50–2.26 (2.34–2.26) | 50–2.05 (2.12–2.05) | 50–2.40 (2.49–2.4) |
| Total No. of measured reflections§ | 169546 (≥13336) | 494573 (≥35913) | 215486 (≥1681) | 163183 (≥14661) | 227584 (≥18692) | 213656 (≥18020) | 217352 (≥19837) | 164368 (≥15531) |
| Unique reflections | 19097 (1795) | 45117 (4250) | 30938 (2844) | 20534 (2008) | 24039 (2373) | 23970 (2282) | 32729 (3202) | 20275 (1989) |
| Multiplicity | 8.9 (8.6) | 11.0 (10.5) | 7.0 (7.2) | 7.9 (7.9) | 9.5 (9.0) | 8.9 (8.7) | 6.6 (6.7) | 8.1 (8.6) |
| Completeness (%) | 96.0 (92.0) | 96.6 (92.1) | 95.6 (89.9) | 99.8 (100) | 99.7 (99.7) | 98.2 (96.2) | 99.7 (99.7) | 99.5 (99.4) |
| Rmerge¶ (%) | 18.6 (53.7) | 7.3 (61.4) | 4.0 (13.9) | 18.6 (65.1) | 9.8 (52.1) | 10.9 (41.6) | 7.3 (67.8) | 20.7 (78.1) |
| Mean I/σ(I) | 8.0 (4.3) | 24.1 (4.1) | 37.6 (14.4) | 9.0 (3.5) | 16.4 (4.7) | 14.5 (5.4) | 18.0 (3.2) | 9.2 (4.6) |
| Overall B from Wilson plot (Å2) | 58 | 32 | 31 | 58 | 50 | 49 | 42 | 54 |
Rigaku FR-E running at 45 kV and 45 mA and equipped with Rigaku VariMaxHR confocal optics.
Beamline 17-ID (IMCA-CAT) at the Advanced Photon Source at Argonne National Laboratory, Argonne, Illinois, USA as configured in 2005–2006.
If any reflections are measured more than four times then one cannot extract from HKL-2000 output the total number of measured reflections in a shell but merely a lower limit, hence the use of the ≥ sign.
R
merge =
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Table 4. Data-collection and processing statistics (monoclinic crystal form).
Values in parentheses are for the highest resolution shell.
| Compound | S2 |
|---|---|
| Source | † |
| Wavelength | 1.54178 |
| Detector | Rigaku R-AXIS IV++ |
| Crystal-to-detector distance (mm) | 200 |
| Rotation range/image (°) | 0.5 |
| Total rotation range (°) | 180 |
| Exposure time per image (min) | 5 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 59.6, b = 74.6, c = 81.9, β = 98.3 |
| Mosaicity range (°) | 0.4–0.5 |
| Resolution range (Å) | 50–2.4 (2.49–2.40) |
| Total No. of measured reflections | 103828 (≥9905) |
| Unique reflections | 26953 (2567) |
| Multiplicity | 3.9 (3.9) |
| Completeness (%) | 97.0 (93.8) |
| Rmerge (%) | 2.8 (10.0) |
| Mean I/σ(I) | 43.0 (14.6) |
| Overall B from Wilson plot (Å2) | 49 |
Rigaku FR-E running at 45 kV and 45 mA and equipped with Rigaku VariMaxHR confocal optics.
If any reflections are measured more than four times then one cannot extract from HKL-2000 output the total number of measured reflections in a shell but merely a lower limit, hence the use of the ≥ sign.
R
merge =
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
3. Results and discussion
The RPTPγ domain 1 protein as isolated was robust. We found that the RPTPγ, RPTPγ(V948I, S970T) and RPTPγ(C858S, S970T) proteins could be stored at 193 K in their respective crystallization buffers without affecting crystallization or crystal quality. Initial crystallization trials with commercially available screens led to the growth of large single crystals under many conditions without optimization. Focused crystallization screens were set up to grow crystals of the three crystal morphologies that were used in this work.
Crystals were harvested from several different crystallization conditions using glycerol as the cryoprotectant and were plunged into liquid nitrogen. However, all crystals that were flash-cooled in this manner resulted in poor diffraction. Following this observation, the orthorhombic crystal form was screened with 30 different cryoprotection conditions using CryoPro (Hampton Research) to find the conditions reported in this paper. Many crystals did not survive cryo-handling and cracked upon transfer. To overcome this fragility, small crystals were combined with a slow serial transfer into increasing concentrations of cryoprotectant. Cryoprotection with sugars performed the best and sucrose was used as the cryoprotectant in all subsequent data collections; however, the system was still not robust. Many crystals needed to be analyzed before crystals suitable for data collection were found. For example, when working with the orthorhombic form it was difficult to find cryoprotection conditions that preserved diffraction without increasing mosaicity. Therefore, optimizing the cryoprotectant was a high priority. During our work on RPTPγ, we tried high-pressure flash-cooling (Kim et al., 2005 ▶). The orthorhombic RPTPγ crystals were the only sample out of six different proteins that produced a mosaicity that was as good as those we had obtained by conventional cryoprotection methods. The high-pressure flash-cooling procedure used oil-coated crystals. Diffraction images from these crystals did not exhibit ice rings as was observed when oil was used alone without soluble cryoprotectant. Thus, the use of oil in the high-pressure procedure led to a modification of our cryoprotection procedure for RPTPγ. The crystals were serially transferred into increasing concentrations of sucrose and were then submerged in oil (see §2.6.2). An additional modification to the flash-cooling procedure made the system more robust: rather than plunging crystals into liquid nitrogen, the crystals were flash-cooled in a cold stream.
The experience with the orthorhombic crystals led to procedures for the other two crystal forms. The trigonal crystal form typically yielded higher resolution and this led to its extensive use. However, compound S2 (Table 1 ▶, Fig. 4 ▶) could not be soaked into either the orthorhombic or trigonal crystal forms. Therefore, we attempted cocrystallization with broad screens to find conditions that led to suitable crystals for this compound, which yielded a third crystal form. The structures resulting from the crystals and data sets described here will be published elsewhere (Sheriff et al., 2011 ▶).
Acknowledgments
We thank Yi Hu of Lexicon Pharmaceuticals Inc. for the DNA plasmid containing the cytoplasmic portion of RPTPγ. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago.
References
- Almo, S. C. et al. (2007). J. Struct. Funct. Genomics, 8, 121–140. [DOI] [PMC free article] [PubMed]
- Appiah, K. K., Kostich, W. A., Gerritz, S. W., Huang, Y., Hamman, B. D., Allen, J., Zhang, W., Lanthorn, T. H., Albright, C. F., Westphal, R., Banks, M. N. & O’Connell, J. C. (2011). J. Biomol. Screen. 16, 476–485. [DOI] [PubMed]
- Barford, D., Flint, A. J. & Tonks, N. K. (1994). Science, 263, 1397–1404. [PubMed]
- Barnea, G., Silvennoinen, O., Shaanan, B., Honegger, A. M., Canoll, P. D., D’Eustachio, P., Morse, B., Levy, J. B., Laforgia, S., Huebner, K., Musacchio, J. M., Sap, J. & Schlessinger, J. (1993). Mol. Cell. Biol. 13, 1497–1506. [DOI] [PMC free article] [PubMed]
- Barr, A. J., Ugochukwu, E., Lee, W. H., King, O. N., Filippakopoulos, P., Alfano, I., Savitsky, P., Burgess-Brown, N. A., Müller, S. & Knapp, S. (2009). Cell, 136, 352–363. [DOI] [PMC free article] [PubMed]
- Hoffmann, K. M. V., Tonks, N. K. & Barford, D. (1997). J. Biol. Chem. 272, 27505–27508. [DOI] [PubMed]
- Kim, C. U., Kapfer, R. & Gruner, S. M. (2005). Acta Cryst. D61, 881–890. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Sack, J. S., Kish, K. F., Pokross, M., Xie, D., Duke, G. J., Tredup, J. A., Kiefer, S. E. & Newitt, J. A. (2008). Acta Cryst. D64, 705–710. [DOI] [PubMed]
- Sheriff, S. et al. (2011). J. Med. Chem. In the press.
- Zhang, Z.-Y., Zhou, B. & Xie, L. (2002). Pharmacol. Ther. 93, 307–317. [DOI] [PubMed]