

Abstract
Background
Paracrine signaling between hepatic stellate cells (HSC) and liver endothelial cells (LEC) modulates fibrogenesis, angiogenesis, and portal hypertension. However, mechanisms regulating these processes are not fully defined. Sorafenib is a receptor tyrosine kinase inhibitor that blocks growth factor signaling in tumor cells but also displays important and not yet fully characterized effects on liver nonparenchymal cells including HSC and LEC. The aim of this study was to test the hypothesis that sorafenib influences paracrine signaling between HSC and LEC and thereby regulates matrix and vascular changes associated with chronic liver injury.
Results
Complementary magnetic resonance elastography, micro-CT, and histochemical analyses indicate that sorafenib attenuates the changes in both matrix and vascular compartments that occur in response to bile-duct ligation induced liver injury in rats. Cell biology studies demonstrate that sorafenib markedly reduces cell to cell apposition and junctional complexes, thus reducing the proximity typically observed between these sinusoidal barrier cells. At the molecular level, sorafenib down-regulates angiopoietin-1 and fibronectin, both released by HSC in a manner dependent on the transcription factor KLF6, suggesting that this pathway underlies both matrix and vascular changes associated with chronic liver disease.
Conclusion
Collectively, our results demonstrate that sorafenib inhibits both matrix restructuring and vascular remodeling that accompany chronic liver diseases and characterize cell and molecular mechanisms underlying this effect. These data may help to refine future therapies for advanced gastrointestinal and liver diseases characterized by abundant fibrosis and neovascularization.
Keywords: portal hypertension, KLF6, sorafenib, cirrhosis, angiopoietin-1
Introduction
Liver cirrhosis is associated with variable changes in architecture of both matrix and vasculature within the sinusoidal tree. Matrix changes are characterized by increased deposition of fibronectin, collagen I, and other fibrillar proteins. Concomitant vascular changes primarily include sinusoidal vasoconstriction, angiogenesis, and pathological remodeling of sinusoids typified by increased mural cell coverage and vigorous wrapping by hepatic stellate cells (HSC) around liver endothelial cells (LEC)(1–3). These vascular changes disrupt integrity and homeostasis of sinusoidal function and in concert with matrix changes, lead to portal hypertension and its clinical complications.
Sorafenib is a multi-kinase inhibitor compound recently approved for use in humans with liver cancer(4). Its recent introduction to the clinic has fueled a plethora of studies aimed at understanding not only its therapeutical potential, but also, possible mechanisms underlying beneficial roles of this drug. In addition to its better known effects on epithelial cancer cell proliferation(5), sorafenib also regulates receptor tyrosine kinase pathways in adjacent stromal cells including myofibroblasts and endothelial cells(6). Although inhibitory effects of sorafenib on these nonparenchymal cell types are less characterized, they are nonetheless likely to significantly contribute to antitumoral efficacy of this drug. Furthermore, since HSC and LEC are integral to development of matrix and vascular changes during liver fibrosis, characterizing effects and mechanisms of action of sorafenib in this disease process is of notable medical importance.
Consequently, in the current study we demonstrate that sorafenib improves liver fibrosis by acting, at least in part, via a novel mechanism which is triggered within HSC and LEC. Our results report a pathway whereby angiopoietin-1 (Ang1) cooperates with fibronectin to regulate remodeling of sinusoids that accompanies liver fibrosis. We find that both Ang1 and fibronectin are regulated by platelet derived growth factor (PDGF) signaling and are functionally linked by a shared transcription factor; the zinc finger protein, KLF6. However, these cooperative Ang1 and fibronectin pathways are readily inhibited by sorafenib through distinct downstream molecular signals that are independent and dependent on Raf, respectively. Complementary in vivo studies reveal a role for these pathways in the process of increased liver stiffness and provide evidence that sorafenib restores sinusoidal homeostasis by limiting injury-induced matrix and angiogenic changes. Collectively, these findings are of significant importance for building the theoretical framework necessary to design new therapies to treat fibrosis in liver and in other gastrointestinal organs susceptible to exuberant fibrogenic responses.
Methods
Detailed Methods are provided in Supplementary Methods.
Cell culture/transfection
Primary murine LEC, human LEC (ScienCell), or a cell line derived from transformed mouse liver endothelial cells (TSEC)(7), were grown with endothelial culture media with 10% serum and 1% endothelial growth supplement. Human HSC (ScienCell) were grown in DMEM with 10% serum.
Isolation of murine LEC
LEC were isolated from whole rat liver by repeated mincing followed by enzymatic digestion and CD-31 based immunomagnetic separation as described previously with modifications (8).
Preparation of HSC derived conditioned media
Human HSC were serum starved, treated with either vehicle or sorafenib in serum free DMEM, and conditioned media (CM) was harvested over 12–24 h.
In vitro LEC tubulogenesis-coculture assays and electron microscopy
Human LEC and HSC were plated on matrigel coated 4-well glass slides and tubulogenesis was visualized to study angiogenic interactions between LEC and HSC in vitro as described previously(3). Transmission electron microscopy (TEM) was performed to visualize vascular connections between human LEC and HSC cultured on Matrigel.
Chemotaxis assay
Chemotaxis of human LEC was measured by Boyden assay in response to CM with additional compounds added to media as indicated in individual experiments.
Confocal immunofluorescence microscopy
Immunofluorescence was performed on murine LEC or TSEC as previously described(9). Murine LEC and TSEC cells were grown to monolayer on collagen coated glass slides and stained for ZO-1. Images were captured using a confocal laser scanning microscope.
Quantitative and semi-quantitative PCR
RNA was isolated from human HSC (RNeasy/Qiagen), reverse-transcribed (Superscript/Invitrogen) and real-time-PCR was carried out (Applied Biosystems 7500).
ChIP assay
Human HSC were transfected with Flag-tagged KLF6 or control vector. After 36h, cells were serum starved for 12 hours, stimulated with/without PDGF for 12h and ChIP was performed (EZ-ChIP kit) as described previously(10).
Bile-duct ligation (BDL) and sorafenib administration in vivo
Sprague Dawley rats were subjected to BDL to induce fibrosis as previously described(11). Rats were injected with vehicle or sorafenib(6) (1.5 mg/kg body weight) for in vivo experiments. Procedures were performed per Mayo Clinic IACUC guidelines.
3D-reconstruction of hepatic vasculature by micro-CT
Animals were injected with a radio-opaque liquid-silicone compound (Microfil®, MV-122; Flow Tech., Inc., Carver, MA) via portal vein (infusion rate: 8–10 ml/min, pressure: 10–12 mmHg). Intact animal was placed under refrigeration at 4°C after perfusion to allow polymerization. Livers were scanned and reconstructed as described previously(12). See supplementary methods for additional details.
Magnetic resonance elastography (MRE)
Animals were subjected to MRE at four weeks after the SHAM or BDL surgery. Liver stiffness was measured by generating longitudinal shear waves through the abdominal wall by a pneumatic driver followed by detection of propagating shear wave displacement pattern by a phase-contrast magnetic resonance imaging system as described previously(13–15).
Immunostaining of liver tissue
Immunohistochemistry was performed on paraffin-embedded and frozen rat liver tissue sections as previously described(16).
Statistical analysis
Data are expressed as means and standard errors of mean (SEM) of at least three independent experiments unless otherwise indicated. Groups were compared by two-tailed Student-t test. P value<0.05 was considered statistically significant.
Results
Sorafenib attenuates injury-induced increases in liver stiffness, matrix deposition, and vascular remodeling in vivo
MRE provides one of the best in vivo estimations of liver stiffness, which is a variable that correlates with both matrix and vascular changes that occur in response to liver injury(17). Furthermore, this technique provides an assessment of stiffness throughout liver, is non-invasive, and ideal for performing sequential imaging studies in individual animals. Thus, in the current study we utilize this technique to evaluate for changes in liver stiffness by sorafenib administration. We administered sorafenib for 4 weeks to sham or BDL rats beginning immediately after surgery. Sorafenib was well tolerated without overt adverse effects (reduced body weight, diarrhea, hemorrhage, or mortality). In BDL rats treated with vehicle, MRE evidenced a time-dependent increase in liver stiffness as compared with sham-operated rats (Figure 1A shows composite data, Figure 1B; top panel shows representative magnetic resonance images, lower panel shows corresponding MRE). Conversely, in BDL rats receiving sorafenib, increase in liver stiffness was attenuated (Figure 1A). Notably, sorafenib also influenced matrix deposition that occurs in response to BDL. This attenuated fibrosis in sorafenib-treated BDL rats was depicted at 4 weeks by reduced Sirius red staining of liver sections compared to BDL rats receiving vehicle (Figure 1C; graph of composite data and 1 D; representative images). Since MRE reflects angioarchitectural changes in addition to matrix changes, we subsequently ascertained the vascular component that could contribute to liver stiffness and its modulation by sorafenib. To investigate the vascular changes in response to sorafenib, we initially stained liver tissue sections from treated animals with an antibody against vWF, a marker of vascular endothelium, especially prominent in actively remodeling vessels in fibrosis(18). Indeed, vWF was increased in vehicle-treated BDL rats as compared to sham-operated rats as previously shown by us and others(16). However, when BDL rats were treated with sorafenib, vWF staining was markedly attenuated (Figure 2A shows composite data and Figure 2B shows representative images), indicating that this drug attenuates vascular changes that occur during liver wound healing response. To complement these studies, we used a micro-CT based approach to assess vascular features in sham and BDL rats in greater detail. This technique provides measurement of total vascular volume (depicted in Figure 2D, top left and middle panels; red indicates selected vascular trunk and associated vascular volume), as well as precise analysis of branching of medium and large hepatic vessels (Figure 2D; right panel), thereby both complementing and corroborating the aforementioned histochemical analyses. Ratio of vascular volume to total liver volume as well as number of branches (primary/secondary/tertiary total) was increased in livers of BDL rats; Figure 2D lower panel shows representative images while Figure 2C depicts composite data). However, BDL rats receiving sorafenib exhibited attenuation in both these micro-CT vascular parameters consistent with the vWF histochemical analysis. Thus, sorafenib effectively attenuates pathobiological vascular changes that occur in response to BDL.
Figure 1.
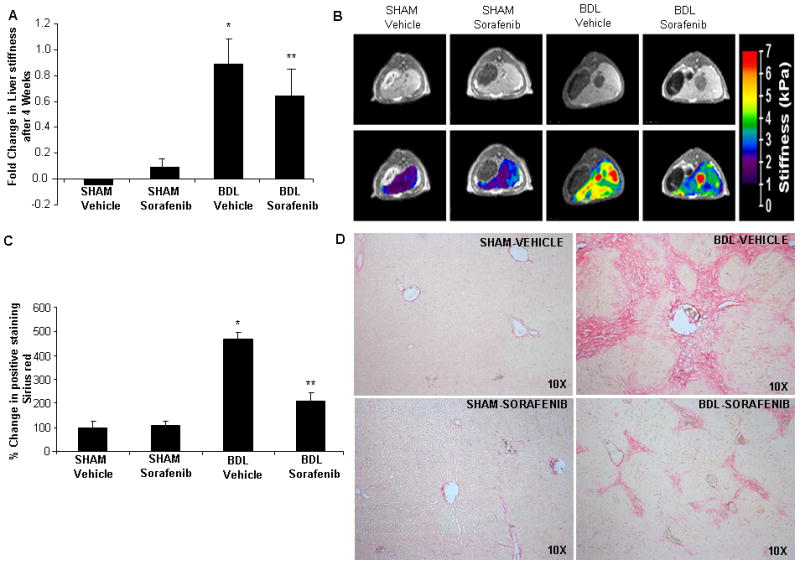
Sorafenib attenuates injury-induced increases in liver stiffness/matrix deposition in vivo. A & B: SHAM and BDL operated rats treated either with vehicle or sorafenib were subjected to MRE at 4 weeks and changes in liver stiffness were assessed. Vehicle-BDL rats showed significant increase in liver stiffness as compared to SHAM rats. This change was significantly attenuated in BDL rats receiving sorafenib, (N>4, mean ± SEM,* p<0.05, sham-vehicle,** BDL-vehicle). Quantitative analysis is depicted graphically in A; images from representative animals are shown in B (upper panels: MRI images and lower panels: MRE images with stiffness-colored heat-map). C & D: Sirius red staining in SHAM and BDL rats treated with sorafenib or vehicle; Sorafenib attenuated matrix deposition in BDL rats as compared to vehicle treated rats (10X; N>4, mean ± SEM, * p<0.05, sham-vehicle,** BDL-vehicle). Quantitative analysis is depicted graphically in C while representative images are shown in D.
Figure 2.
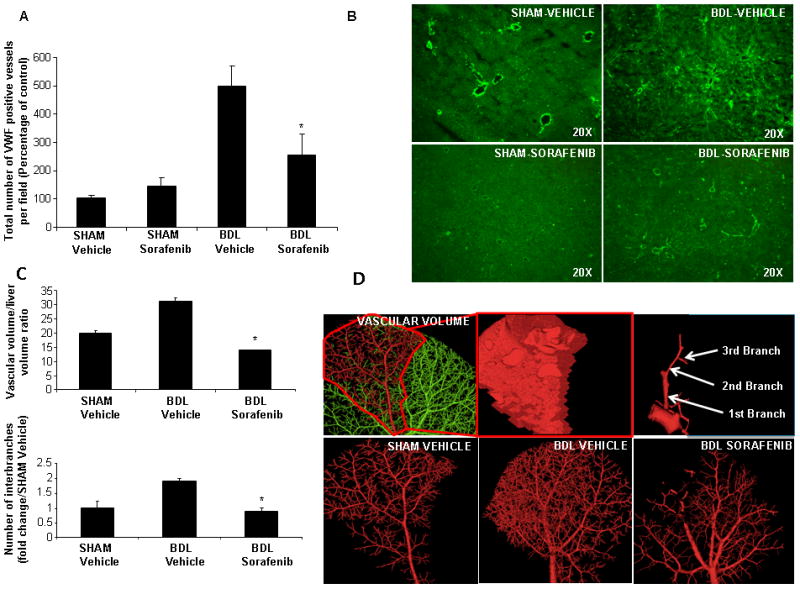
Sorafenib attenuates injury-induced hepatic vascular remodeling in vivo. A&B: vWF immunofluorescence of liver was performed in SHAM and BDL rats treated with vehicle or sorafenib. Analysis depicts attenuated neo-capillary formation in BDL-sorafenib rats as compared to BDL-vehicle (20X, n>3, mean ± SEM, * p<0.05, BDL-vehicle). Quantitative analysis is depicted in A; representative images are shown in B. C & D: SHAM and BDL-rats treated with vehicle or sorafenib were injected with Microfil®; specific liver lobes were scanned to measure vascular volume (left - middle upper panels; D). Images were also digitally reconstructed to quantify vascular branching (right upper panel; D). Quantitative analysis of vascular volume/liver ratio and number of interbranches is depicted graphically in C; images from representative animals are shown in lower panels of D. BDL-rats treated with vehicle evidenced increased total liver vascular space, as measured by the vascular volume/liver volume ratio, and higher interbranch numbers. Sorafenib in BDL rats was associated with significant decrease of total vascular space (n>4, mean ± SEM, * p=0.01, BDL-vehicle) and decrease of primary/secondary/tertiary branches (* p<0.05, BDL-vehicle).
Regulation of stellate and endothelial cell interactions constitutes a key cellular mechanism underlying the beneficial effect of sorafenib on vascular restitution
Since sorafenib reverses vascular defects that are induced during liver injury in vivo, we next investigated cellular mechanisms underlying these effects using a reconstituted cell system composed of human derived HSC and LEC, cell populations which are responsible for matrix and vessel changes in response to injury. Since fibrotic sinusoids are associated with increased HSC wrapping of LEC lined sinusoids(2), we specifically examined whether sorafenib reverses this processes. To explore this concept we incubated human HSC with sorafenib or vehicle for 24 hours and transferred these cells into co-culture on matrigel with human LEC. Each cell type was labeled with either YFP or DS-Red plasmids to distinguish them and facilitate monitoring of their behavior. Using this optimized cell culture system, we found that HSC and LEC formed robust vascular networks where both cells juxtapose and form wrapping contacts; however HSC pre-treated with sorafenib showed a stark contrast with compromised ability of HSC to effectively establish wrapping interactions with LEC in this assay (Figure 3A, 10X magnification in upper panel with zoomed Z-stack in lower panel and quantification of intercellular connections of LEC, HSC, and LEC-HSC juxtaposition, right panel graphs). These fluorescent microscopy experiments were complemented with time-lapse video microscopy to observe temporal kinetics of these observations in real-time (Supplementary Figure 1-Movie). Furthermore, reduced juxtaposition of sorafenib-treated HSC with LEC was confirmed by electron microscopy which revealed increased gaps and decreased apposition between LEC and HSC when HSC were pre-incubated with sorafenib (Figure 3B; see dashed arrow). Thus, together these in vitro assays using combinations of light and electron microscopy as well as time-lapse real-time imaging suggest that disruption of exuberant endothelial-to-stellate cell wrapping and contacts form part of the cellular mechanisms underlying the beneficial effect of sorafenib on vascular structure.
Figure 3.
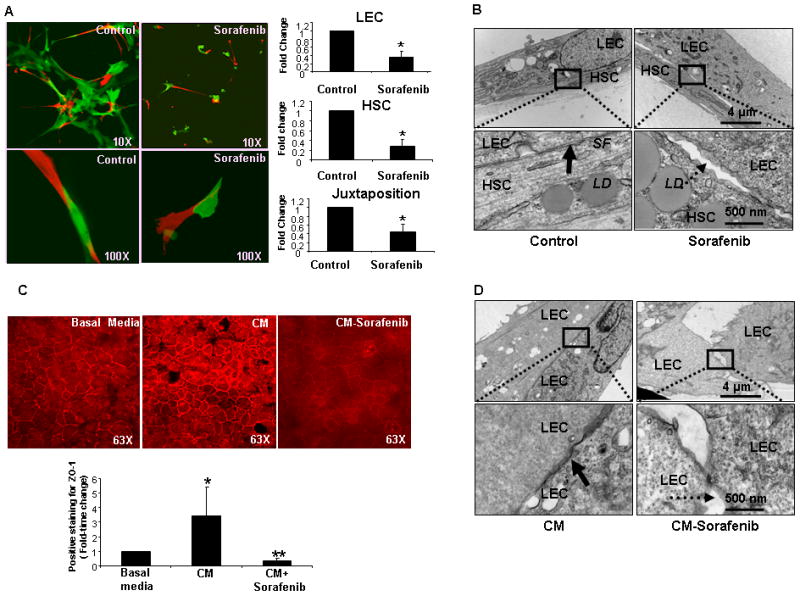
Sorafenib inhibits endothelial cell-stellate cell interactions and communications in vitro. A: Human HSC and LEC were labeled with DS-Red or YFP respectively and cocultured in Matrigel. Vascular tube networks formed between HSC (red) or LEC (green) which was significantly inhibited when HSC were pre-treated with sorafenib prior to coculturing with LEC as evidenced by reduced juxtapositions of LEC and HSC. (Left: representative images from sorafenib or control-vehicle groups; 10X in upper panels and Z-stack zoom in lower panel, 100X. Right: composite data of vascular connections between LEC, between HSC, and juxtapositions between the two cell types (n=3, mean ± SEM, * p<0.05-control). B: TEM of human LEC and HSC cocultured in Matrigel with HSC pretreated with control vehicle or sorafenib (upper panel: 10,000X and lower panel: 50,000X); Solid arrow denotes tight juxtaposition between the two cell-types observed with control-vehicle but disrupted in sorafenib-treated cells as denoted by dashed arrow. C: ZO-1 staining (red) evidences junctions between TSEC. ZO-1 showed modest staining in TSEC treated with basal media, whereas plasma membrane staining was robust with conditioned media (CM) derived from vehicle-treated HSC; ZO-1 staining was significantly decreased when treated with CM derived from sorafenib treated HSC (n=3, mean ± SEM, * p<0.05 - basal,** p<0.05 - CM). Upper, denotes representative images. Lower, depicts data in composite graph. D: TEM of human LEC incubated on Matrigel with CM from HSC treated with vehicle or sorafenib (upper, 10,000X, lower, 50,000X); Arrow in left panel denotes junctional complexes and close apposition of LEC when treated with CM derived from vehicle-treated HSC. Dashed arrow denotes disrupted junctions between LEC when CM derived from HSC pre-treated with sorafenib.
Sorafenib disrupts endothelial-to-endothelial cell junctional interactions in vitro
In addition to increased wrapping of contractile stellate cells around endothelial cells, vascular remodeling in cirrhosis is also typified by increased assembly of endothelial-to-endothelial cell junctions that lead to cell motility and angiogenesis. This is in contradistinction to normal hepatic sinusoids which are discontinuous with limited endothelial-to-endothelial cell interactions. Hence, we next investigated whether treatment of HSC with sorafenib regulates junctional structures between LEC and angiogenesis. To test this model, we plated and cultured endothelial cells (TSEC) under conditions in which they form prominent junctions and incubated them with conditioned media derived from HSC pre-treated either with sorafenib or control vehicle. Cells were immunostained with a ZO-1 antibody to label junctional structures between endothelial cells. We found that ZO-1 staining was prominent in TSEC incubated with conditioned media derived from vehicle-treated HSC, while staining was significantly decreased in cells incubated with media derived from sorafenib stimulated HSC (Figure 3C), suggesting that this drug modulates formation of cell-cell junctions amongst endothelia. These results initially observed in TSEC cells were also confirmed in primary murine LEC (Supplementary Figure 2). We utilized transmission electron microscopy, which showed an increased number of intercellular junctions between human LEC incubated with conditioned media derived from vehicle-treated HSC (Figure 3D). In contrast, junctional structures revealed by this high resolution technique were markedly reduced when LEC were incubated with conditioned media derived from sorafenib stimulated HSC (Figure 3D). Thus, this data demonstrates that sorafenib modulates the structural basis of junctional complexes that can be formed between endothelial cells which are the foundation of vascular remodeling, and subsequently led us to define signaling cascades which can modulate these processes at the molecular level.
Sorafenib regulates vascular and matrix remodeling through complementary signaling pathways
Prior studies have delineated a critical role of PDGF on vascular function especially its ability to regulate pericytic and myofibroblastic mural wall cells such as HSC through PDGF receptor β (PDGFR-β)(3, 19). As a first step to better define effects of sorafenib on PDGFR signaling in HSC, we examined integrity of this signaling pathway in human derived HSC stimulated with PDGF and/or sorafenib. Congruent with its function as a tyrosine kinase inhibitor, sorafenib abolished PDGF induced PDGFR-β phosphorylation. PDGF-induced Raf and Akt phosphorylation were also inhibited by this drug indicating that sorafenib inhibits several canonical downstream pathways of PDGF (Figure 4A). We next determined specific vascular molecules that may reside downstream of these sorafenib signaling targets in HSC. To this end, we performed expression analysis using a pathway specific angiogenesis array in human HSC, which revealed that PDGF induces expression of both angiopoetin-1 (Ang1) and fibronectin in HSC, and that sorafenib reverses this effect (Supplementary figure 3). Congruent with microarray results, fibronectin protein levels were decreased in HSC, after a 24-hour treatment with sorafenib (Figure 4A). In absence of a reliable antibody, changes in Ang1, were confirmed by quantitative reverse-transcription (RT-PCR), which revealed up-regulation of Ang-1 mRNA levels after incubation with PDGF, in a temporal manner with maximal Ang-1 mRNA levels observed after 24-hours (Figure 4B). Conversely, sorafenib as well as the mechanistically linked compound, imatinib, blocked the inducing effect of PDGF on Ang-1 mRNA levels (Figure 4C). To extend our analyses of signaling pathways responsible for PDGF induced Ang1 production in HSC, we treated HSC with U0126, a MEK inhibitor or wortmannin, a PI3K/Akt pathway inhibitor, in presence or absence of PDGF, and assessed mRNA levels of Ang1 by RT-PCR. Whereas wortmannin markedly inhibited Ang1 synthesis, U0126 did not (Figure 4D). Moreover, Ang1 synthesis was not impaired in Raf-silenced HSC (Figure 4E). Conversely, Western blot analyses revealed that fibronectin expression was inhibited in Raf-silenced HSC (Figure 4A, lower right panel). Hence, Ang1 expression in HSC occurs through a PDGFR and PI3K/Akt dependent but Raf-independent mechanism, while fibronectin expression in HSC occurs through canonical PDGFR and Raf pathways. Thus, these results suggest that expression of genes that participate in remodeling of vasculature is regulated by key molecular pathways that are downstream of tyrosine kinase receptors, such as PDGF, and that sorafenib effectively inhibits these events.
Figure 4.
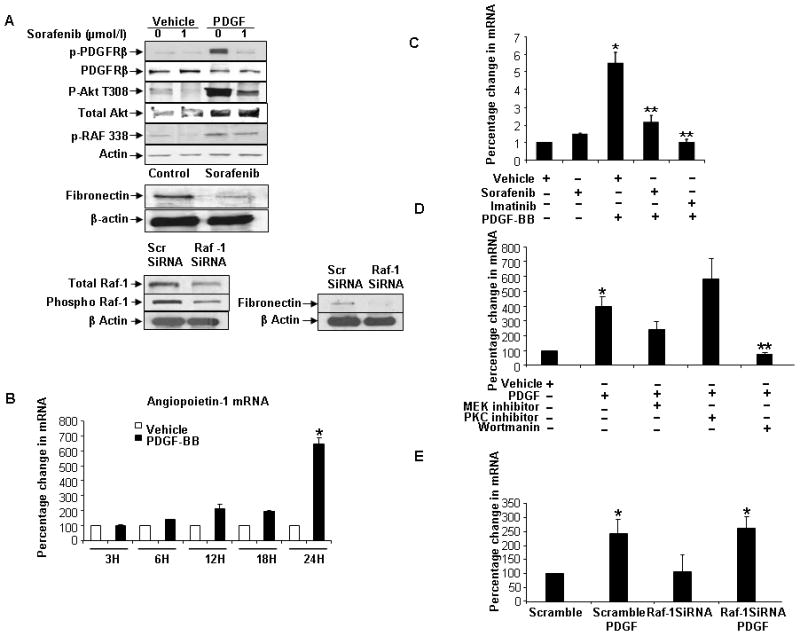
Sorafenib inhibits the release of angiopoietin-1 (Ang1) and fibronectin from HSC. A: HSC were treated with sorafenib (2 μmol/l) for 1h, stimulated with PDGF for 10 minutes then lysed for Western blot. Sorafenib inhibited phosphorylation of PDGFRβ and downstream molecules Akt and Raf-1, and fibronectin synthesis in HSC. Lower, show effects of Raf-1 knockdown in HSC using siRNA. Fibronectin synthesis was reduced in Raf-1-silenced HSC. Representative blots are depicted. B: Time-course of Ang1 synthesis by q-PCR in HSC after PDGF. Ang1 mRNA levels were increased in temporally with maximum at 24h after PDGF (n=3, mean ± SEM, * p<0.05-basal). C: Sorafenib and imatinib inhibit PDGF-induced Ang1 synthesis in HSC. (n=3, mean ± SEM, * p<0.05-vehicle,** PDGF). D: Ang1 mRNA levels after PDGF stimulation were decreased when HSC were treated with the PI3-K inhibitor wortmannin, but not the MEK inhibitor U012 (n=3, mean ± SEM, * p<0.05-vehicle). E: Cells were transfected with Raf-1 siRNA, treated with/without PDGF and lysed 48h after transfection. PCR for Ang1 mRNA in Raf-1 silenced HSC after PDGF treatment shows no significant change compared to scrambled-siRNA/PDGF (n=3, mean ± SEM, versus scramble-PDGF).
KLF6 mediates PDGF-induced Ang1 and fibronectin expression
We next investigated how this signaling cascade converges on nuclear transcription factors that may regulate expression of these angiogenic factors. We focused on KLF proteins since this family of proteins have emerged as key regulators of HSC function and phenotype(20–23). A systematic and family-wide screening approach of KLF proteins revealed that KLF6, KLF7, KLF8, KLF9 and KLF15 were repressed in cells pre-treated with sorafenib (data not shown). Of these, KLF6 is a molecule prominently implicated in fibrosis thus drawing our attention to this specific KLF protein. Indeed, RT-PCR revealed a significant up-regulation of KLF6 after incubation with PDGF, an effect that was abrogated by sorafenib (Figure 5A). To further explore participation of KLF6 in regulation of fibronectin and Ang1 in HSC, we performed RNAi-based knock-down in HSC. Indeed, downregulation of KLF6 abolished PDGF-induced induction of Ang1 mRNA and fibronectin protein levels (Figure 5B/C, respectively). Corroborative cell biological studies also demonstrated that tubulogenesis of LEC was significantly decreased after their incubation with conditioned media from KLF6 siRNA transfected HSC (Figure 5D) similar to that which was previously observed with conditioned media derived from HSC treated with sorafenib, suggesting that this transcription factor regulates intracellular events that participate in active endothelial tubulogenesis. Lastly, KLF6 as a direct regulator of angiogenic genes was firmly established by ChIP assay, which demonstrated that this transcription factor occupies the Ang1 promoter in cultured HSC (Figure 5E). Thus, these experiments describe a novel pathway whereby KLF6 mediates PDGF signaling in HSC that leads to production of both vascular proteins such as Ang1, and matrix proteins such as fibronectin, which are established players in angioarchitectural changes that accompany fibrosis.
Figure 5.
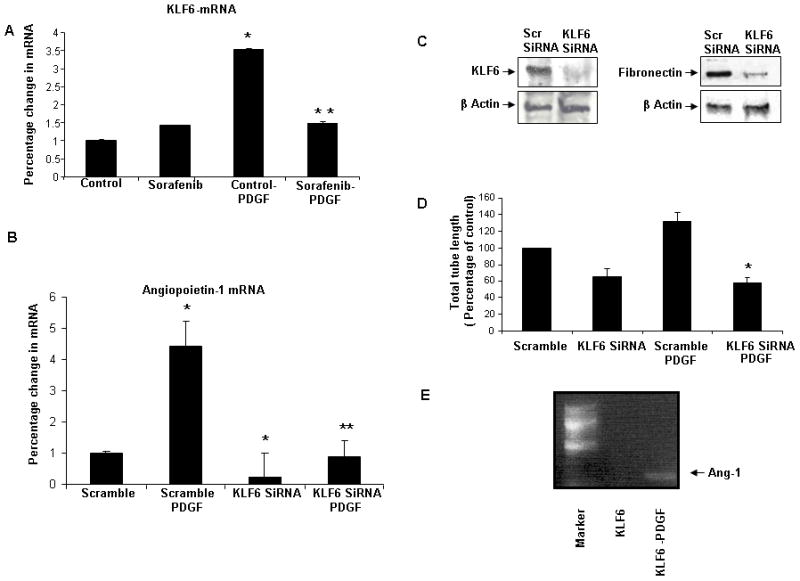
KLF6 mediates PDGF-induced angiopoietin-1 (Ang1) synthesis. A: KLF6 mRNA levels are increased after PDGF-stimulation of HSC. PDGF stimulation followed by 24h with sorafenib reduced KLF6 mRNA levels compared to PDGF alone (n=3, mean ± SEM, * p<0.005-control, ** control-PDGF). B: PDGF stimulation of Ang1 mRNA levels was abolished when KLF6 was knocked-down using siRNA (n=3, mean ± SEM,* p<0.005-scramble, ** scramble-PDGF). C: Right, fibronectin protein levels were reduced in KLF6-silenced HSC. Left, depicts effective knock-down of KLF6 by siRNA. D: LEC tubulogenesis in CM derived from KLF6 silenced, PDGF-stimulated HSC was significantly decreased as compared to CM derived from PDGF-stimulated HSC in absence of KLF6 siRNA (* scramble-PDGF). E: ChIP assay to assess the binding of KLF6 to Ang1 promoter. E depicts increased binding of Ang-1 to the overexpressed flag-tagged KLF6 with PDGF vs control.
HSC derived angiopoietin-1 disrupts vascular homeostasis
We then returned to our in vitro models to ascertain a functional role for these molecular findings. Ang1 contributes importantly to vessel maturation(24). However, excessive Ang1 may disrupt normal vessels and lead to vascular restructuring and angiogenesis which characterizes cirrhosis. Hence we first investigated whether Ang1 may increase junctional structures between LEC. Thus, we plated TSEC, an LEC cell line that forms exuberant junctions at confluence, and immunostained cells with ZO-1Ab to identify junctional structures. To specifically implicate Ang1 in this process, in some experimental groups we examined ZO-1 staining after incubating TSEC with HSC conditioned media containing Ang1 neutralizing antibody or supplemental recombinant Ang1. As previously depicted in Figure 3C, ZO-1 staining was significantly increased in conditioned-media treated group; this effect was abolished when treated with conditioned media derived from sorafenib-treated HSC (Figure 6A). Additionally, sorafenib-induced inhibition of junction formation between cells was reversed upon addition of recombinant Ang1 in HSC-derived media (Figure 6A). A similar pattern to sorafenib was observed when TSEC were incubated with conditioned media pretreated with Ang-1 neutralizing antibody (Figure 6A). Those findings were corroborated by transmission electron microscopy which also revealed a reduction in junctional complexes in LEC upon addition of Ang1 neutralizing Ab to HSC-derived conditioned media (Figure 6B). These morphological analyses also revealed a reversal of sorafenib-induced inhibition of junctional complexes between cells by addition of recombinant Ang1 to HSC conditioned media (Figure 6B). Thus, these results demonstrate that HSC derived Ang1 promotes intercellular junctions in LEC, events which could contribute to sinusoidal remodeling and angiogenesis that characterizes fibrotic vasculature.
Figure 6.
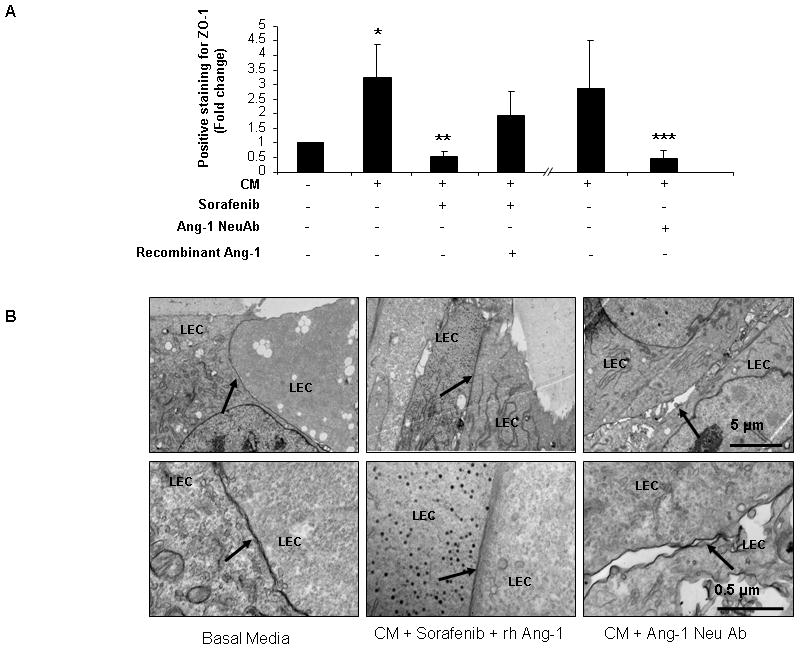
Angiopoietin-1 (Ang1) release by HSC promotes ability of LEC to form junctions in vitro. A: ZO-1 staining of TSEC was performed to assess junctions between LEC in response to CM and molecules as previously depicted in Figure 3C. ZO-1 staining was significantly increased in the control group in which LEC were incubated with conditioned media from HSC. ZO-1 staining increase was abolished when LEC were treated with CM derived from sorafenib-treated HSC. Inhibitory effect of sorafenib was partially reversed by adding human recombinant (rh)-Ang1 to CM (4-fold rise in ZO-1 staining). Inhibition of ZO-1 staining was also observed in a manner similar to sorafenib when LEC were incubated in HSC CM that contained Ang1-neutralizing antibody (n=3, mean ± SEM, * p<0.05-basal media, ** CM, *** CM). B: Human LEC were cultured on Matrigel with HSC derived CM treated with vehicle or sorafenib or with addition of Ang1 neutralizing antibody and samples were prepared for TEM. LEC incubated with CM showed close apposition and junctional complexes as did cells incubated with CM containing rh-Ang1 in addition to sorafenib (CM with sorafenib group was previously depicted in Figure 3D). Addition of Ang1 neutralizing antibody to CM resulted in gaps and disruption of LEC-to-LEC apposition as depicted by the arrows (Upper panel: 10000X, lower panel: 50000X; arrows point to sites of LEC-LEC contact).
Lastly, we extended these cell morphological observations using functional assays of vascular maturation that require LEC junctional complexes. Congruent with the morphological studies, Ang1 neutralizing-antibody attenuated tubulogenesis of LEC that occurs in response to conditioned media from HSC stimulated with PDGF (Figure 7A). Similarly, LEC tubulogenesis was restored by adding recombinant Ang1 to conditioned media derived from sorafenib-stimulated HSC, highlighting the decisive role of Ang1 and its regulation by sorafenib in this three-dimensional tubulogenic process (Figure 7B). These experiments were complemented using chemotactic assays that require cellular guidance cues and cell motility machinery. In this regard, conditioned media from sorafenib stimulated HSC or those treated with Ang1 neutralizing antibody significantly reduced the ability of LEC to migrate as compared to relevant control groups (Figure 7C). Also, similar to the three-dimensional tubulation studies, addition of recombinant Ang1 to conditioned media derived from sorafenib-treated HSC rescued LEC migration (Figure 7C). These findings provide a functional hierarchy to the pathways described here by indicating that Ang1 is both required and sufficient for effects on vascular remodeling.
Figure 7.
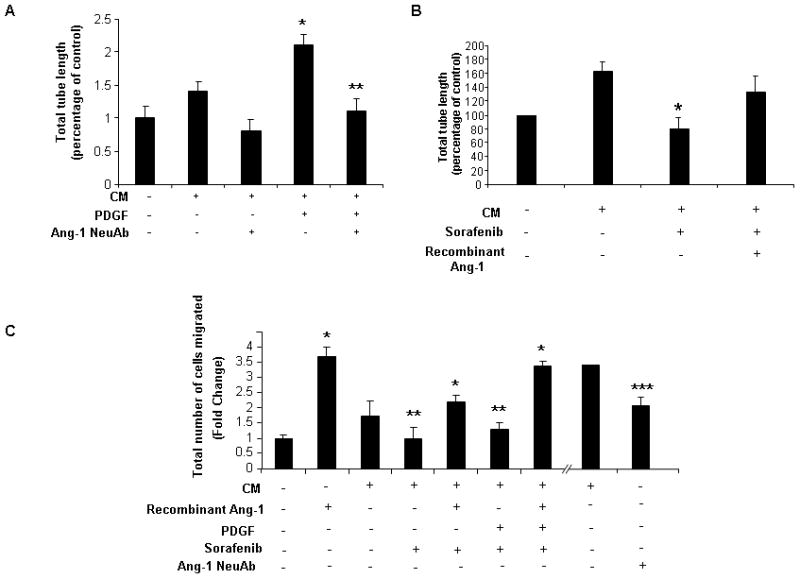
Release of angiopoietin-1 by HSC promotes LEC tubulogenesis and migration; A: Release of angiopoietin-1 by HSC promotes LEC tubulogenesis; Tube formation of human LEC on Matrigel was decreased when cells were incubated in PDGF stimulated HSC derived CM with addition of Ang1 neutralizing antibody as compared to IgG control (* p<0.05-CM, ** CM-PDGF). B: Tube formation of human LEC on Matrigel was decreased when cells were incubated with CM derived from HSC pre-treated with sorafenib (* p<0.05-CM). Addition of rh-Ang1 to the media partially rescued the inhibitory effect of sorafenib. C: Migration of human LEC was assessed in response to chemotactic responses to CM with interventions as described. LEC migration was decreased when stimulated with CM derived from sorafenib-treated HSC; effect was reversed by addition of rh-Ang1 to the media (* p<0.05-basal media, ** CM). LEC migration was decreased when incubated with CM treated with Ang1-neutralizing antibody (*** p<0.05-CM).
Discussion
Development of cirrhosis requires changes in matrix composition and turnover as well as conspicuous changes in intrahepatic vasculature that require orchestrated interaction between nonparenchymal liver cells, especially endothelial and stellate-cells. These vascular changes significantly contribute to the morbid complication of portal hypertension that accompanies advanced fibrosis. In the current study, we focused on identifying novel cellular and molecular pathways underlying angio-matrix changes that occur during liver fibrosis and defining how sorafenib, a compound that shows promising clinical utility in patients with cirrhosis and liver cancer, affects these pathways. In this regard, the present studies reveal several novel cellular and molecular phenomena that shed further light on angioarchitectural changes that accompany fibrosis (see Figure 8 for schematic model of signaling supported by these studies). First, we demonstrate that HSC secrete Ang1 which behaves as a key contributor to fibrosis-associated vascular changes. We show that excessive HSC-derived Ang1 disrupts sinusoidal homeostasis by promoting increased wrapping interactions between HSC and LEC as well as increased junctional connections amongst LEC. These phenomena culminate in a sinusoidal remodeling process that enhances HSC contraction around sinusoids as well as increased angiogenesis. Surprisingly, Ang1 production requires PI3K/Akt activation though it is independent of Raf, which is the classical target of sorafenib in hepatoma cells (4, 25), thus demonstrating that sorafenib uses distinct pathways to exert its changes in epithelial versus mesenchymal cells. These vascular changes are also coordinated with matrix remodeling as shown by an increase in Raf dependent fibronectin production, which like Ang1 production relies upon integrity of the KLF6 transcriptional pathway, thus revealing a remarkable coordination of vascular and matrix changes that contribute to cirrhosis. Lastly, we provide clear evidence that the multikinase inhibitor sorafenib inhibits the KLF6-Ang1-fibronectin molecular triad thereby attenuating angioarchitectural changes that typify cirrhosis. These observations also suggest that the function of sorafenib in cancer and cirrhosis might have distinct differences which can be exploited for tailoring different concentration responses that can achieve beneficial effects in the two conditions. However, it should be noted that therapies that target intrahepatic angiogenesis in human cirrhosis have not been evaluated in any systematic fashion and thus any beneficial or even harmful effect of such an intervention can not be reliably predicted at this time(2).
Figure 8.
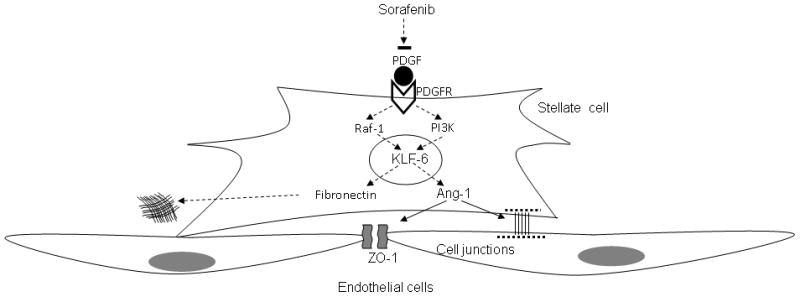
Schematic depiction of 1) signaling pathways by which PDGF regulates Ang1 and fibronectin production in HSC, 2) effects of pathways on vascular and matrix changes, respectively, and 3) how sorafenib inhibits these pathways.
To define nuclear events that regulate Ang1 expression we examined the promoter of this gene for cis-regulatory sequences which can potentially bind to relevant transcription factors. Interestingly, the Ang1 promoter contains several sequences that conform to the consensus for SP/KLF proteins. This family of transcription factors is composed of 24 different proteins, with many emerging as key regulators of gastrointestinal and hepatic cell biology and pathobiology(26). Indeed, we performed a family-wide screen to define which KLF protein regulates Ang1 expression. Using this approach we demonstrate that KLF6 occupies the promoter of Ang1 and that Ang1 expression is inhibited by siRNA knock down of KLF6. This is significant since KLF6 is the only member of this family whose function in liver fibrosis has been well established. For instance, KLF6 has been associated to liver wound healing(22, 27). Therefore, these findings when taken in context of prior studies indicate that KLF6 may be an operator of angioarchitectural changes that accompany fibrosis by virtue of its ability to drive a membrane-to-nucleus pathway in HSC that begins with activation of PDGFR and culminates in binding of KLF6 to the Ang1 promoter.
Our study utilizes state-of-the-art imaging techniques such as MRE and micro-CT for monitoring changes that occur microscopically but impact the organ as a whole. In this regard, our study can also be considered as a preclinical assessment of these innovative methodologies, the importance of which is underscored by the fact that MRE is gaining increasing attention as a potentially useful diagnostic modality in noninvasive assessment of liver fibrosis. Interestingly, our MRE assessment of rat liver during BDL-induced fibrosis revealed increased stiffness. Previous studies have indicated that such an increased stiffness reflects changes in matrix remodeling(28). However, increasing evidence, including results reported in this study, indicates that stiffness also reflects other processes that frequently accompany matrix deposition including inflammation, edema, and even vascular structure and portal pressure changes(15). Indeed, complementary utilization of high resolution micro-CT allowed us to define that abnormal MRE signals were indeed accompanied by prominent vascular changes. These observations were complimented by histological examinations which corroborated increases in vascular density after BDL. Thus, this powerful combination of MRE and micro-CT allow us to resolve two different important components of liver cirrhosis, namely matrix changes and vascular remodeling.
Some, though not all, recent studies suggest that therapeutic approaches that target aberrant vasculature structure in cirrhosis could have a beneficial effect on portal hypertension(18, 29–31). Congruent with this idea, it has been recently proposed that multikinase receptor tyrosine kinase inhibitors such as sorafenib, decrease portal hypertension in animal models of cirrhosis(18, 29, 30), although detailed molecular mechanisms responsible for this effect have warranted further investigation. We here extend our current understanding of how this targeted therapy drug may have a potential beneficial role in treatment of liver fibrosis and portal hypertension by virtue of its effects on sinusoidal cell crosstalk centered on Ang1(32–34). Indeed, HSC secrete Ang1 to promote formation of junctional complexes between LEC, a key step for angiogenesis and vascular restructuring within a mechanically stressed fibrotic microenvironment(1, 18, 35). Importantly, the capillary response regulated by Ang1 in diseased liver in vivo appears to be congruent with molecular mechanisms described here. For example, while normal liver sinusoids are characterized by a discontinuous phenotype, in cirrhosis these delicate vascular structures undergo what is commonly referred to as “capillarization,” with more durable stellate cell coverage of more closely interconnected endothelial cells. These phenotypic changes coincide with known functions of Ang1 as a stabilizer of vessels(36). Therefore, our results may also help to explain how sinusoidal vasculature adopts distinct phenotypic changes in cirrhosis and utilize this knowledge for designing future therapeutic interventions targeting this pathway. In total, this study underscores the importance of considering both, vasculature and matrix as combined therapeutic targets of therapies aimed to ameliorate cirrhosis and its complications.
Supplementary Material
Acknowledgments
The authors wish to thank Helen Hendrickson for her managerial support in the laboratory and Terri Johnson for her secretarial assistance.
Financial Support: DK59615-06 (Shah), P30DK084567 (LaRusso), EB000305 (Ritman)
Abbreviations
- HSC
hepatic stellate cells
- LEC
Liver endothelial cells
- TSEC
transformed liver endothelial cells
- BDL
bile duct ligation
- MRE
Magnetic Resonance Elastography
- Micro-CT
micro-computed tomography
- TEM
Transmission electron microscopy
- CM
Stellate cell derived conditioned media
Contributor Information
Dominique Thabut, Email: dthabut@gmail.com.
Chittaranjan Routray, Email: routray.chittaranjan@mayo.edu.
Gwen Lomberk, Email: lomberk.gwen@mayo.edu.
Uday Shergill, Email: ushergill2@gmail.com.
Kevin Glaser, Email: glaser.kevin@mayo.edu.
Robert Huebert, Email: huebert.robert@mayo.edu.
Leena Patel, Email: leena_patel200@hotmail.com.
Tetyana Masyuk, Email: masyuk.tetyana@mayo.edu.
Boris Blechacz, Email: blechacz.boris@mayo.edu.
Andrew Vercnocke, Email: vercnocke.andrew@mayo.edu.
Erik Ritman, Email: ritman.erik@mayo.edu.
Richard Ehman, Email: ehman.richard@mayo.edu.
Raul Urrutia, Email: urrutia.raul@mayo.edu.
Vijay Shah, Email: shah.vijay@mayo.edu.
References
- 1.Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, Kodama Y, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–1738. doi: 10.1053/j.gastro.2008.07.065. [DOI] [PubMed] [Google Scholar]
- 2.Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? J Hepatol. 2010;53:976–980. doi: 10.1016/j.jhep.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671–679. doi: 10.1053/j.gastro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Martinelli E, Troiani T, Morgillo F, Rodolico G, Vitagliano D, Morelli MP, Tuccillo C, et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin Cancer Res. 2010;16:4990–5001. doi: 10.1158/1078-0432.CCR-10-0923. [DOI] [PubMed] [Google Scholar]
- 6.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 7.Huebert RC, Jagavelu K, Liebl AF, Huang BQ, Splinter PL, Larusso NF, Urrutia RA, et al. Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis. Lab Invest. 2010;90:1770–1781. doi: 10.1038/labinvest.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 9.McNeil E, Capaldo CT, Macara IG. Zonula occludens-1 function in the assembly of tight junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell. 2006;17:1922–1932. doi: 10.1091/mbc.E05-07-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das A, Fernandez-Zapico ME, Cao S, Yao J, Fiorucci S, Hebbel RP, Urrutia R, et al. Disruption of an SP2/KLF6 repression complex by SHP is required for farnesoid X receptor-induced endothelial cell migration. J Biol Chem. 2006;281:39105–39113. doi: 10.1074/jbc.M607720200. [DOI] [PubMed] [Google Scholar]
- 11.Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology. 1998;114:344–351. doi: 10.1016/s0016-5085(98)70487-1. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen SM, Demirkaya O, Ritman EL. Three-dimensional imaging of vasculature and parenchyma in intact rodent organs with X-ray micro-CT. Am J Physiol. 1998;275:H1103–1114. doi: 10.1152/ajpheart.1998.275.3.H1103. [DOI] [PubMed] [Google Scholar]
- 13.Muthupillai R, Lomas DJ, Rossman PJ, Greenleaf JF, Manduca A, Ehman RL. Magnetic resonance elastography by direct visualization of propagating acoustic strain waves. Science. 1995;269:1854–1857. doi: 10.1126/science.7569924. [DOI] [PubMed] [Google Scholar]
- 14.Manduca A, Oliphant TE, Dresner MA, Mahowald JL, Kruse SA, Amromin E, Felmlee JP, et al. Magnetic resonance elastography: non-invasive mapping of tissue elasticity. Med Image Anal. 2001;5:237–254. doi: 10.1016/s1361-8415(00)00039-6. [DOI] [PubMed] [Google Scholar]
- 15.Talwalkar JA, Yin M, Fidler JL, Sanderson SO, Kamath PS, Ehman RL. Magnetic resonance imaging of hepatic fibrosis: emerging clinical applications. Hepatology. 2008;47:332–342. doi: 10.1002/hep.21972. [DOI] [PubMed] [Google Scholar]
- 16.Jagavelu K, Routray C, Shergill U, O’Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52:590–601. doi: 10.1002/hep.23739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dechene A, Sowa JP, Gieseler RK, Jochum C, Bechmann LP, El Fouly A, Schlattjan M, et al. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology. 2010;52:1008–1016. doi: 10.1002/hep.23754. [DOI] [PubMed] [Google Scholar]
- 18.Tugues S, Fernandez-Varo G, Munoz-Luque J, Ros J, Arroyo V, Rodes J, Friedman SL, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46:1919–1926. doi: 10.1002/hep.21921. [DOI] [PubMed] [Google Scholar]
- 19.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 20.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 21.Rippe RA, Almounajed G, Brenner DA. Sp1 binding activity increases in activated Ito cells. Hepatology. 1995;22:241–251. [PubMed] [Google Scholar]
- 22.Ratziu V, Lalazar A, Wong L, Dang Q, Collins C, Shaulian E, Jensen S, et al. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc Natl Acad Sci U S A. 1998;95:9500–9505. doi: 10.1073/pnas.95.16.9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 25.Iyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: a clinical and pharmacologic review. Expert Opin Pharmacother. 2010;11:1943–1955. doi: 10.1517/14656566.2010.496453. [DOI] [PubMed] [Google Scholar]
- 26.Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, Abate M, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. 2008;135:282–291. e281. doi: 10.1053/j.gastro.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narla G, DiFeo A, Yao S, Banno A, Hod E, Reeves HL, Qiao RF, et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65:5761–5768. doi: 10.1158/0008-5472.CAN-05-0217. [DOI] [PubMed] [Google Scholar]
- 28.Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–1154. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 29.Hennenberg M, Trebicka J, Stark C, Kohistani AZ, Heller J, Sauerbruch T. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol. 2009;157:258–270. doi: 10.1111/j.1476-5381.2009.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mejias M, Garcia-Pras E, Tiani C, Miquel R, Bosch J, Fernandez M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology. 2009;49:1245–1256. doi: 10.1002/hep.22758. [DOI] [PubMed] [Google Scholar]
- 31.Patsenker E, Popov Y, Stickel F, Schneider V, Ledermann M, Sagesser H, Niedobitek G, et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. 2006;98:1014–1023. doi: 10.1161/01.RES.0000218275.54089.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. 2000;87:603–607. doi: 10.1161/01.res.87.7.603. [DOI] [PubMed] [Google Scholar]
- 34.Fukuhara S, Sako K, Noda K, Nagao K, Miura K, Mochizuki N. Tie2 is tied at the cell-cell contacts and to extracellular matrix by angiopoietin-1. Exp Mol Med. 2009;41:133–139. doi: 10.3858/emm.2009.41.3.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, Vizzutti F, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 36.Wang YL, Hui YN, Guo B, Ma JX. Strengthening tight junctions of retinal microvascular endothelial cells by pericytes under normoxia and hypoxia involving angiopoietin-1 signal way. Eye (Lond) 2007;21:1501–1510. doi: 10.1038/sj.eye.6702716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.