

Abstract
Objective
Docetaxel causes cell death through induction of apoptosis; however, cell death characteristics for docetaxel have not yet been fully elucidated. We examined the role of focal adhesion kinase (FAK) cleavage in docetaxel-mediated apoptosis.
Methods
FAK degradation after treatment with docetaxel was determined in both taxane-sensitive (HeyA8 and SKOV3) and taxane-resistant (HeyA8-MDRand SKOV3-TR) ovarian cancer celllines by Westernblot analysis. Cell growth was determined with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. FAK-targeting small interfering RNA (siRNA) was used to decrease FAK expression. Apoptosis and caspase activity were determined using commercially available kits.
Results
SKOV3 and HeyA8 celllines were both sensitive to docetaxel (IC50 levels,1–6.2 nmol/L), whereas the SKOV3-TR and HeyA8-MDR cells were resistant (IC50 ≥250 nmol/L for both). Docetaxel induced high rates of apoptosis in SKOV3 and HeyA8 cells (84% and 66% apoptosis, respectively) but minimal apoptosis (5–8%) in SKOV3-TR and HeyA8-MDR cells. Similarly, FAK was cleaved in SKOV3 and HeyA8 cells in response to docetaxel treatment but unchanged in the resistant cells. Caspase-3 and caspase-8 activity also increased significantly in docetaxel-treated SKOV3 and HeyA8 cells but not in the taxane-resistant cells. DEVD-fmk (caspase-3 blocker) was able to block both FAK cleavage and apoptosis mediated by docetaxel in SKOV3 and HeyA8 cells. FAK siRNA transfection resulted in 70% to 90% decrease in FAK levels in all cell lines within 72 hours. FAK silencing augmented docetaxel-mediated growth inhibition (5- to 8-fold increase) and apoptosis in both of the taxane-sensitive and taxane-resistant cell lines.
Conclusions
Docetaxel induces FAK cleavage, mediated through activation of caspase-3, in taxane-sensitive ovarian cancer cells but not in taxane-resistant cells. The absence of FAK degradation may contribute to cell survival in taxane-resistant cells. FAK silencing promotes the in vitro efficacy of docetaxel in both taxane-sensitive and taxane-resistant cell lines and may serve as a novel therapeutic approach.
Ovarian cancer is the leading cause of death from a gynecologic malignancy (1). The high mortality is mainly due to advanced stage at presentation in most patients. Despite the standard therapy of surgical cytoreduction and systemic platinum and taxane combination, most patients develop recurrent disease and eventually succumb to their disease (2). Therefore, novel therapeutic approaches are urgently needed for ovarian carcinoma.
The taxanes docetaxel and paclitaxel have assumed an important role in both the primary and salvage treatment of ovarian cancer (3). Taxanes promote both polymerization (tubulin assembly in microtubules) and inhibit depolymerization of microtubules causing a mitotic arrest (4). However, the molecular events leading to apoptosis by taxanes are not fully understood. It has been shown that paclitaxel inhibits mitosis in metaphase by stabilizing microtubule dynamics instead of altering microtubule polymer mass (5). In addition to a G2-M arrest, paclitaxel-induced apoptosis can also be initiated in the S phase of the cell cycle (6). Modulation of several apoptosis-related proteins, including p53, p21, Bcl-2, and Bax, has been shown upon treatment with paclitaxel (7–10).
Extracellular matrix molecules regulate cell function by acting as both structural support and signaling mediators (11). Focal adhesion kinase (FAK) is a multifunctional nonreceptor protein tyrosine kinase that localizes to sites of attachment to the extracellular matrix and participates in cell adhesion–induced signaling (12, 13). FAK phosphorylation at Tyr397, a residue lying immediately NH2 terminal to the catalytic domain, is vital for its biochemical and biological functions (12, 13). FAK plays a role in cell migration, invasion, and proliferation (14–17). FAK also functions in the transmission of a cell adhesion–dependent cell survival signal (18, 19). Recent evidence suggests that FAK is also protective against apoptosis induced by a variety of agents (20–24). The antiapoptotic functions of FAK are mediated, in part, via mitogen-activated protein kinase and protein kinase B/Akt activation (25–27). FAK is proteolytically cleaved during induction of apoptosis, and caspases have been suggested to be involved (21, 28). Sasaki et al. have shown that cisplatin-induced FAK cleavage and processing is in part mediated by caspase-3 (29). However, the effect of docetaxel on FAK and its potential relevance for cell survival are not known in ovarian carcinoma.
We have previously shown that FAK overexpression in ovarian carcinoma is predictive of poor clinical outcome, and FAK plays a functionally significant role in ovarian cancer migration and invasion (30). In the present study, we examined the functional role of FAK in docetaxel-mediated apoptosis, and whether silencing FAK sensitizes ovarian cancer cells to docetaxel.
Materials and Methods
Cell culture
The human epithelial ovarian cancer cell lines SKOV3 and HeyA8 (31, 32) and the taxane-resistant counterparts of these cell lines, SKOV3-TR (SKOV3 taxane resistant, a kind gift of Dr. Michael Seiden, Department of Medicine, Massachusetts General Hospital, Boston, MA; ref. 33) and HeyA8-MDR (a kind gift of Dr. Isaiah J. Fidler, Department of Cancer Biology, M.D. Anderson Cancer Center, Houston, TX; ref. 34), were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 0.1% gentamicin sulfate (Gemini Bioproducts, Calabasas, CA) in 5% CO2/95% air at 37°C. In vitro experiments were done with 70% to 80% confluent cultures.
Small interfering RNA
Two small interfering RNA (siRNA) constructs were purchased from Qiagen (Germantown, MD): a control sequence with no homology to any human mRNA (as determined by BLAST search; target sequence 5′-AATTCTCCGAACGTGTCACGT-3′) and a sequence designed to target FAK mRNA (target sequence 5′-AAC-CACCTGGGCCAGTATTAT-3′), previously shown to down-regulate FAK (27). Lyophilized product was reconstituted in the provided buffer [100 mmol/L potassium acetate, 30 mmol/L HEPES potassium hydroxide, 2 mmol/L magnesium acetate (pH 7.4)], heated to 90°C for 60 seconds, incubated at 37°C for 60 minutes, and stored at −20°C until used.
Transfection of small interfering RNA
Cells (5 × 105) were plated in 35-mm plates and allowed to adhere for 24 hours. On the day of transfection, 15 μL of RNAiFect Transfection Reagent (Qiagen) was added to 100 nmol/L siRNA (12 μL) in EC-R buffer (73 μL; Qiagen) to give a final volume of 100 μL, incubated at room temperature for 15 minutes, and added to plates in serum-containing media for 16 hours. Cells were collected as lysates or subjected to the indicated experiment.
Western blot analysis
Cells were lysed in modified radioimmunoprecipitation assay buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% triton, 0.5% deoxycholate plus 25 μg/mL leupeptin, 10 μg/mL aprotinin, 2 mmol/L EDTA, and 1 mmol/L sodium orthovanadate; Sigma Chemical Co., St. Louis, MO) as previously described (30). Cells were removed by scraping and centrifuged at 12,500 rpm for 30 minutes. The protein concentration of the supernatant was determined using a bicinchoninic acid protein assay reagent kit (Pierce, Rockford, IL), and whole-cell lysates were analyzed by 7.5% SDS-PAGE. Samples were transferred to nitrocellulose by semidry transfer (Bio-Rad Laboratories, Hercules, CA), blocked with 5% nonfat milk, and incubated with 0.25 μg/mL anti-FAK antibody (Biosource International, Camarillo, CA) for 1 hour at room temperature. Antibody was detected with 0.167 μg/mL horseradish peroxidase–conjugated anti-mouse secondary antibody (The Jackson Laboratory, Bar Harbor, ME) and developed with an enhanced chemiluminescence detection kit (Pierce). Equal loading was confirmed by detection of β-actin. Densitometric analysis was done by Scion Imaging software (Scion Corp., Frederick, MD).
For experiments regarding FAK processing, 1.5 × 106 cells were plated in a six-well plate overnight. On the following day, the medium was replaced with serum-free medium containing MITO+ (serum extender for serum-free conditions; BD Biosciences, San Jose, CA). Docetaxel was added at IC50 concentrations for 48 to 72 hours. At the time of harvest, both the detached and attached cells were removed and lysed with modified radioimmunoprecipitation assay buffer, as described above. For experiments regarding the role of caspase-3, DEVD-fmk (20 μmol/L; BD Biosciences/Clontech, Palo Alto, CA) was added 1 hour before adding docetaxel.
Cytotoxicity assay
To determine the IC50 concentration of cytotoxic drugs under different conditions, 2 × 103 cells were seeded onto 96-well plates and incubated overnight at 37°C, after which either control or FAK-targeting siRNA was added. The medium was exchanged after 48 hours with increasing concentrations of docetaxel (obtained from Aventis Pharma, Bridgewater, NJ) dissolved in ethanol (final concentration range, 0.1–10,000 nmol/L prepared in medium) or cisplatin (purchased from LKT Laboratories, Inc., St. Paul, MN) dissolved in water. After a 96-hour incubation, 50 μL of 0.15% 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added to each well and incubated for 2 hours. The supernatant was removed, and cells were dissolved in 100 μL DMSO. The absorbance at 570 nm was recorded using a FALCON microplate reader (Becton Dickinson Lab ware, Bedford, MA), and cell survival expressed as a percent increase or decrease above control conditions, after subtracting blank A 570 readings. The IC50 concentration was determined by finding the A 570 reading midpoint between maximal and minimal readings and finding the chemotherapy concentration that intersects the growth curve at that A 570 reading.
Terminal deoxynucleotidyl transferase–mediated nick end labeling assay
Terminal deoxynucleotidyl transferase – mediated nick end labeling–positive cells were detected using Dead End Fluorometric terminal deoxynucleotidyl transferase–mediated nick end labeling system (APO-DIRECT, BD Biosciences/PharMingen, San Diego, CA) according to the manufacturer’s instructions. Briefly, ovarian cancer cells in culture were treated with varying concentrations of docetaxel and harvested after 12, 24, 48, or 96 hours. Cells were fixed with 4% paraformaldehyde solution for 25 minutes on ice. Intracellular DNA fragments were then labeled by exposing cells to fluorescein-12-dUTP, treated with propidium iodide and RNase A solution, and analyzed by flow cytometry (EPICS XL, Beckman Coulter, Miami, FL). The percentages of apoptotic cells were averaged over three consecutive experiments.
Caspase activity
Ovarian cancer cells (1 × 106) in culture (six-well plates) were treated with or without docetaxel at the cell line specific IC50 or IC90 concentration for 24 hours. After washing with PBS, cells were lysed in 50 μL of cell lysis buffer provided. Caspase-3, caspase-8, and caspase-9 activities were measured with the appropriate apoptosis detection kit (BD Biosciences/Clontech, Palo Alto, CA), using substrate DEVD-AFC for caspase-3, IETD-AFC for caspase-8, and LEHD-AMC for caspase-9.
Reagents
Docetaxel was purchased from Sanofi-Aventis (Bridgewater, NJ). Cisplatin was purchased from LKT Laboratories. Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Statistical analysis
Differences were analyzed using Student’s t test, and P < 0.05 was considered significant. All experiments were done at least in triplicate. The Statistical Package for the Social Sciences (SPSS, Inc., Chicago, IL) was used for all analyses.
Results
In vitro sensitivity of ovarian cancer cell lines to docetaxel
We first analyzed the effect of docetaxel on the growth of ovarian cancer cell lines by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay at doses ranging from 0.1 to 10,000 nmol/L. Growth of ovarian cancer cells was inhibited in a dose-dependent manner (Fig. 1). The IC50 levels for the SKOV3 and HeyA8 cells were 6.2 and 1 nmol/L, respectively. The IC50 levels for SKOV3-TR and HeyA8-MDR were 225 and 450 nmol/L, respectively (Fig. 1). Therefore, the SKOV3-TR and HeyA8-MDR cell lines were 36- and 300-fold more resistant to docetaxel, respectively.
Fig. 1.

Effect of docetaxel on ovarian cancer cell growth: (A) HeyA8 and HeyA8-MDR or (B) SKOV3 or SKOV3-TR cells were plated in 96-well plates and subsequently incubated with increasing concentrations of docetaxel for 96 hours, and cell viability was determined. Points, mean of three independent experiments; bars, SE.
Caspase activity increases with docetaxel therapy
Caspase activity is known to play an important role in cisplatin-mediated apoptosis, with caspase-8 and caspase-9 as initiators and caspase-3 and caspase-7 as executors (35). Thus, we assayed caspase-3, caspase-8, and caspase-9 by testing the conversion of the substrates DEVD-AFC (caspase-3), IETD-AFC (caspase-8), and LEHD-AMC (caspase-9) into free AFC (for caspase-3 and caspase-8) and AMC (for caspase-9) after treatment with docetaxel using both IC50 and IC90 levels (Fig. 2). Cisplatin was used as a positive control. Caspase-3 activity increased in a dose-dependent manner in the SKOV3 (2.8-fold with IC50 and 5.4-fold with IC90 levels of docetaxel) and HeyA8 (1.8-fold with IC50 and 2.5-fold with IC90 levels of docetaxel) cell lines. Caspase-8 activity increased by 2.8- to 3-fold in SKOV3 cells and by 2.8- to 2.9-fold in HeyA8 cells. Caspase-9 activity increased by 1.35- to 1.64-fold in SKOV3 and 1.5- to 1.6-fold in HeyA8 cells. In contrast, docetaxel treatment (using IC50 and IC90 levels for sensitive cell lines) was not able to induce caspase activity in the resistant cell lines SKOV3-TR and HeyA8-MDR.
Fig. 2.

Effect of docetaxel on caspase-3 (A), caspase-8 (B), and caspase-9 (C) activity. Ovarian cancer cells were treated with docetaxel for 24 hours followed by fluorometric profiling of caspase activity using a commercially available kit. Columns, means of three independent experiments; bars, SE.
Focal adhesion kinase is cleaved in response to docetaxel
It has been reported that FAK may be cleaved by caspase-3 (21, 29, 36). Based on our observations regarding modulation of caspase levels by docetaxel in ovarian cancer cells, we next examined whether FAK is cleaved after treatment with docetaxel and whether there are differences in extent of FAK cleavage between the taxane-sensitive and taxane-resistant cell lines. In SKOV3 cells, FAK was cleaved after 48 and 72 hours of exposure to docetaxel (Fig. 3A). The 71-kDa fragments increased after treatment with docetaxel, whereas the 125-kDa fragments decreased. Similar findings were observed with the HeyA8 cells (data not shown). However, taxane-resistant SKOV3-TR and HeyA8-MDR cells did not show any increase in FAK cleavage products after treatment with docetaxel (Fig. 3B). To test the dependence of FAK cleavage on caspase-3, the effect of DEVD-fmk (caspase-3 inhibitor) was examined. In the presence of DEVD-fmk, docetaxel-induced FAK cleavage was blocked. These results suggest that docetaxel-mediated FAK cleavage occurs via caspase-3.
Fig. 3.

Docetaxel treatment results in FAK cleavage. The taxane-sensitive (A) and taxane-resistant (B) SKOV3 cells were cultured in the presence of docetaxel for 48 or 72 hours. Western blot analysis for FAK in SKOV3 and SKOV3-TR cells in the presence or absence of the caspase-3 inhibitor DEVD-fmk.
Focal adhesion kinase is down-regulated with focal adhesion kinase–specific small interfering RNA
Next, we sought to examine whether FAK silencing using siRNA will sensitize ovarian cancer cells to docetaxel. We first optimized the transfection conditions for these cells lines by testing various ratios of rhodamine-conjugated nonsilencing siRNA to RNAifect Reagent (μg/μL) and determined that a ratio of 1:6 was optimal and resulted in about 90% transfection efficiency. Treatment with FAK siRNA, but not control (nonsilencing) siRNA, resulted in a detectable decrease in FAK expression after 24 hours of treatment and maximal suppression of 90% after 72 hours of transfection (Fig. 4A). Actin expression was unaffected by either control or FAK siRNA treatment, indicating that nonspecific down-regulation of protein expression did not occur.
Fig. 4.
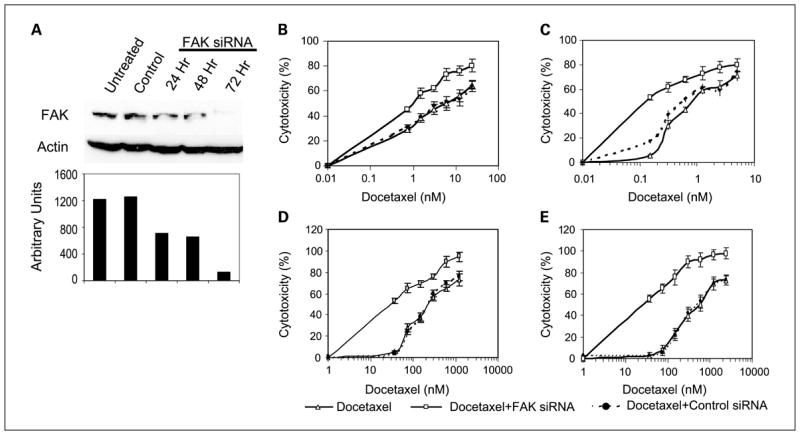
Effect of FAK silencing on docetaxel-sensitivity in ovarian cancer cell lines. A, Western blot analysis of SKOV3 whole-cell lysates probed with anti-FAK and anti-actin monoclonal antibodies. Bottom, densitometry results. Control siRNA did not significantly affect FAK expression compared with untreated cells whereas FAK-specific siRNA resulted in >90% suppression of FAK expression by 72 hours. Effects of docetaxel on (B) SKOV3, (C) HeyA8, (D) SKOV3-TR, and (E) HeyA8-MDR growth were tested alone or in combination with FAK siRNA or control siRNA. Points, means of three independent experiments; bars, SE.
Focal adhesion kinase down-regulation sensitizes ovarian cancer cells to docetaxel
The effect of FAK silencing using siRNA on in vitro sensitivity of human ovarian cancer cells to docetaxel was determined. Ovarian cancer cells were exposed to FAK siRNA or a nonsilencing siRNA for 48 hours before treatment with increasing concentrations of docetaxel. The fold differences were calculated at the point where 50% cytotoxicity was observed. Docetaxel cytotoxicity was almost 5-fold greater after FAK down-regulation in SKOV3 and 8-fold greater in HeyA8 cells (Fig. 4B and C). Next, we examined the effects of FAK silencing in the taxane-resistant SKOV3-TR and HeyA8-MDR cells. Similar findings were observed with the resistant cell lines (4.8-fold increase in sensitivity in SKOV3-TR and 5.5-fold increase in HeyA8-MDR cells; Fig. 4D and E). Control siRNA treatment did not affect sensitivity of ovarian cancer cells to docetaxel.
We then examined caspase-3 activity in response to FAK silencing and docetaxel treatment. Caspase-3 activity was enhanced by docetaxel after FAK down-regulation in the taxane-sensitive cell lines (Fig. 5A) but not in the resistant cell lines (Fig. 5B). Treatment with FAK siRNA or control siRNA alone did not potentiate the activity of caspase-3 in either taxane-sensitive or taxane-resistant cell lines (Fig. 5A and B). Caspase-3 activity was 6.1-fold higher in the SKOV3 cells and 4.8-fold higher in the HeyA8 cells after treatment with FAK siRNA in combination with docetaxel (both Ps < 0.001 compared with treatment with vehicle alone). Caspase-3 activity was also enhanced by docetaxel after FAK down-regulation in the resistant cell lines (Fig. 5B). Thus, down-regulation of FAK alone does not increase caspase-3 activity, but FAK inhibition enhances the docetaxel-mediated induction of caspase-3 activity.
Fig. 5.

FAK gene silencing potentiates docetaxel-induced caspase-3 activity in (A) SKOV3 and HeyA8 and (B) SKOV3-TR and HeyA8-MDR cells. Columns, means of three experiments; bars, SE. *, P < 0.01; **, P < 0.001.
Focal adhesion kinase silencing enhances docetaxel-induced apoptosis
We examined the effect of FAK suppression on docetaxel-mediated apoptosis. We first characterized induction of apoptosis by docetaxel alone in the ovarian cancer cells using the terminal deoxynucleotidyl transferase–mediated nick end labeling assay. Cells were treated with either IC50 or IC90 levels of docetaxel for the taxane-sensitive cell lines and harvested at time intervals ranging from 12 to 96 hours. The percentage of apoptosis in the control cells remained low over time in all cell lines (Fig. 6). After docetaxel treatment for 96 hours, the proportion of apoptotic cells increased to 84% in the SKOV3 cells (Fig. 6A) and 66% in the HeyA8 cells (Fig. 6B). To determine whether docetaxel-induced apoptosis is indeed caspase mediated, we used the caspase-3 inhibitor DEVD-fmk, which significantly reduced the proportion of apoptotic cells (Fig. 6C) in response to docetaxel after 72 hours (72–7% in SKOV3 and 56–6% in HeyA8 cells). The level of apoptosis remained low in the taxane-resistant cell lines despite treatment with docetaxel and was not affected by DEVD-fmk (Fig. 6D).
Fig. 6.

Effects of docetaxel on (A) SKOV3 and SKOV3-TR or (B) HeyA8 and HeyA8-MDR ovarian cancer cells. The percentage of apoptosis was determined by terminal deoxynucleotidyl transferase – mediated nick end labeling. Cells were treated with or without IC90 concentration of docetaxel for the taxane-sensitive cell lines. Points, means of three different experiments; bars, SE. Effect of docetaxel with or without the caspase-3 inhibitor (DEVD-fmk) on (C) SKOV3 and SKOV3-TR and (D) HeyA8 and HeyA8-MDR ovarian cancer cell apoptosis. Columns, means of three independents experiments; bars, SE.
To test whether apoptosis might be affected by FAK silencing, we used the IC20 levels of docetaxel. Ovarian cancer cells were treated with FAK siRNA for 48 hours, and then docetaxel was added. At the IC20 levels of docetaxel, about 41% apoptosis was observed in the SKOV3 cells and 42% in the HeyA8 cells (Fig. 7). However, in combination with FAK siRNA, apoptosis increased to 83% in the SKOV3 cells and 84% in the HeyA8 cells (both Ps < 0.01 when compared with docetaxel alone). Next, we examined the effects of FAK silencing with or without docetaxel on apoptosis in the taxane-resistant cells. Although neither agent alone induced much apoptosis, the combination resulted in a marked increase in apoptosis in the SKOV3-TR and HeyA8-MDR cells (Fig. 7B). Control siRNA did not enhance apoptosis in combination with docetaxel (Fig. 7A–B). These findings suggest that FAK silencing in combination with docetaxel has at least an additive effect on induction of apoptosis.
Fig. 7.

Docetaxel-mediated apoptosis with or without FAK siRNA in (A) SKOV3 and HeyA8 and (B) SKOV3-TR and HeyA8-MDR ovarian cancer cells. Columns, means of three independent experiments; bars, SE. Doc, docetaxel.
Discussion
The major findings from our study are that FAK becomes degraded in ovarian cancer cells after docetaxel treatment, which does not occur in taxane-resistant cells. Furthermore, FAK gene silencing promotes docetaxel-mediated apoptosis and growth inhibition. FAK suppression improved the efficacy of docetaxel in both taxane-sensitive and taxane-resistant cell lines. These findings suggest that FAK is a substrate for docetaxel via caspase-3, and its inhibition potentiates docetaxel’s apoptotic effects on ovarian cancer cells. Docetaxel is being increasingly used in clinical settings due to its greater potency compared with paclitaxel (37, 38) and lower risk of neuropathy (39, 40). We and others have shown that for adjuvant therapy of ovarian carcinoma, docetaxel is at least as effective as paclitaxel in combination therapy with platinum but may have a better side effect profile (39, 40). Although taxanes are known to inhibit depolymerization of microtubules (4) and modulate some apoptosis-related genes, such as p53, p21, Bcl2, and Bax (8–10, 41), other molecular and biological effects have not been well characterized.
Cell adhesion to extracellular matrix substrates regulates many aspects of normal cell physiology, including proliferation, migration, and cell survival (42, 43). FAK is a nonreceptor protein tyrosine kinase that is a critical mediator of signaling events between cells and their extracellular matrix (12, 13). FAK is tyrosine phosphorylated upon integrin binding or downstream of ligand binding by growth factor receptors (25, 44). The major site of phosphorylation of FAK is Tyr397, which is vital for the biochemical and biological functions of FAK. FAK activation at focal adhesion sites leads to cytoskeletal reorganization, cellular adhesion, and survival. FAK is overexpressed by many human tumors including colon (45, 46), breast (45), thyroid (46), ovarian (30, 47), and head and neck cancers (48), making FAK a rational target for therapeutic intervention. We have previously shown that FAK overexpression is associated with poor outcome in patients with ovarian carcinoma (30). Furthermore, FAK plays a functionally important role in ovarian cancer migration and invasion (30). FAK overexpression has been reported to protect cells from stressors, including UV irradiation, hydrogen peroxide exposure, and etoposide treatment (23). Although the mechanisms underlying such protective roles are not fully understood, they may involve activation of nuclear factor-κB and induction of inhibitor of apoptosis proteins (23).
FAK’s role in protecting tumor cells from apoptosis has been addressed previously (29, 49). Tamura et al. have shown that malignant astrocytoma/glioblastoma and breast cancer cells are resistant to apoptosis in nonadherent suspension culture conditions through a mechanism involving FAK/phosphatidylinositol 3-kinase/Akt signaling (50). Phosphatidylinositol 3-kinase binds to the phosphorylated Y397 residue in FAK, and this association may promote phosphatidylinositol 3-kinase activity (51). FAK’s role in promoting cancer cell survival is likely affected by multiple cellular and molecular factors, including state of cell attachment, Src activity, and PTEN activity. The isolation and propagation of transfected cancer cells that overexpress FRNK, a protein that functions as a dominant negative by either inhibiting localization or competing for binding partners, indicated that suppression of FAK activity may not in and of itself induce apoptosis (52).
Because FAK has been shown to play a role in promoting cell survival, it is not surprising that certain stimuli, such as UV irradiation or growth factor deprivation, can promote the cleavage and degradation of FAK during the early stages of cell apoptosis (13). The cleavage of FAK coincides with the loss of FAK from focal contacts, cell rounding, and redistribution of FAK to apoptotic membrane protrusions (53). Based on its amino acid sequence, FAK contains caspase-3 cleavage sites, which are conserved in many species, including human, chicken, and frog FAK cDNA sequences (13, 54). The caspase cleavage sites in FAK serve to separate the kinase domain from the COOH-terminal focal adhesion targeting sequences and may accelerate the cell death process. Jones et al. have shown that inhibition of FAK localization to focal adhesions by expression of the FAT domain resulted in increased sensitivity to apoptosis-inducing agents (55). Caspases are activated during chemotherapy-induced apoptosis in various cell types, including ovarian cancer cells (10, 29). Furthermore, caspase-3-mediated FAK cleavage has been previously shown with platinum-based chemotherapy (20, 29, 36). These findings raised the possibility that docetaxel-induced apoptosis may involve disruption of cell attachment by caspase-mediated FAK degradation. Cytotoxic agents, such as S-(1, 2-dicholorovinyl)-L-cysteine and cisplatin, have been shown to result in cleavage of FAK and other cytoskeletal proteins, resulting in cell detachment and apoptosis (20, 56). In our experiments, there was a differential effect of docetaxel on taxane-sensitive and taxane-resistant cells with regard to caspase activation and FAK degradation, which occurred in the former but not in the latter. FAK suppression alone by siRNA did not induce substantial apoptosis or inhibit proliferation in monolayer culture. However, FAK down-regulation in the presence of docetaxel-potentiated apoptosis in both taxane-sensitive and taxane-resistant cells. These results suggest that FAK is an important survival factor for ovarian cancer cells.
Because of the pivotal role of FAK in many processes associated with cancer progression, the inhibition of its function may present significant therapeutic opportunities. Strategies aimed at inhibition of critical protein-protein interactions or inhibition of the kinase activity of FAK are being devised (57). However, the importance of FAK kinase activity versus other aspects of FAK activity in cancer development and progression needs to be shown. Small molecule inhibitors are also being developed that act as competitors for ATP binding at the catalytic site. We and others are using siRNA approaches for down-regulating FAK, which may hold promise for clinical use.
In summary, we have shown that FAK is degraded during docetaxel-induced, caspase-mediated apoptosis. Furthermore, our results indicate that FAK down-regulation can sensitize ovarian cancer cells to docetaxel chemotherapy. These results suggest that FAK suppression in combination with docetaxel chemotherapy may be an attractive therapeutic combination in ovarian carcinoma, particularly in patients with shown resistance to conventional chemotherapy.
Acknowledgments
Grant support: NIH R01 grant CA110793-01, University of Texas M.D. Anderson Cancer Center Specialized Programs of Research Excellence in Ovarian Cancer grant 1P50CA83639, and Sanofi-Aventis Pharmaceuticals (A.K. Sood).
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. [comment] N Engl J Med. 1996;334:1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 3.Eisenhauer EA, Vermorken JB. The taxoids. Comparative clinical pharmacology and therapeutic potential. Drugs. 1998;55:5–30. doi: 10.2165/00003495-199855010-00002. [DOI] [PubMed] [Google Scholar]
- 4.Lavelle F, Bissery MC, Combeau C, Riou JF, Vrignaud P, Andre S. Preclinical evaluation of docetaxel (Taxotere) Semin Oncol. 1995;22:3–16. [PubMed] [Google Scholar]
- 5.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–25. [PubMed] [Google Scholar]
- 6.Lieu CH, Chang YN, Lai YK. Dual cytotoxic mechanisms of submicromolar taxol on human leukemia HL-60 cells. Biochem Pharmacol. 1997;53:1587–96. doi: 10.1016/s0006-2952(97)82450-1. [DOI] [PubMed] [Google Scholar]
- 7.Blagosklonny MV, Schulte T, Nguyen P, Trepel J, Neckers LM. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer Res. 1995;56:1851–4. [PubMed] [Google Scholar]
- 8.Tishler RB, Lamppu DM, Park S, Price BD. Microtubule-active drugs taxol, vinblastine, and nocodazole increase the levels of transcriptionally active p53. Cancer Res. 1995;55:6021–5. [PubMed] [Google Scholar]
- 9.Zaffaroni N, Silvestrini R, Orlandi L, Bearzatto A, Gornati D, Villa R. Induction of apoptosis by taxol and cisplatin and effect on cell cycle-related proteins in cisplatin-sensitive and -resistant human ovarian cells. Br J Cancer. 1998;77:1378–85. doi: 10.1038/bjc.1998.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolfschoten GM, Hulscher TM, Duyndam MC, Pinedo HM, Boven E. Variation in the kinetics of caspase-3 activation, Bcl-2 phosphorylation and apoptotic morphology in unselected human ovarian cancer celllines as a response to docetaxel. Biochem Pharmacol. 2002;63:733–43. doi: 10.1016/s0006-2952(01)00895-4. [DOI] [PubMed] [Google Scholar]
- 11.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 12.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 13.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 14.Cary LA, Chang JF, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–94. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- 15.Zhao JH, Reiske H, Guan JL. Regulation of the cell cycle by focal adhesion kinase. J Cell Biol. 1998;143:1997–2008. doi: 10.1083/jcb.143.7.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsia DA, Mitra SK, Hauck CR, et al. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–67. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sieg DJ, Hauck CR, Ilic D, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 18.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–9. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu LH, Owens LV, Sturge GC, et al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–8. [PubMed] [Google Scholar]
- 20.van deWater B, Nagelkerke JF, Stevens JL. Dephosphorylation of focal adhesion kinase (FAK) and loss of focal contacts precede caspase-mediated cleavage of FAK during apoptosis in renal epithelial cells. J Biol Chem. 1999;274:13328–37. doi: 10.1074/jbc.274.19.13328. [DOI] [PubMed] [Google Scholar]
- 21.Gervais FG, Thornberry NA, Ruffolo SC, Nicholson DW, Roy S. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J Biol Chem. 1998;273:17102–8. doi: 10.1074/jbc.273.27.17102. [DOI] [PubMed] [Google Scholar]
- 22.Grossmann J, Artinger M, Grasso AW, et al. Hierarchical cleavage of focal adhesion kinase by caspases alters signal transduction during apoptosis of intestinal epithelial cells. Gastroenterology. 2001;120:79–88. doi: 10.1053/gast.2001.20879. [DOI] [PubMed] [Google Scholar]
- 23.Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J Biol Chem. 2000;275:16309–15. doi: 10.1074/jbc.275.21.16309. [DOI] [PubMed] [Google Scholar]
- 24.Bellas RE, Harrington EO, Sheahan KL, Newton J, Marcus C, Rounds S. FAK blunts adenosine-homocysteine-induced endothelial cell apoptosis: requirement for PI 3-kinase. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1135–42. doi: 10.1152/ajplung.00174.2001. [DOI] [PubMed] [Google Scholar]
- 25.Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol. 1998;8:151–7. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- 26.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–93. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery. 2004;135:555–62. doi: 10.1016/j.surg.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 28.Wen LP, Fahrni JA, Troie S, Guan JL, Orth K, Rosen GD. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272:26056–61. doi: 10.1074/jbc.272.41.26056. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki H, Kotsuji F, Tsang BK. Caspase 3-mediated focal adhesion kinase processing in human ovarian cancer cells: possible regulation by X-linked inhibitor of apoptosis protein. Gynecol Oncol. 2002;85:339–50. doi: 10.1006/gyno.2002.6632. [DOI] [PubMed] [Google Scholar]
- 30.Sood AK, Coffin JE, Schneider GB, et al. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol. 2004;165:1087–95. doi: 10.1016/S0002-9440(10)63370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sood AK, Seftor EA, Fletcher MS, et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol. 2001;158:1279–88. doi: 10.1016/S0002-9440(10)64079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apte SM, Fan D, Killion JJ, Fidler IJ. Targeting the platelet-derived growth factor receptor in antivascular therapy for human ovarian carcinoma. Clin Cancer Res. 2004;10:897–908. doi: 10.1158/1078-0432.ccr-1151-3. [DOI] [PubMed] [Google Scholar]
- 33.Lamendola DE, Duan Z, Yusuf RZ, Seiden MV. Molecular description of evolving paclitaxel resistance in the SKOV-3 human ovarian carcinoma cell line. Cancer Res. 2003;63:2200–5. [PubMed] [Google Scholar]
- 34.Thaker PH, Yazici S, Nilsson MB, et al. Antivascular therapy for orthotopic human ovarian carcinoma through blockade of the vascular endothelial growth factor and epidermal growth factor receptor. Clin Cancer Res. 2005;11:4923–33. doi: 10.1158/1078-0432.CCR-04-2060. [DOI] [PubMed] [Google Scholar]
- 35.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 36.Carragher NO, Fincham VJ, Riley D, Frame MC. Cleavage of focal adhesion kinase by different proteases during SRC-regulated transformation and apoptosis. Distinct roles for calpain and caspases. J Biol Chem. 2001;276:4270–5. doi: 10.1074/jbc.M008972200. [DOI] [PubMed] [Google Scholar]
- 37.Ringel I, Horwitz SB. Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. JNatl Cancer Inst. 1991;83:288–91. doi: 10.1093/jnci/83.4.288. [DOI] [PubMed] [Google Scholar]
- 38.Barasoain I, De Ines C, Diaz F. Interaction of tubulin and cellular microtubules with Taxotere (RP56976), a new semisynthetic analog of Taxol. Proc Am Assoc Cancer Res. 1991;32:329. [Google Scholar]
- 39.Hsu Y, Sood AK, Sorosky JI. Docetaxel versus paclitaxel for adjuvant treatment of ovarian cancer: case-control analysis of toxicity. Am J Clin Oncol. 2004;27:14–8. doi: 10.1097/01.coc.0000045849.95834.6b. [DOI] [PubMed] [Google Scholar]
- 40.Vasey PA, Atkinson R, Coleman R, et al. Docetaxel-carboplatin as first line chemotherapy for epithelial ovarian cancer. Br J Cancer. 2001;84:170–8. doi: 10.1054/bjoc.2000.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blagosklonny MV, Schulte TW, Nguyen P, Mimnaugh EG, Trepel J, Neckers L. Taxol induction of p21WAF1 and p53 requires c-raf-1. Cancer Res. 1995;55:4623–6. [PubMed] [Google Scholar]
- 42.Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- 43.Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E. Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science. 1996;271:499–502. doi: 10.1126/science.271.5248.499. [DOI] [PubMed] [Google Scholar]
- 44.Casamassima A, Rozengurt E. Insulin-like growth factor I stimulates tyrosine phosphorylation of p130(Cas), focal adhesion kinase, and paxillin. Role of phosphatidylinositol 3′-kinase and formation of a p130(Cas). Crk complex. J Biol Chem. 1998;273:26149–56. doi: 10.1074/jbc.273.40.26149. [DOI] [PubMed] [Google Scholar]
- 45.Cance WG, Harris JE, Iacocca MV, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6:2417–23. [PubMed] [Google Scholar]
- 46.Owens LV, Xu L, Craven RJ, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55:2752–5. [PubMed] [Google Scholar]
- 47.Judson PL, He X, Cance WG, Van Le L. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 1999;86:1551–6. doi: 10.1002/(sici)1097-0142(19991015)86:6<1551::aid-cncr23>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 48.Kornberg LJ. Focal adhesion kinase expression in oral cancers. Head Neck. 1998;20:634–9. doi: 10.1002/(sici)1097-0347(199810)20:7<634::aid-hed10>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 49.Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochem Biophys Res Commun. 2003;311:786–92. doi: 10.1016/j.bbrc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- 50.Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 51.Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 1994;91:10148–52. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van deWater B, Houtepen F, Huigsloot M, Tijdens IB. Suppression of chemically induced apoptosis but not necrosis of renal proximal tubular epithelial (LLC-PK1) cells by focal adhesion kinase (FAK). Role of FAK in maintaining focal adhesion organization after acute renal cell injury. J Biol Chem. 2001;276:36183–93. doi: 10.1074/jbc.M102091200. [DOI] [PubMed] [Google Scholar]
- 53.Levkau B, Herren B, Koyama H, Ross R, Raines EW. Caspase-mediated cleavage of focal adhesion kinase pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J Exp Med. 1998;187:579–86. doi: 10.1084/jem.187.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaller M, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK, a structurally unique protein tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–6. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones G, Machado J, Jr, Tolnay M, Merlo A. PTEN-independent induction of caspase-mediated cell death and reduced invasion by the focal adhesion targeting domain (FAT) in human astrocytic brain tumors which highly express focal adhesion kinase (FAK) Cancer Res. 2001;61:5688–91. [PubMed] [Google Scholar]
- 56.Kruidering M, van deWater B, Zhan Y, et al. Cisplatin effects on F-actin and matrix proteins precede renal tubular cell detachment and apoptosis in vitro. Cell Death Differ. 1998;5:601–14. doi: 10.1038/sj.cdd.4400392. [DOI] [PubMed] [Google Scholar]
- 57.McLean GW, Avizienyte E, Frame MC. Focal adhesion kinase as a potential target in oncology. Expert Opin Pharmacother. 2003;4:227–34. doi: 10.1517/14656566.4.2.227. [DOI] [PubMed] [Google Scholar]