

Abstract
A Phase I/II observer-blind, randomized, controlled trial evaluated the safety and immunogenicity of a dengue virus (DENV) vaccine candidate in healthy Thai infants (aged 12–15 months) without measurable pre-vaccination neutralizing antibodies to DENV and Japanese encephalitis virus. Fifty-one subjects received two doses of either DENV (N = 34; four received 1/10th dose) or control vaccine (N = 17; dose 1, live varicella; dose 2, Haemophilus influenzae type b). After each vaccine dose, adverse events (AEs) were solicited for 21 days, and non-serious AEs were solicited for 30 days; serious AEs (SAEs) were recorded throughout the study. Laboratory safety assessments were performed at 10 and 30 days; neutralizing antibodies were measured at 30 days. The DENV vaccine was well-tolerated without any related SAEs. After the second dose, 85.7% of full-dose DENV vaccinees developed at least trivalent and 53.6% developed tetravalent neutralizing antibodies ≥ 1:10 to DENV (control group = 0%). This vaccine candidate, therefore, warrants continued development in this age group (NCT00322049; clinicaltrials.gov).
Introduction
Dengue is a febrile illness caused by one of four antigenically distinct dengue virus (DENV) serotypes (DENV-1, DENV-2, DENV-3, or DENV-4) of the family Flaviviridae and the genus Flavivirus.1 DENV infection can lead to outcomes that include asymptomatic infection, a non-specific febrile illness, dengue fever, and the life-threatening dengue hemorrhagic fever (DHF).2 People, particularly children, living in hyperendemic areas who have circulating antibody from an earlier DENV infection are at increased risk for severe disease (e.g., DHF) when subsequently infected by a heterotypic DENV serotype. This has been postulated to be primarily because of circulating, non-neutralizing, heterotypic antibody from a prior infection facilitating viral entry into target cells (macrophages and monocytes) causing an immune-based enhancement of disease.3,4 There are an estimated 36 million cases of dengue fever, 2.1 million cases of DHF, and 21,000 DENV related deaths annually. Approximately 3.6 billion people (55% of the global population) in 124 countries are at risk of DENV infection.5 Thailand is endemic for all four serotypes; usually one or two serotypes predominate at any given time.6 Prevention of dengue through vaccination is an important objective of the World Health Organization,7 and it has gained the attention of national health authorities of most dengue-endemic countries.8,9
The Walter Reed Army Institute of Research (WRAIR) and GlaxoSmithKline Biologicals (GSK) have developed a live-attenuated tetravalent DENV vaccine candidate to protect children and adults against dengue.10–12 The vaccine candidate comprises four monovalent live virus strains representing each of the four serotypes attenuated by serial passage in primary dog kidney (PDK) cells (Table 1). A well-tolerated and immunogenic formulation of the DENV vaccine candidate was identified in a phase 2 trial conducted in US adult volunteers.13 A similar formulation was tested in an open-label trial in seven flavivirus-naïve Thai children between the ages of 6 and 9 years. The vaccine was well-tolerated without serious adverse events (SAEs) or alert clinical laboratory values. Solicited symptoms were more frequent after dose 1; the symptoms occurring in at least four of seven subjects were pain, redness, and swelling at the injection site, headache, and temperature ≥ 37.5°C. Three subjects had DENV-4 vaccine viremia, in two cases associated with transient fever. Thirty days after dose 1, DENV-2 and DENV-4 responses predominated, with 50% of the cohort seroconverting to each type with geometric mean titers (GMTs) of 16 and 30, respectively. Thirty days after dose 2, 100% of the cohort had seroconverted to all DENV serotypes, with an observed boost in GMT for each DENV serotype (DENV-1, from less than 10 to 55; DENV-2, from 16 to 475; DENV-3, from 6 to 350; DENV-4, from 30 to 171).12
Table 1.
DENV vaccine potency: DENV strains, PDK passage, and assayed viral concentration of each monovalent DENV component in the blended tetravalent vaccine preparations retained from each day of vaccine dosing
| Origin | No. of PDK passages | Viral concentration (log10 FFU/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| First dose | Second dose | |||||||||
| A (dil) | A (und) | B | C | A (dil) | A (und) | B | C | |||
| DENV-1 (45AZ5) | West Pacific | PDK 27 | 5.0 | 6.2 | 6.1 | 6.0 | 4.9 | 6.1 | 6.2 | 6.2 |
| DENV-2 (S16803) | Thailand | PDK 50 | 5.3 | 6.3 | 6.3 | 6.2 | 5.3 | 6.3 | 6.2 | 6.3 |
| DENV-3 (CH53489) | Thailand | PDK 20 | 4.1 | 4.9 | 4.8 | 4.8 | 3.9 | 5.2 | 5.0 | 4.7 |
| DENV-4 (341750) | Colombia | PDK 6 | 5.0 | 6.0 | 5.8 | 6.1 | 5.3 | 6.2 | 6.1 | 5.9 |
Cohort A (dil 1:10 and und): dose 1 on February 29, 2004 and dose 2 on August 29, 2004.
Cohort B: dose 1 on May 9, 2004 and dose 2 on November 14, 2004.
Cohort C: dose 1 was split between July 18 and 25, 2004, dose 2 was split between January 9 and 14, 2005, and the titers were averaged. Dil 1:10 = 1.0 mL of the full-dose preparation was diluted in 9.0 mL diluent (1/10th dose). UnD = undiluted (full-dose).
Herein, we report the reactogenicity, safety and immunogenicity profile of the DENV vaccine candidate administered as two doses to flavivirus-naive Thai subjects aged 12 to 15 months. These subjects are being followed to assess long-term persistence of anti-DENV neutralizing antibodies and evaluate the safety and reactogenicity of a booster dose administered 3 years after primary vaccination (to be published separately).
Materials and Methods
Study design.
This was a phase I/II randomized controlled trial to evaluate the safety and immunogenicity of two doses of live-attenuated tetravalent DENV vaccine administered on a 0- and 6-month schedule. Subjects were enrolled and randomized using a computer-generated sequence of vaccine assignments to receive either DENV or control vaccine in a 2:1 ratio. Vaccine assignments were concealed at the time of allocation; vaccines were administered so that subjects, parents, healthcare providers, data collectors, outcome adjudicators, and data analysts were blinded to their identity (i.e., there was observer blinding).
The study was conducted between February 29, 2004 and March 27, 2005 in accordance with Good Clinical Practice (GCP) guidelines, the provisions of the 1996 version of the Declaration of Helsinki, and both US and Thai regulations. The clinical protocol, subsequent protocol amendments, informed consent form, and other information that required pre-approval were reviewed and approved by the ethical review committees of the Royal Thai Army, the Thai Ministry of Public Health, and the US Army Office of the Surgeon General. The US Army and GSK monitored the conduct of the trial and the veracity of the data. A Thai independent data monitoring committee (IDMC) that included the Study Medical Monitor reviewed the adverse events (AEs). Written informed consent was obtained from each parent before the performance of any study-specific procedures.
Role of the sponsors.
The study was designed by the US Army and GSK. Investigators from Phramongkutklao (PMK) Hospital and the Armed Forces Research Institute of Medical Sciences (AFRIMS) performed the study; Thai investigators collected the data. The US Army Medical Research and Materiel Command (USAMRMC), as the sponsor of this study, monitored and reported on subject safety. A GSK statistician analyzed the data according to a pre-specified and mutually approved plan. All the authors reviewed the manuscript and vouch for its accuracy and completeness. This study was funded by USAMRMC and GSK.
Study subjects.
Parents of potential study subjects were briefed on the study details during well-baby check-ups at the Department of Pediatrics, PMK Hospital, Bangkok, Thailand. Subjects were considered for enrolment if their weight to height ratio was greater than the fifth percentile compared with the standards for similar gender and age of Thai children.14 After parental written consent, subjects aged 12–15 months at dose 1 were screened as previously described.12
After a review of medical history and physical examination (subjects were to be in good general health), screening tests included a complete blood count (CBC with white blood cell differential and platelet count), alanine aminotransferase (ALT), aspartate aminotransferase (AST), hepatitis B surface antigen, antibody to human immunodeficiency virus (HIV), and antibody to hepatitis C virus. Those with normal test results were further screened to exclude those with pre-existing antibody to DENV-1, -2, -3, or -4 or Japanese encephalitis virus (JEV) by hemagglutination inhibition (HAI) and 50% plaque reduction neutralization test (PRNT50). Screening procedures were conducted 15–21 days before initial study vaccination.
Subjects were excluded from the study if they had received any vaccination within 30 days before or 30 days after any protocol-specified vaccine administration or measles–mumps–rubella (MMR) vaccination within 60 days before or 30 days after the first study vaccination. Other key exclusion criteria were history of varicella disease or invasive Haemophilus influenzae B (Hib) disease or previous vaccination against a flavivirus, varicella virus or with a booster dose of Hib during the second year of life.
Study cohorts.
Sequential vaccinations of dose-escalating cohorts (cohorts A, B, and C) were planned with increasing numbers of subjects per cohort. To ensure the safety of subjects, an initial cohort of six infants was initially assigned to cohort A to receive either low-dose DENV vaccine or control vaccine. Fifteen and thirty infants were assigned, respectively, to cohorts B and C to receive either full-dose DENV vaccine or control vaccine. After administration of dose 1 for a given cohort, the 21-day post-dose 1 reactogenicity data were reviewed, preserving the blind, by the IDMC and the US Food and Drug Administration. Based on acceptable safety profiles, the IDMC permitted vaccinations of the following cohort.
Cohort A received a low-dose (one-tenth of full dose) DENV vaccine candidate (N = 4) or control vaccine (N = 2). Cohort B received full-dose DENV vaccine (N = 10) or control vaccine (N = 5). Cohort C received full-dose DENV vaccine (N = 20) or control vaccine (N = 10).
Vaccines.
DENV vaccine candidate.
The tetravalent DENV vaccine candidate was prepared before administration from rehydrated freeze-dried monovalent vaccines (Table 1) that were mixed in equal volumes in a sterile glass vial for delivery of up to six doses. The monovalent vaccines were produced for each DENV serotype at the Salk Institute, Swiftwater, PA.10 For the one-tenth dose, 1.0 mL full-dose preparation was diluted in 9.0 mL diluent of Eagle's Minimum Essential Medium. Each DENV vaccine dose was administered as a 1.0-mL subcutaneous injection by an unblinded vaccination team according to written procedures; these personnel also administered the control vaccines and did not participate in the evaluation of subjects thereafter. Quality control personnel and external observers monitored these processes.
Freeze-dried monovalent DENV vaccines were transported on dry ice from WRAIR, Silver Spring, MD to AFRIMS, Bangkok, Thailand, where they were stored at −70°C until reconstitution on the morning of vaccinations. All DENV vaccine preparations were held on ice through all dilution, reconstitution, and blending procedures to prepare the tetravalent vaccine. The monovalent vaccines were returned to −70°C storage immediately after blending to make the tetravalent vaccine. The tetravalent vaccines remained on ice until completion of vaccinations (< 8 hours from reconstitution); at that time, they were stored at −70°C.
All infants assigned to a given cohort were scheduled for vaccination over a 1- or 2-day period. Vaccination required formulation of tetravalent vaccine from lyophilized monovalent vaccines on eight occasions, including two occasions when additional 10-fold dilutions were conducted. On each of the days that the DENV vaccine was formulated, retained samples of tetravalent product were frozen at −70°C, subsequently transported to WRAIR on dry ice, and returned to −70°C storage until thawed for potency testing using an immunofocus assay (IFA) as previously described.12
Control vaccines.
Varicella (Varilrix; GSK, Rixensart, Belgium) and H. influenzae type B (Hiberix; GSK, Rixensart, Belgium) vaccines(0.5-mL dose vial) were used as control vaccines for DENV vaccine doses 1 and 2, respectively. Both were reconstituted and administered according to the prescribing information: the former by subcutaneous injection and the latter by intramuscular injection.
JEV vaccine.
Two doses of JEV vaccine (inactivated Beijing strain; Thailand Government Pharmaceutical Organization, Bangkok, Thailand) were administered as 0.25-mL subcutaneous injections at study months 7 and 7.5 to all study subjects in accordance with national immunization recommendations.
AEs.
Subjects were evaluated in person at 2, 4, 10, 14, 21, and 30 days after each DENV/control vaccination and by telephone at 1, 3, 5, 6, 7, 9, 13, and 18 days. During clinic visits, physical examinations included assessments for rash, conjunctival hemorrhage, conjunctival injection, mucosal hemorrhage, lymphadenopathy, hepatomegaly, and splenomegaly. Parents of study subjects recorded the axillary temperature, injection site and general symptoms on diary cards for 21 days after each DENV/control vaccination and for 7 days after each JEV vaccination. Injection site symptoms included pain, redness, and swelling; general symptoms included cough, decreased activity, drowsiness, irritability, loss of appetite, vomiting, and fever (axillary temperature ≥ 37.5°C). If the subject had a fever with axillary temperature ≥ 38°C or illness, parents were instructed to return for an evaluation. The intensities of AEs were scored. Grade 3 AEs were defined as those symptoms preventing normal daily activity, fever > 39°C, or solicited injection site reactions such as spontaneous pain (subject cries when limb is moved), swelling, or redness > 20 mm.
SAEs, defined as medically significant events, including those resulting in hospitalization, disability, or death, were recorded throughout the study. Non-SAEs were collected for 30 days after each DENV/control vaccination. Safety laboratory assessments (including absolute neutrophil count [ANC], platelet count [PLT], ALT, and AST) were performed on days 10 and 30 after DENV/control vaccinations and with parental permission if subjects presented with temperature elevations of ≥ 38°C; alert values were defined as shown in the footnote to Table 2.12 All laboratory, emergency room, outpatient clinic, and physician office visits unrelated to the study were recorded throughout the study.
Table 2.
Subjects with alert laboratory values
| Subject no. | Vaccine group | Alert value | Time point for alert value | Outcome |
|---|---|---|---|---|
| 3 | Cohort A | ANC = 787 cells/mm3 | Day 0 | PLT (day 0) = 223,000 cells/mm3, repeat ANC on day 10 = 3,310 cells/mm3, DENV/JE IgM (days 0 and 10) was negative |
| 12 | Cohort B | AST = 149 U/L, ALT = 181 U/L, ALT = 232 U/L, AST = 237 U/L, ALT = 289 U/L | Day 0, Day 10 post-dose 1, Day 10 post-dose 2 (DEN-4 viremia) | AST = 46 U/L, ALT = 35 U/L at end of study; see text for more details |
| 13 | Cohort B | ANC = 934 cells/mm3, PLT = 37,000 cells/mm3 | Day 0 | DENV/JE IgM ELISA (day 0) was negative, ANC (day 2) = 1,640 cells/mm3, PLT (day 2) = 304,000 cells/mm3 |
| 34 | Cohort C | ANC = 468 cells/mm3, PLT = 46,000 cells/mm3, PLT = 530,000 cells/mm3 | Day 30 post-dose 1, Day 30 post-dose 1, Day 0 pre-dose 2 | DENV PCR (day 30) was negative, ANC (day 32) = 2,740 cells/mm3, PLT (day 32) = 389,000 cells/mm3 |
| 35 | Cohort C | PLT = 17,000 cells/mm3 | Day 180 post-dose 1 (pre-dose 2) | PLT (day 183) = 318,000 cells/mm3, DENV PCR (day 180 post-dose 1) was negative, DEN (IgM) ELISA (day 183) was negative |
| 46 | Cohort C | AST = 83 U/L, AST = 153 U/L | Day 0 pre-dose 1, Day 0 pre-dose 2 | AST decreased but remained above the normal range with ALT normal until last visit, when it was twice normal |
| 19 | Control | PLT = 84,300 cells/mm3 | Day 0 | Normal at all other time points |
| 27 | Control | PLT = 75,700 cells/mm3 | Day 10 post-dose 1 | Normal at all other time points |
Alert value for ANC was < 1,000 cells/mm3. Alert value for PLT count was < 100,000 cells/mm3. Alert value for AST was > 2.5 times the upper limit of normal (i.e., > 142 U/L). Alert value for ALT was > 2.5 times the upper limit of normal (i.e., > 135 U/L).
Assays for immune response, viremia, and vaccine potency.
Antibodies to DENV and JEV wild-type (Nakayama strain) and vaccine viruses were measured at screening in Bangkok using the AFRIMS PRNT50 assay. Subsequently, antibody responses after DENV/control vaccine dose were measured on day 0 (before dose 1), day 10 (after dose 1), month 1 (after dose 1), month 6 (before dose 2), day 30 (after dose 2 and before administration of JEV vaccine dose 1), and 30 days after JEV vaccine dose 2 using a WRAIR PRNT assay.12,15 Each serum was screened initially at a dilution of 1:10 against all four dengue serotypes using wild-type strains (i.e., DENV-1 WP74, DENV-2 S16803, DENV-3 CH53489, and DENV-4 TVP360). Specimens that were neutralizing antibody-positive by virtue of showing a 50% or greater reduction in virus plaques were endpoint titered against the respective virus serotype. Subsequent serial PRNT testing was conducted at WRAIR using an attenuated JEV strain (SA14-14-2). Seroconversions for dengue and JEV were defined for each serotype as an increase in neutralizing antibody from less than 1:10 to greater than or equal to 1:10.
On day 10 after each DENV/control vaccination, regardless of symptoms, specimens were tested for dengue viremia using a nested dengue reverse transcriptase-polymerase chain reaction (RT-PCR).16 At other times during the 21 days after each DENV/control vaccine dose, if subjects presented ill with fever (≥ 38°C), viremia testing was conducted if parental permission for additional venipuncture was obtained. DENV isolates were molecularly characterized to distinguish viremia induced by vaccine from that of natural infection.12,17 The AFRIMS DENV/JEV immunoglobulin M (IgM)/IgG enzyme-linked immunosorbent assay (ELISA)18 was used to corroborate DENV infections. Viremia detected by RT-PCR was further characterized by quantitative RT-PCR (qRT-PCR) and quantitation of infectivity in inoculated Toxorhynchites splendens mosquitoes (a quantal response assay to determine 50% mosquito infectious doses per 1 mL [MID50/mL] of serum). Although the limit of detection (LOD) for the qRT-PCR is 5 RNA copies for DENV-1, -2, and -3 and 25 RNA copies for DENV-4, no LOD has been defined for the quantal response assay given its inherent biological variability.3
Data analysis.
All statistical analyses were performed using SAS software (version 8.2; SAS, Cary, NC). Safety analyses were performed on the per-protocol cohort and stratified by treatment cohort (cohorts A, B, and C and control) with the full-dose DENV vaccine groups (cohorts B and C) pooled.
The overall percentages of subjects reporting an AE after vaccine administration (21 days for solicited AEs after each DENV/control vaccination, 7 days for solicited AEs after JEV vaccination, and 30 days after any vaccine dose for spontaneously reported symptoms) were tabulated with exact 95% confidence intervals (CIs) by type of AE, intensity (any grade and grade 3), and relationship to vaccination. The verbatim reports of unsolicited symptoms were coded using matched Medical Dictionary for Regulatory Activities (MedDRA)19 terms. All SAEs occurring during the study were listed for each treatment group. The proportion of subjects with abnormal physical examination findings detected up to 30 days after each DENV/control vaccination was tabulated with exact 95% CI.
The per-protocol cohort for immunogenicity included all evaluable subjects (i.e., those who met all eligibility criteria and complied with protocol-defined procedures with no elimination criteria during the study) for whom data concerning immunogenicity endpoint measures were available (subjects for whom assay results were available for at least one laboratory test after DENV vaccination). Seroconversion rates and geometric means of anti-DENV neutralizing antibody titer (GMTs with exact 95% CIs) for each DENV vaccine type were calculated for each vaccine dose. The seroconversion rate (with exact 95% CIs) of anti-JEV neutralizing antibody titer (assay uses the Nakayama JEV strain) was also calculated after JEV vaccine dose 2. The GMT calculations were performed by taking the anti-log of the mean of the log titer transformation; values less than 1:10 were coded as 5.0. The proportion of subjects with dengue viremia (10 days after vaccination) was tabulated for each DENV vaccine dose.
Results
Study population.
Of 96 subjects screened, 15 were excluded, because they were flavivirus-antibody positive by either PRNT or HAI assays to DENV and/or JEV. The first 51 subjects meeting eligibility criteria were assigned sequentially to one of three cohorts. Their mean age was 13.6 months (range from 12 to 15 months), and 28 (55%) were males; the treatment groups were similar with respect to age. All subjects were included in the safety analysis. Two subjects were excluded from the per-protocol immunogenicity analysis. One subject in the cohort C control group relocated out of the study area after dose 1 and could not be contacted. Another subject in the cohort B DENV group was excluded from the per-protocol immunogenicity analysis because of developing wild-type DENV-4 infection after dose 1 (described below). This subject subsequently received dose 2.
DENV vaccine potency.
The potency of the monovalent components of the administered vaccines was determined using blended tetravalent vaccine preparations retained from each of the DENV vaccination days. Differences among retained samples for any serotype were small, and the majority was consistent, with the known test error of log 0.3 focus-forming units per 1 mL (FFU/mL) for this assay (data are shown for tetravalent vaccines only in Table 1; no important differences were observed for retains of monovalent vaccines used to blend the tetravalent products). The potencies for each DENV serotype in diluted vaccine administered to cohort A confirmed that the one-tenth dilution target was achieved. Importantly, there were no notable differences in potency of the DENV vaccine administered to cohorts B and C.
DENV vaccine safety.
A diary card was completed and collected from the parents of all but two subjects (both control vaccinees) for all doses received: 99.5% and 98.5% compliance for returning diary cards in the DENV and control groups, respectively.
After dose 1, solicited local symptoms of redness and swelling appeared more frequently in the DENV full-dose group compared with control and low-dose DENV groups (Table 3). Redness typically lasted 1 day and never lasted longer than 3 days. There seemed to be no notable differences in injection site symptoms after dose 2.
Table 3.
Incidence of any or grade 3 solicited injection site symptoms 21 days after DENV or control vaccination (total vaccinated cohort)
| Symptom | Type | Control group | 1/10 dose DENV cohort A | Full-dose DENV cohorts B and C | |||
|---|---|---|---|---|---|---|---|
| n | Percent (95% CI) | n | Percent (95% CI) | n | Percent (95% CI) | ||
| Dose 1 | N = 17 | N = 4 | N = 30 | ||||
| Pain | All | 3 | 17.6 (3.8–43.4) | 1 | 25.0 (0.6–80.6) | 11 | 36.7 (19.9–56.1) |
| Pain | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Redness | All | 1 | 5.9 (0.1–28.7) | 2 | 50.0 (6.8–93.2) | 21 | 70.0 (50.6–85.3) |
| Redness | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 7 | 23.3 (9.9–42.3) |
| Swelling | All | 2 | 11.8 (1.5–36.4) | 1 | 25.0 (0.6–80.6) | 16 | 53.3 (34.3–71.7) |
| Swelling | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 3 | 10.0 (2.1–26.5) |
| Dose 2 | N = 16 | N = 4 | N = 30 | ||||
| Pain | All | 2 | 12.5 (1.6–38.3) | 1 | 25.0 (0.6–80.6) | 13 | 43.3 (25.5–62.6) |
| Pain | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Redness | All | 3 | 18.8 (4.0–45.6) | 1 | 25.0 (0.6–80.6) | 13 | 43.3 (25.5–62.6) |
| Redness | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 6 | 20.0 (7.7–38.6) |
| Swelling | All | 1 | 6.3 (0.2–30.2) | 1 | 25.0 (0.6–80.6) | 8 | 26.7 (12.3–45.9) |
| Swelling | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
Redness after dose 1: cohort A with two grade 1 symptoms and zero grade 2 symptoms; cohorts B and C with fifteen grade 1 symptoms and six with grade 2 symptoms. Swelling after dose 1: cohort A with one grade 1 symptom and zero grade 2 symptoms; cohorts B and C with fourteen grade 1 symptoms and two grade 2 symptoms. N = number of subjects having received at least one dose of either vaccine; n/% = number/percentage of subjects reporting a specified symptom; 95% CI (LL, UL) = 95% confidence interval (lower and upper limits).
There were no clinically important differences in reported general symptoms among groups other than irritability, which was more common in the one-tenth dose group after dose 1 and in the control group after dose 2 (Table 4). Fever was the only symptom reported more frequently in the DENV full-dose group after dose 2 (53.3%) than dose 1 (26.7%). However, the incidence of fever after dose 2 in the control group was 50.0%. Grade 3 solicited general symptoms were uncommon.
Table 4.
Incidence of any or grade 3 solicited general symptoms 21 days after DENV or control vaccination (total vaccinated cohort)
| Symptom | Type | Control group | 1/10 dose DENV cohort A | Full-dose DENV cohorts B and C | |||
|---|---|---|---|---|---|---|---|
| n | Percent (95% CI) | n | Percent (95% CI) | n | Percent (95% CI) | ||
| Dose 1 | N = 17 | N = 4 | N = 30 | ||||
| Cough | All | 7 | 41.2 (18.4–67.1) | 2 | 50.0 (6.8–93.2) | 13 | 43.3 (25.5–62.6) |
| Cough | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Decreased activity | All | 2 | 11.8 (1.5–36.4) | 1 | 25.0 (0.6–80.6) | 5 | 16.7 (5.6–34.7) |
| Decreased activity | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Drowsiness | All | 4 | 23.5 (6.8–49.9) | 0 | 0.0 (0.0–60.2) | 9 | 30.0 (14.7–49.4) |
| Drowsiness | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Irritability | All | 3 | 17.6 (3.8–43.4) | 3 | 75.0 (19.4–99.4) | 12 | 40.0 (22.7–59.4) |
| Irritability | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Loss of appetite | All | 4 | 23.5 (6.8–49.9) | 1 | 25.0 (0.6–80.6) | 9 | 30.0 (14.7–49.4) |
| Loss of appetite | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Axillary temperature (°C) | All | 8 | 47.1 (23.0–72.2) | 3 | 75.0 (19.4–99.4) | 8 | 26.7 (12.3–45.9) |
| Axillary temperature (°C) | ≥ 39.1 | 1 | 5.9 (0.1–28.7) | 0 | 0.0 (0.0–60.2) | 1 | 3.3 (0.1–17.2) |
| Vomiting | All | 5 | 29.4 (10.3–56.0) | 1 | 25.0 (0.6–80.6) | 11 | 36.7 (19.9–56.1) |
| Vomiting | Grade 3 | 0 | 0.0 (0.0–19.5) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Dose 2 | N = 16 | N = 4 | N = 30 | ||||
| Cough | All | 8 | 50.0 (24.7–75.3) | 2 | 50.0 (6.8–93.2) | 14 | 46.7 (28.3–65.7) |
| Cough | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Decreased activity | All | 9 | 56.3 (29.9–80.2) | 1 | 25.0 (0.6–80.6) | 6 | 20.0 (7.7–38.6) |
| Decreased activity | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 1 | 3.3 (0.1–17.2) |
| Drowsiness | All | 8 | 50.0 (24.7–75.3) | 0 | 0.0 (0.0–60.2) | 9 | 30.0 (14.7–49.4) |
| Drowsiness | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 1 | 3.3 (0.1–17.2) |
| Irritability | All | 10 | 62.5 (35.4–84.8) | 0 | 0.0 (0.0–60.2) | 11 | 36.7 (19.9–56.1) |
| Irritability | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 1 | 3.3 (0.1–17.2) |
| Loss of appetite | All | 9 | 56.3 (29.9–80.2) | 1 | 25.0 (0.6–80.6) | 12 | 40.0 (22.7–59.4) |
| Loss of appetite | Grade 3 | 0 | 0.0 (0.0–20.6) | 0 | 0.0 (0.0–60.2) | 0 | 0.0 (0.0–11.6) |
| Axillary temperature (°C) | All | 8 | 50.0 (24.7–75.3) | 0 | 0.0 (0.0–60.2) | 16 | 53.3 (34.3–71.7) |
| Axillary temperature (°C) | ≥ 39.1 | 1 | 6.3 (0.2–30.2) | 0 | 0.0 (0.0–60.2) | 2 | 6.7 (0.8–22.1) |
| Vomiting | All | 7 | 43.8 (19.8–70.1) | 1 | 25.0 (0.6–80.6) | 13 | 43.3 (25.5–62.6) |
| Vomiting | Grade 3 | 1 | 6.3 (0.2–30.2) | 0 | 0.0 (0.0–60.2) | 1 | 3.3 (0.1–17.2) |
Irritability after dose 1: cohort A with one grade 1 symptom and two grade 2 symptoms; cohorts B and C with four grade 1 symptoms and eight grade 2 symptoms. Irritability after dose 2: cohorts B and C with three grade 1 symptoms and eight grade 2 symptoms. N = number of subjects having received at least one dose of either vaccine; n/% = number/percentage of subjects reporting a specified symptom; 95% CI (LL, UL) = 95% confidence interval (lower and upper limits).
A summary of reported fevers (≥ 38°C) within 21 days of DENV/control vaccination is provided in Table 5. Of 20 DENV recipients who were febrile (≥ 38°C), four subjects were tested for dengue viremia, because no more probable alternative cause could be identified for their fevers; all four tested negative. One subject with fever outside the 21-day window after DENV/control vaccination (subject 18) tested positive for wild-type DENV-4 viremia by PCR and sequencing.
Table 5.
Onset, duration, DENV PCR testing (when performed), and presumptive diagnosis in subjects with fever (≥ 38°C) during the 21-day period after each DENV/control vaccine dose by treatment group
| Full-dose DENV | 1/10 dose DENV | Control | |
|---|---|---|---|
| Dose 1 | N = 6 | N = 1 | N = 3 |
| Fever onset | |||
| Mean in days (range) | 6.7 (1–19) | 19 (19) | 17.0 (12–20) |
| Fever duration | |||
| Mean in days (range) | 1.8 (1–3) | 1 (1) | 2.0 (1–4) |
| Presumptive diagnosis | |||
| Isolated fever* | 3 | 0 | 1 |
| Acute pharyngitis | 1 | 0 | 2 |
| Acute gastritis/gastroenteritis | 1 | 1 | – |
| Influenza/ILI | 1 (PCR negative) | 0 | – |
| Dose 2 | N = 13 | N = 0 | N = 6 |
| Fever onset | |||
| Mean in days (range) | 10.4 (1–20) | – | 12.8 (5–19) |
| Fever duration | |||
| Mean in days (range) | 2.3 (1–7) | – | 1.8 (1–3) |
| Presumptive diagnosis | |||
| Isolated fever* | 4 | – | – |
| Acute pharyngitis | 8 (2 PCR negative) | – | 3 |
| Acute gastritis/gastroenteritis | 1 | – | 1 |
| Acute otitis media | – | – | 2 (1 PCR negative) |
Isolated fever cases had fever for 1 day without symptoms, which led to an organ-based diagnosis, and fever was presumed due to vaccine if the subject was in the DENV treatment group.
The most frequent physical examination finding during the 30-day follow-up period after each vaccine dose was lymphadenopathy (cervical, inguinal, or both), the prevalence of which did not increase from baseline and was similar between full-dose DENV vaccine and control vaccine groups. Lymphadenopathy in young children is a common but non-specific finding. Neither hepatomegaly nor splenomegaly was reported. There were no reports of hemorrhage or conjunctival hemorrhage after any dose of the full-dose DENV vaccine. One subject in cohort A had a mucosal hemorrhage associated with trauma after dose 1 that was not attributed to vaccine. After dose 2, three full-dose DENV vaccinees (10%) had conjunctival injection as did one subject (5.9%) in the control group.
In the DENV full-dose group, six subjects (20%) had a rash after dose 1, and two subjects (6.7%) had a rash after dose 2. In the control group (N = 17 for dose 1; N = 16 for dose 2), one subject (5.9%) had a rash after dose 1 (Varilrix), and two subjects (12.5%) had a rash after dose 2 (Hiberix). In two subjects in the DENV full-dose vaccine group, rash was associated with fever after dose 1. One subject developed a rash with fever (temperature of 37.8°C on day 8 after vaccination), and another subject developed a generalized rash 11 days after vaccination with grade 3 fever (temperature of 40.2°C on day 5 after vaccination).
There were four SAEs reported: one traumatic fracture of the left forearm (cohort B DENV vaccinee), two cases of acute gastroenteritis with febrile seizure that occurred approximately 5 months after dose 1 (both cohort C control vaccinees), and one case of viral gastritis with fever, vomiting, and dehydration 21 days after dose 1 (cohort C control vaccinee). None of the SAEs were attributed to vaccination by the investigator.
Six of thirty-four subjects in the DENV vaccine group (17.6%) and two of seventeen subjects in the control vaccine group had at least one alert laboratory value during the study period. Five instances were before vaccination with either dose 1 or dose 2 and thus, were unrelated to the study vaccine; all cases normalized during this trial or its extension. These findings are summarized in Table 2. Only the findings for one subject (subject 12), summarized in the paragraph below, were considered to be both clinically significant and possibly related to the DENV vaccine candidate.
Subject number 12 in the cohort B DENV full-dose group exhibited varying transaminase elevations. AST/ALT levels were documented as follows: day 0 dose 1 (149/181 U/L), 10 days post-dose 1 (86/232 U/L) with return of AST/ALT to normal range by 30 days after dose 1; day 0 dose 2 (56/77 U/L), 10 days after the second dose (237/289) associated with an ANC of 672 cells/mm3 and DENV-4 viremia (Table 6). During the subsequent days, the subject had 1 day of fever and elevated levels of AST (highest recorded value of 648) and ALT (highest recorded value of 566 U/L); these values returned to normal by 30 days after dose 2. Retained serum from screening was tested for antibody to human herpes virus 6 (HHV-6) as well as other possible causes of elevated transaminases (parvovirus, leptospirosis, scrub typhus, Epstein–Barr, cytomegalovirus, hepatitis A virus, hepatitis B virus, and hepatitis C virus). These investigations only revealed an elevated IgG antibody titer (≥ 1:256) to HHV-6, both at the time of screening and at several time points during the study. Ultrasound of the liver and biliary system on day 23 after dose 2 was normal. A consulting hepatologist concluded that there was no evidence of any underlying liver disease and that, although non-specific transaminase elevations can be associated with many viral infections, given the temporal relationship of the elevations to administration of the live DENV vaccine, these liver enzyme elevations were probably DENV vaccine-induced.
Table 6.
DENV viremias observed on day 10 after administration of either of two doses of tetravalent DENV vaccine at 0 and 6 months
| Subject number and group | Nested PCR | Infectivity quantification and phylogenicity | |||
|---|---|---|---|---|---|
| Mosquito inoculation | Sequence | Quantitative RNA (log genome equivalents/mL) | |||
| log MID50/mL | 95% CI | ||||
| Day 10 | |||||
| 17; cohort B | DENV-4 | 4.6 | 4.27–4.89 | Vaccine* | 5.9 |
| 24; cohort C | DENV-2 | 2.2 | N/A | Negative | 3.0 |
| 26; cohort C | DENV-2 | 2.2 | N/A | Negative | 3.2 |
| 34; cohort C | DENV-4 | 7.2 | 6.88–7.53 | Vaccine* | 7.9 |
| 46; cohort C | DENV-4 | 3.7 | 3.19–4.02 | Vaccine* | 5.0 |
| Day 190 | |||||
| 12; cohort B | DENV-4 | Not assessed | Not assessed | Not assessed | Undetectable |
| 28; cohort C | DENV-4 | Not assessed | Not assessed | Vaccine* | 4.2 |
Day 10 = 10 days post-dose 1; day 190 = 10 days post-dose 2.
Virus isolate DENV-4 genotype IIB.
Case history of dengue fever after dose 1 of DENV vaccine.
One subject in the cohort B DENV vaccine group experienced dengue fever caused by wild-type DENV-4 infection during the interval between doses 1 and 2. This subject had no detectable neutralizing antibodies to any DENV serotype 30 days post-dose 1 (Table 7). Three months after Dose 1, the subject experienced fever (up to 39.2°C) for 3 days, flushed face, and generalized erythema accompanied by neutropenia (ANC of 762) but a normal platelet count (213,000). She remained at home. Her fever responded to acetaminophen; she remained active and playful. Her only symptoms or signs other than fever were loss of appetite for 1 week and a convalescent rash on the third day. Both parents and investigators deemed the illness as not severe. DENV-4 viremia was detected by RT-PCR; E-gene sequencing identified it as wild-type virus. Dengue IgM/IgG ELISA testing (positive cut-off ≥ 40 units) on 1- and 6-month samples after dose 1 revealed non-diagnostic levels of IgM but rising levels of IgG to DENV (0 to 52 units). A response limited to anti-DENV IgG after a DENV infection suggests that this subject was immunologically primed by vaccine dose 1 despite having no detectable neutralizing antibodies to any DENV serotype 30 days after vaccination. This was a case of breakthrough infection after an undetectable response to a single dose of DENV vaccine. The wild-type infection did not result in prolonged fever, plasma leakage, hemorrhage, or any other severe manifestation of dengue. This subject tolerated a second dose of DENV vaccine without difficulty.
Table 7.
DENV neutralizing antibody titers for subject 18 who sustained dengue fever between DENV vaccine doses 1 and 2 and was eliminated from the per-protocol immunogenicity analysis
| Study day | Neutralizing antibodies to | |||
|---|---|---|---|---|
| DENV-1 | DENV-2 | DENV-3 | DENV-4 | |
| 0; pre-dose 1 | < 10 | < 10 | < 10 | < 10 |
| 30 days post-dose 1 | < 10 | < 10 | < 10 | < 10 |
| 180 days post-dose 1 and pre-dose 2 | 158 | 160 | 49 | 254 |
| 30 days post-dose 2 | 73 | 80 | 31 | 117 |
| 75 days post-dose 1 JEV vaccine | 79 | 81 | 25 | 131 |
DENV vaccine viremia.
Circulating DENV RNA and viremia titers were assessed by nested RT-PCR and mosquito inoculation, respectively. Seven subjects, all of whom received DENV vaccine, were viremic on day 10 post-vaccination (Table 6) with DENV-4 (N = 5) or DENV-2 (N = 2) viremia. All were asymptomatic. In four of five cases of DENV-4 viremia, the phylogeny based on the DENV envelope (E) gene's sequence indicated that the virus was of vaccine origin; in the other three cases, because no sequence analysis was feasible, the viruses were presumed to be of vaccine origin. There was positive correlation between the quantitation by molecular and biologic assays; they found a range of more than 5.0 between the minimum and maximum log viremia levels detected. Notably, even vaccine viremia (DENV-4) of log 7.9 per 1 mL did not cause illness.
Alignment of E-gene nucleotide sequences from four of five DENV-4 isolates showed one point mutation at nucleotide position 1,147, which corresponds to domain III. In synonymous substitution, the vaccine strain contained adenine at amino acid position 383, whereas viruses collected from subjects uniformly contained guanine.
Of 20 DENV recipients who were febrile (≥ 38°C) within 21 days of DENV/control vaccination, four subjects were bled for DENV PCR testing based on physician recommendation to safeguard subject safety, and parents agreed to the extra blood draw; all four PCRs were negative. One subject (subject 18) with fever outside the 21-day window (3 months after full-dose DENV-1 vaccination) had wild-type DENV-4 viremia (Table 7).
DENV vaccine immunogenicity.
The PRNT50 responses of subjects included in the per-protocol immunogenicity cohort by vaccination cohort are shown in Table 8 and summarized in Figure 1. All subjects were flavivirus-naïve on day 0 pre-dose 1 as indicated by lack of DENV and JEV neutralizing antibodies on day 0 and the absence of a secondary response detected by DENV IgM:IgG testing of serum specimens collected 30 days after DENV vaccine dose 1 (data not shown). One month after dose 1 dengue vaccination, 25% of subjects in the low-dose DENV group (one bivalent responder to DENV-2 and DENV-4) and 37.9% of the subjects in the full-dose DENV group were seropositive for DENV-2 and DENV-4 neutralizing antibodies (12 monovalent, 4 bivalent, and 1 trivalent responders). Recipients of control vaccines developed no measurable neutralizing antibodies to DENV during the study.
Table 8.
Seropositivity rates and GMT by PRNT50 to each DENV serotype (per-protocol immunogenicity cohort)
| Serotype and group | Timing | N | ≥ 10 Dil | GMT (95% CI) | |
|---|---|---|---|---|---|
| n | Percent (95% CI) | ||||
| DEN-1 | |||||
| Control | PI(M1) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Control | PII(M7) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Dengue low dose | PI(M1) | 4 | 0 | 0.0 (0.0–60.2) | UD (UD–UD) |
| Dengue low dose | PII(M7) | 4 | 1 | 25.0 (0.6–80.6) | 6.1 (3.3–11.4) |
| Dengue full dose | PI(M1) | 29 | 1 | 3.4 (0.1–17.8) | 5.2 (4.8–5.5) |
| Dengue full dose | PII(M7) | 29 | 16 | 55.2 (35.7–73.6) | 20.7 (11.4–37.7) |
| DEN-2 | |||||
| Control | PI(M1) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Control | PII(M7) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Dengue low dose | PI(M1) | 4 | 1 | 25.0 (0.6–80.6) | 8.8 (1.4–54.4) |
| Dengue low dose | PII(M7) | 4 | 4 | 100 (39.8–100) | 173.6 (50.1–601.5) |
| Dengue full dose | PI(M1) | 29 | 11 | 37.9 (20.7–57.7) | 13.7 (7.7–24.5) |
| Dengue full dose | PII(M7) | 28 | 28 | 100 (87.7–100) | 239.6 (163.6–351.1) |
| DEN-3 | |||||
| Control | PI(M1) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Control | PII(M7) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Dengue low dose | PI(M1) | 4 | 0 | 0.0 (0.0–60.2) | UD (UD–UD) |
| Dengue low dose | PII(M7) | 4 | 0 | 0.0 (0.0–60.2) | UD (UD–UD) |
| Dengue full dose | PI(M1) | 29 | 0 | 0.0 (0.0–11.9) | UD (UD–UD) |
| Dengue full dose | PII(M7) | 29 | 25 | 86.2 (68.3–96.1) | 29.2 (18.6–45.7) |
| DEN-4 | |||||
| Control | PI(M1) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Control | PII(M7) | 16 | 0 | 0.0 (0.0–20.6) | UD (UD–UD) |
| Dengue low dose | PI(M1) | 4 | 1 | 25.0 (0.6–80.6) | 15.7 (0.4–600.1) |
| Dengue low dose | PII(M7) | 4 | 3 | 75.0 (19.4–99.4) | 31.9 (2.5–408.2) |
| Dengue full dose | PI(M1) | 29 | 11 | 37.9 (20.7–57.7) | 22.5 (10.0–50.3) |
| Dengue full dose | PII(M7) | 28 | 27 | 96.4 (81.7–99.9) | 52.5 (29.0–95.0) |
GMTs calculated on all subjects. PI(M1) = blood sampling 1 month after dose 1 at study month 1; PII(M7) = blood sampling 1 month after dose 2 at study month 7; N = number of subjects with available data; Dil = dilution; n/% = number/percentage of subjects with titer within the specified range; 95% CI = 95% confidence interval; LL = lower limit; UL = upper limit; UD = undetectable level.
Figure 1.

Percentage of subjects per group with neutralizing antibodies to each DENV serotype 30 days after dose 2 (per-protocol immunogenicity cohort).
After the second DENV vaccination, subjects in the low-dose DENV group (cohort A) exhibited modest responses to DENV-2 and DENV-4 but almost no response to DENV-1 and no response to DENV-3. Among these four subjects, there was one monovalent, two bivalent, and one trivalent response 30 days after dose 2.
After the second DENV vaccination, subjects in the full-dose DENV group appeared to respond more vigorously than subjects receiving the low-dose DENV vaccine. Notably, subjects in cohort B appeared to exhibit better seroconversion rates (N = 9, 11.1% trivalent and 89.9% tetravalent responses) than subjects in cohort C (N = 14, 42.1% trivalent and 36.8% tetravalent responses; P = 0.02 by Fisher exact test for DENV-1). We elected to pool data for both cohorts despite this apparent heterogeneity, because between both cohorts, there was no significant difference in having at least trivalent seroconversion (100% versus 79%; P = 0.27 by Fisher exact test). Comparison of GMTs found no significant differences at day 30 post-doses 1 and 2, and investigation of vaccine potency (below) did not reveal a difference in the vaccines administered. The pooled seroconversion rates 30 days after dose 2 were 55.2% for DENV-1, 100% for DENV-2, 86.2% for DENV-3, and 96.4% for DENV-4 neutralizing antibodies (Figure 2) with 1 monovalent, 3 bivalent, 9 trivalent, and 15 (53.6%) tetravalent responders (one subject's serum was of inadequate volume to test for antibodies to DENV-2; this subject had at least a bivalent response). In the pooled full-dose DENV vaccine groups, the highest GMT was to DENV-2 (239.6), and the next highest GMT was to DENV-4 (52.5); GMTs were lowest to DENV-3 (29.2) and DENV-1 (20.7). GMTs declined in the range of 1.6- to 1.8-fold over the next 45 days. At 1 year after DENV vaccine dose 2, the DENV-2 seropositivity rate remained > 90%, whereas the rates for the other three serotypes substantially declined.
Figure 2.
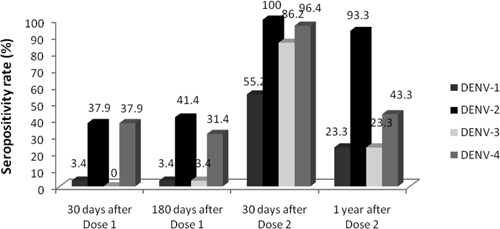
Percentage of subjects in the pooled full-dose DENV group with neutralizing antibodies to each DENV serotype at four time points after vaccination (per-protocol cohort).
JEV vaccine safety and immunogenicity.
The administration of JEV vaccine as recommended by the Thai national immunization program allowed us to make a preliminary assessment of the impact of preceding DENV vaccination. There were no clinically important differences in the occurrence of injection site or solicited general symptoms after JEV vaccination between the DENV and control groups (data not shown).
Thirty days after the second dose of JEV vaccine (given 1 and 1.5 months after dose 2 of DENV/control vaccine), 9 of 28 (32.1%) in the full-dose DENV vaccine group, 4 of 4 (100%) in the low-dose DENV vaccine group, and 9 of 16 (56.3%) in the control group had JEV antibodies titers ≥ 1:10. Respectively, the GMTs were 12.4, 19.3, and 16.8. Although use of the heterologous Nakayama JEV strain in the PRNT assay may have modestly underestimated immunogenicity, the differences among groups were not statistically significant (P > 0.05).
Discussion
We observed in this pilot study that the WRAIR/GSK tetravalent DENV vaccine candidate, when administered as two doses over 6 months to flavivirus-naive infants 12 to 15 months of age, was well-tolerated and moderately immunogenic. We considered this age range as the earliest time point to elicit a potentially protective immune response unmodified by anti-DENV maternal antibodies, which have been determined to wane by 12 months of age.20
The study used sequential escalation of dose and cohort size to minimize risk to subjects. Before advancing to a subsequent cohort, safety data from the preceding cohort were reviewed by an IDMC. There were no safety issues that affected conduct of the study. The study compliance rate was high with > 98% of participants returning the diary cards after vaccination; 96% were included in the per-protocol immunogenicity analysis.
The full-dose DENV vaccine was well-tolerated; its overall safety profile was similar to that of the control vaccines that were both live and inactivated. The low-dose DENV vaccine did not confer any safety advantages compared with the full-dose DENV vaccine. There were no DENV vaccine-related SAEs reported during the study. Mild to moderate injection site redness was the most frequently reported solicited local reaction after DENV vaccination. General symptom reporting was similar from doses 1 to 2, except for elevated temperature, which was reported more frequently after dose 2.
Physical examination of subjects did not detect findings indicative of overt DENV infection except for rash in a minority of subjects, some of whom had transient fever. One subject who developed a generalized rash 11 days after dose 1 also had grade 3 fever (that lasted for 1 day).
One subject had evidence of transient and asymptomatic liver injury manifested as an increase in AST and ALT (with an elevated AST and ALT before dose 1) in the immediate post-vaccination periods. The evidence of injury was most notable after dose 2 and was associated with detection of low-level DENV-4 viremia. Timing of the injury suggests association with the vaccine; future studies should assess this phenomenon.
One subject developed dengue fever because of natural infection with DENV-4 approximately 3 months after dose 1 of full-dose DENV vaccine. The subject was immunologically primed by vaccine dose 1 (secondary antibody response to wild-type DENV infection) despite no detection of neutralizing antibodies to any DENV serotype 30 days post-dose 1. Although the vaccine-related priming did not prevent dengue fever, we cannot know whether it altered the ultimate severity of the illness. However, the subject did not experience prolonged fever, plasma leakage, hemorrhage, or other hallmarks of severe dengue.
All subjects were evaluated for viremia on day 10 after vaccinations, as this is the most likely time point for vaccine-related viremia. Additionally, blood was drawn from four febrile subjects within 21 days of each DENV/control vaccination. The investigators acknowledge that practical considerations limiting more frequent venipunctures in infants invariably led to an underestimate of the true occurrence of dengue viremia in DENV vaccine recipients. Asymptomatic viremia caused by DENV-4 and DENV-2, measured on day 10 after the full-vaccine dose, appeared in seven subjects: five after dose 1 (three DENV-4 and two DENV-2) and two after dose 2 (DENV-4). Four of five instances of DENV-4 viremias were determined to be caused by vaccine viruses according to phylogenetic analyses; presumably, the low virus titers for the fifth DENV-4 instance and the two DENV-2 instances precluded molecular assessment of phylogeny. This viremia experience is consistent with that reported in older Thai children receiving the DENV vaccine candidate.12
Molecular analyses of four DENV-4 vaccine viremia isolates recovered at sufficient titer to permit consensus sequence analysis revealed a consistent amino acid change, among others, at amino acid position 383 in domain III of the E-genome identical to that observed in three subjects in our first pediatric trial.12 In our cumulative pediatric experience, despite levels of viremia ranging from 4.2 to 7.9 log genome equivalents per 1 mL, the subjects were not ill with dengue. In response to these vigorous infections, the four subjects developed neutralizing antibodies to DENV-4 that exceeded the GMT for the full-dose vaccine group as a whole. No further analysis of these viremia isolates was performed, and we do not know whether this E383 variant existed at a low level as a quasispecies in the uncloned virus seed used to prepare the vaccine. Nevertheless, this question is less relevant for future development of this vaccine candidate, because the virus seeds have been rederived by transfection of purified RNA to enhance their characterization. In recently completed clinical trials of the vaccine candidate made with the post-transfection seeds, DENV-4 viremia occurred uncommonly (two instances in 122 adult subjects) and at low levels (1.7 and 2.1 log genome equivalents per 1 mL), despite frequent sampling (Thomas SJ, personal communication).
Of the seven viremias, all were positive by nested PCR. One viremia (subject 12) was positive by the nested PCR but not by qRT-PCR and mosquito inoculation. Although this may represent artifact, our previous experience suggests that the nested PCR is more sensitive than qRT-PCR for DENV-4 alone.17 Consequently, this likely represents a very low-grade viremia.
Of interest and consistent with previous studies, DENV-4 vaccine viremias were uniformly higher than DENV-2 viremias.
Low-dose DENV vaccine administered to only four subjects (cohort A) was well-tolerated but seemed to offer reduced immunogenicity, suggesting that reductions in vaccine potency to levels administered to cohort A may provide ineffective immunization.
The full-dose DENV vaccine given two times, 6 months apart, elicited either trivalent or tetravalent seroconversions in 85.7% of vaccinees at 30 days after the second dose. We observed a possible cohort effect between cohorts B and C; immune responses especially to DENV-1 appeared to be reduced in cohort C compared with those responses in cohort B (Figure 1). DENV vaccine potency measured by IFA was consistently within the expected range for all serotypes. All subjects were flavivirus-naive based on both the HAI and PRNT50 assays against DENV and JE virus conducted at screening and the PRNT50 assay on the day that vaccine dose 1 was administered. Demographic differences, mean subject ages (13.3 versus 13.6 months, respectively), and differences in maternal breastfeeding (on retrospective questioning) could not explain the cohort effect between cohorts B and C. Possible explanations include (1) variability in the PRNT assay, (2) chance variation for one endpoint among many tested with no correction for multiplicity, (3) uncertainty inherent with the use of small group sizes, and (4) undefined host differences between cohorts.
Despite the potential heterogeneity between cohorts B and C, we believe that the presentation of pooled data is justified for an exploratory study. The immunogenicity elicited was generally consistent with that determined in prior studies with flavivirus-naive adults and children (i.e., tetravalent neutralizing antibodies in 88–100% of vaccine recipients with strong immune responses to DENV-2 and DENV-4 components and lower responses to DENV-1 and DENV-3 components).12
Immune responses were highest 30 days after dose 2 and decreased less than twofold in the next 45 days. By 1 year after dose 2, we observed a notable decrease in seropositivity rates for neutralizing antibodies to all DENV serotypes except DENV-2. The drop in seroconversions to DENV-4, despite evidence of DENV-4 replication (multiple day 10 viremias), suggests that these 1-year seroconversions underestimate the homotypic DENV-4 immunity elicited by live virus vaccinations. Live virus vaccination likely evokes broad-based antigenic priming to the full complement of structural and non-structural proteins, resulting in a memory response upon subsequent exposure to wild-type dengue viruses, regardless of serotype. Long-term persistence of anti-DENV neutralizing antibodies in this cohort is being evaluated further and will be reported separately. The impact of declining levels of neutralizing antibodies on protective immunity is uncertain, but it may reflect reduced immunity, as described below.
The measure of immunogenicity used in this clinical trial was the PRNT, which measures the ability of serum immunoglobulin to neutralize the infectivity of a virus inoculum in vitro by binding to surface proteins of the DENV, principally the envelope protein. Although there are serotype-specific epitopes on the envelope protein, there are also more immunodominant, broadly reactive epitopes as well. Given the lack of PRNT serotype specificity, it is impossible using only this test to confirm that all four viruses in the vaccine replicated to a degree adequate to elicit a protective immune response. Moreover, any inference of protection based on a serum neutralizing antibody response is unreliable, because the minimum level of neutralizing antibody correlated with protection from disease is unknown. However, it is reasonable to postulate that the risk of breakthrough disease in an immunized individual is proportional to the level of homotypic neutralizing antibody induced shortly after live virus vaccination, because the neutralizing antibody response is presumably correlating with the total adaptive immune response, including all humoral and cellular mechanisms. Thus, higher levels of neutralizing antibody 1 month after live virus vaccination portend better protection than lower levels. It is possible that the presence of detectable neutralizing antibody to a given DENV serotype 6 to 12 months after vaccination may be a more specific indicator of the induction of homologous protective immunity compared with that detected 1 month after vaccination, because maturation of the adaptive immune response preserves production of the highest affinity antibodies by long-lived plasma cells in the bone marrow. Nevertheless, the increased specificity of this late antibody readout is speculative and also likely increases the proportion of vaccine recipients judged falsely to be non-responders. These uncertainties regarding the correlation of the acute post-vaccination antibody response to long-term protection highlight the importance of evaluating the durability of long-term protective effect of vaccination based on clinical endpoints during the development of any DENV vaccine candidate.
All participants received a first dose of inactivated JEV vaccine at 19 to 22 months of age in accordance with the Thai national immunization program. To minimize the risk of Japanese encephalitis, subjects were asked not to travel outside of Bangkok, a low-risk area for JEV transmission, before receiving JEV vaccination. Prior receipt of DENV vaccine did not seem to influence the reactogenicity of JEV vaccine, but the seroconversion rate and GMTs achieved after two JEV vaccine doses seemed to be reduced in recipients of the full-dose DENV vaccine. Although not achieving statistical significance, these findings should be further evaluated in subsequent studies of any DENV vaccine candidate intended for infants in countries where both DENV and JEV cocirculate and where JEV vaccination is recommended.
The safety and reactogenicity results from this study support development of the vaccine candidate in the 12- to 15-month age group. Vaccination at this early age has potential advantages in reducing the overall pediatric disease burden and preventing secondary transmission within the household.
ACKNOWLEDGMENTS
The authors thank the parents and their infants for their outstanding cooperation with the study requirements. We acknowledge the dedicated support of the staff of the Department of Pediatrics, Phramongkutklao Hospital, Department of Virology, Armed Forces Research Institute of Medical Sciences (AFRIMS), and Pilot Bioproduction Facility and Department of Virus Diseases, Walter Reed Army Institute of Research (Silver Spring, MD). We express gratitude to the AFRIMS vaccination team led by Dr. Chusak Pimgate. The authors thank the study's medical monitor, Dr. Suchitra Nimmannitya, and the members of the IDMC–Dr. Prayura Kunasol (chairperson), Dr. Nadhirat Sangkawibha, Dr. Pricha Singharaj, Dr. Kamnuan Ungchusak, and Dr. Pote Aimpun. We acknowledge support from Dr. Charles English and Dr. Wellington Sun from the US Army and Dr. Beth-Ann Coller, Dr. Yves Lobet, Dr. Jean-Francois Toussaint, Dr. Yanee Hutagalung, and Ms. Celia Barberousse from GlaxoSmithKline during study conduct.
Disclaimer: Some of the authors are employed by GlaxoSmithKline and hold stock in the company. J.R.P., K.H.E. and B.L.I. are named as inventors on the dengue live attenuated vaccine patents. These statements are made in the interest of full disclosure and not because the authors believe that there is a conflict of interest. The opinions or assertions contained herein are the private views of the authors and are not to be construed as reflecting the official views of the US Department of Defense or the Royal Thai Army. Priya Pavithran, Dr. Roselynn Tien, and Ms. Julia Donnelly (GlaxoSmithKline) assisted in writing this report. Varilix and Hiberix are trademarks of the GlaxoSmithKline group of companies.
Footnotes
Financial support: This work was funded by US Army Medical Research and Materiel Command (Fort Detrick, MD) and GlaxoSmithKline (Rixensart, Belgium).
Authors' addresses: Veerachai Watanaveeradej, Sriluck Simasathien, Rudiwilai Samakoses, and Angkool Kerdpanich, Department of Pediatrics, Phramongkutklao Hospital, Bangkok, Thailand, E-mails: veerachaiw@yahoo.com, ssriluck@hotmail.com, rudiwilai_samakoses@hotmail.com, and akpmk@yahoo.com. Ananda Nisalak, Richard G. Jarman, Stephen J. Thomas, Robert V. Gibbons, and Sumetha Hengprasert, Department of Virology, Armed Forces Research Institute of Medical Sciences, Bangkok, Thailand, E-mails: ananda.nisalak@afrims.org, richard.jarman@afrims.org, Stephen.thomas@afrims.org, Robert.gibbons@afrims.org, and sumethah@afrims.org. Timothy P. Endy, Infectious Disease Division, Department of Medicine, State University of New York, Upstate Medical University, Syracuse, NY, E-mail: endyt@upstate.edu. Bruce L. Innis and David W. Vaughn, GlaxoSmithKline, King of Prussia, PA, E-mails: bruce.2.innis@gsk.com and david.w.vaughn@gsk.com. J. Robert Putnak, Division of Viral Diseases, Walter Reed Army Institute of Research, Silver Spring, MD, E-mail: robert.putnak@amedd.army.mil. Kenneth H. Eckels and Rafael De La Barrera, Pilot Bioproduction Facility, Division of Regulated Activities, Walter Reed Army Institute of Research, Silver Spring, MD, E-mails: kenneth.eckels@amedd.army.mil and rafael.delabarrera1@us.army.mil. Mammen P. Mammen Jr., ATTN: Dengue Product Manager, Pharmaceutical Systems Program Management Office, U.S. Army Medical Materiel Development Activity, Fort Detrick, MD, E-mail: mammen.mammen@us.army.mil.
References
- 1.Forattini OP. Epidemiology and phylogenetic relationships of dengue viruses. Dengue Bull. 2003;27:91–94. [Google Scholar]
- 2.Monath TP. Dengue: the risk to developed and developing countries. Proc Natl Acad Sci USA. 1994;91:2395–2400. doi: 10.1073/pnas.91.7.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhry S, Swaminathan S, Khanna N. Viral genetics as a basis of dengue pathogenesis. Dengue Bull. 2006;30:121–132. [Google Scholar]
- 5.Pediatric Dengue Vaccine Initiative (PDVI) Global Burden of Dengue. 2010. http://www.pdvi.org/about_dengue/GBD.asp Available at. Accessed April 12, 2010.
- 6.Nisalak A, Endy TP, Nimmannitya S, Kalayanarooj S, Thisayakorn U, Scott RM, Burke DS, Hoke CH, Innis BL, Vaughn DW. Serotype-specific dengue virus circulation and dengue disease in Bangkok, Thailand from 1973 to 1999. Am J Trop Med Hyg. 2003;68:191–202. [PubMed] [Google Scholar]
- 7.World Health Organization . Fact Sheet Number 117. Geneva, Switzerland: World Health Organization; 2009. [Google Scholar]
- 8.Whitehead SS, Blaney JE, Durbin AP, Murphy BR. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5:518–528. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 9.DeRoeck D, Deen J, Clemens JD. Policymakers' views on dengue fever/dengue haemorrhagic fever and the need for dengue vaccines in four southeast Asian countries. Vaccine. 2003;22:121–129. doi: 10.1016/s0264-410x(03)00533-4. [DOI] [PubMed] [Google Scholar]
- 10.Eckels KH, Dubois DR, Putnak R, Vaughn DW, Innis BL, Henchal EA, Hoke CH., Jr Modification of dengue virus strains by passage in primary dog kidney cells: preparation of candidate vaccines and immunization of monkeys. Am J Trop Med Hyg. 2003;69:12–16. doi: 10.4269/ajtmh.2003.69.12. [DOI] [PubMed] [Google Scholar]
- 11.Innis BL, Eckels KH. Progress in development of a live-attenuated, tetravalent dengue virus vaccine by the United States Army Medical Research and Materiel Command. Am J Trop Med Hyg. 2003;69:1–4. doi: 10.4269/ajtmh.2003.69.6_suppl.0690001. [DOI] [PubMed] [Google Scholar]
- 12.Simasathien S, Thomas SJ, Watanaveeradej V, Nisalak A, Barberousse C, Innis BL, Sun W, Putnak RJ, Eckels KH, Hutagalung Y, Gibbons RV, Zhang C, De La Berrera R, Jarman RG, Chawachalasai W, Mammen MP., Jr Safety and immunogenicity of a tetravalent live-attenuated dengue vaccine in flavivirus naive children. Am J Trop Med Hyg. 2008;78:426–433. [PubMed] [Google Scholar]
- 13.Sun W, Cunningham D, Wasserman SS, Perry J, Putnak JR, Eckels KH, Vaughn DW, Thomas SJ, Kanesa-Thasan N, Innis BL, Edelman R. Phase 2 clinical trial of three formulations of tetravalent live-attenuated dengue vaccine in flavivirus-naive adults. Hum Vaccin. 2009;5:33–40. doi: 10.4161/hv.5.1.6348. [DOI] [PubMed] [Google Scholar]
- 14.Nutrition Division, Department of Health, Ministry of Public Health . National Growth References for Thai Children Under 20 Years of Age. Bangkok, Thailand: War Veterans Organization; 1999. pp. 28–83. [Google Scholar]
- 15.Russell PK, Nisalak A, Suhhavachana P, Vivona S. A plaque reduction test for dengue neutralization antibodies. J Immunol. 1967;99:285–290. [PubMed] [Google Scholar]
- 16.Klungthong C, Gibbons RV, Thaisomboonsuk B, Nisalak A, Kalayanarooj S, Thirawuth V, Nutkumhang N, Mammen MP, Jr, Jarman RG. Dengue virus detection using whole blood for reverse transcriptase PCR and virus isolation. J Clin Microbiol. 2007;45:2480–2485. doi: 10.1128/JCM.00305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadon N, Delers A, Jarman RG, Klungthong C, Nisalak A, Gibbons RV, Vassilev V. A new quantitative RT-PCR method for sensitive detection of dengue virus in serum samples. J Virol Methods. 2008;153:1–6. doi: 10.1016/j.jviromet.2008.06.023. [DOI] [PubMed] [Google Scholar]
- 18.Innis BL, Nisalak A, Nimmannitya S, Kusalerdchariya S, Chongswasdi V, Suntayakorn S, Puttisri P, Hoke CH., Jr An enzyme-linked immunosorbent assay to characterize dengue infections where dengue and Japanese encephalitis co-circulate. Am J Trop Med Hyg. 1989;40:418–427. doi: 10.4269/ajtmh.1989.40.418. [DOI] [PubMed] [Google Scholar]
- 19.International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) Medical Dictionary for Regulatory Activities and Maintenance and Support Services Organization. 2011. 2011. http://www.meddramsso.com Version 14.0 01, March. Available at. Accessed May 26.
- 20.Watanaveeradej V, Endy TP, Samakoses R, Kerdpanich A, Simasathien S, Polprasert N, Aree C, Vaughn DW, Ho C, Nisalak A. Transplacentally transferred maternal-infant antibodies to dengue virus. Am J Trop Med Hyg. 2002;69:123–128. [PubMed] [Google Scholar]