

Abstract
The coupling of enolates through single electron oxidation is one of the most direct routes to generating 1,4-dicarbonyls. Recent work on the intermolecular heterocoupling of equimolar amounts of two different enolates through single electron oxidation has shown that synthetically useful yields beyond those statistically predicted can be obtained. To determine the underlying basis for the selective formation of heterocoupled products, kinetic, 7Li NMR, and synthetic studies were performed. The collection of data obtained from these experiments show that the selective formation of heterocoupled products is a consequence of heteroaggregation of lithium enolates.
Keywords: Lithium enolate, aggregation, single electron oxidation, oxidative coupling
The coupling of enolates through single electron oxidation is the most direct route to generating 1,4-dicarbonyls that are important precursors or structural components in a variety of natural products.1 Cyclizations are achieved through the intramolecular coupling of enolates derived from diesters2 and diketones3 as well as the intramolecular oxidative cross coupling of enolates derived from two different carbonyl precursors.1b, d Intermolecular homocoupling reactions of enolates are straightforward and have a long history in organic chemistry.2–5 Conversely, bimolecular heterocoupling of equimolar amounts of two enolates through single electron oxidation is more difficult and at best should result in 50% yield of the product. Successful approaches for the synthesis of unsymmetric 1,4-dicarbonyls require the use of superstoichiometric amounts of one enolate relative to the other,5 or the use of silyl bis-enol ethers.6
Unlike other synthetic routes to 1,4-dicarbonyls, the single electron oxidative coupling of equimolar amounts of enolates can afford the same products while requiring no prefunctionalization steps. As a result, the development of efficient enolate oxidative coupling reactions has the potential to lead to improved overall atom economy in multistep syntheses. Despite previous studies on the single electron oxidation of enolates, the ability to selectively heterocouple two different enolates through single electron oxidation remained elusive until recently, when Baran et al. reported the intermolecular oxidative heterocoupling of enolates.7 In all reported cases, when equimolar amounts of two different enolates were oxidized with Fe(III)- or Cu(II)-based oxidants, the heterocoupled products were obtained in greater than 50% yields with some products being obtained in greater than 70% yields. Subsequent synthetic studies on these coupling reactions revealed that best results were obtained in THF.7b Additionally, the presence of α-carbonyl radicals was established through radical clock studies.7b While these studies demonstrate several factors important in the oxidative heterocoupling of enolates, they do not address the underlying basis for the selective formation of heterocoupled product from an equimolar mixture of two different enolates. Herein we present spectroscopic and mechanistic data showing that the selective formation of heterocoupled products is a consequence of the heteroaggregation of lithium enolates.
A considerable body of mechanistic work in our group has demonstrated that selective single electron oxidation or reduction of one component in an equimolar mixture of two unique substrates is responsible for successful cross-coupling of different functional groups.8 In the case of enolate heterocoupling, if two enolates of different stabilities are present, one enolate may be preferentially oxidized to a radical as shown in Scheme 1. Faster oxidation of enolate 1 leads to radical 3. Preferential reaction of 3 with enolate 2 (as opposed to homodimerization) provides the intermediate 4. A second single electron oxidation leads to heterodimer 5. The oxidation of several enolates derived from the reaction of ketones, esters, and amides with lithium hexamethyldisilazide (LiHMDS) was examined with ceric tetra-n-butylammonium nitrate (CTAN) using stopped flow spectrophotometry. Surprisingly, all reactions were too fast to monitor and occurred in the mixing time of the instrument even at reduced temperatures. Although these experiments did not provide the expected results, the data suggested that differential rates of oxidation may not provide the basis for the selectivity observed in these oxidative enolate heterocouplings.
Scheme 1.
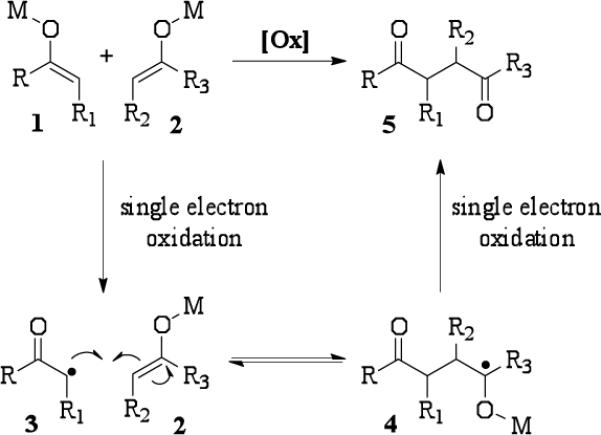
Selective formation of heterocoupled products through preferential oxidation.
Careful inspection of the literature describing successful enolate coupling through oxidation reveals that in most reactions, lithium bases are employed. Lithium coordination to anions, alkoxides, and carbanions often times leads to highly ordered aggregates in solution. The work of Reich,9 Seebach,10 and Collum11 has demonstrated that the unique coordination chemistry of lithium is responsible for the reactivity observed when lithium bases are employed as reagents in many bond-forming reactions. Interestingly, Collum et al. have shown that equimolar mixtures of two different enolates in tetramethylethylenediamine (TMEDA)/toluene preferentially formed heteroaggregated dimers depending on the steric congestion of the carbonyl precursors.12 The formation of heteroaggregated dimers is due to unfavorable steric interactions in the homodimer of the bulky carbonyl precursor.12 Based on these findings, could lithium enolate aggregation play a mechanistic role in the non-statistical formation of heterocoupled products?
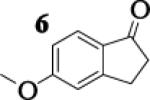
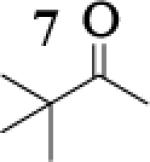
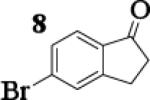
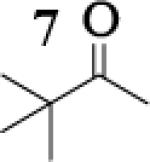
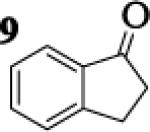
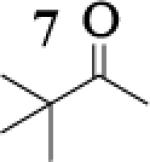
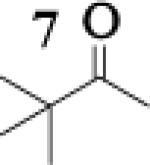
Many successful oxidative couplings of enolates are performed in THF.2–5, 7 Collum's work on the impact of solvent on lithium aggregation shows that enolates are tetrameric in THF.13 Given the complexity of the system, we chose to study the lithium enolate of pinacolone with an equimolar amount of lithium enolates derived from a series of cyclic ketones. Ketones with similar pKa values were chosen so that rates of enolization and stabilities were comparable. As a consequence, the relative rates of oxidation should be similar as well.14 Pinacolone was selected as one of the ketone partners since it is sterically bulky and has been shown previously to preferentially form lithium heteroaggregate dimers in TMEDA/toluene.12 To determine the impact of structure on heteroaggregation of equimolar mixture of two different lithium enolates, 7Li NMR experiments were performed on a series of ketone-ketone mixtures (Table 1). In these experiments, the lithium enolate of pinacolone was mixed with an equal amount of another lithium enolate derived from cyclic aryl ketones.
Table 1.
| Ketone A | Ketone B | |
|---|---|---|
|
|
15.7 : 1 |
|
|
14.7 : 1 |
|
|
14.3 : 1 |
|
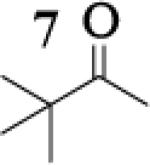
|
8.5 : 1 |
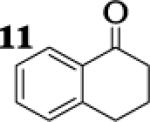
|
|
4.4 : 1 |
Distributions obtained by integrating 7Li NMR spectra at −30 °C.
[A] = [B] = 0.15 M and [LiHMDS] = 0.304 M in 2.0 M THF:Toluene
The results of the 7Li NMR experiments revealed several important features of the aggregation of lithium enolates in THF. For all the equimolar enolate mixtures of ketone-ketone partners examined, the lithium aggregates were ensembles of homoaggregated and heteroaggregated tetramers (A4 : A3B1 : A2B2 : A1B3 : B4) consistent with those reported by Collum et al.13 As illustrated in Figure 1 (Spectrum 1), when lithium enolates of 7 and 9 were generated separately and mixed at 1−78 °C, the homotetramer of 9 (A4) as well as smaller amounts of other aggregates including the homotetramer of 7 (B4) were the predominant species indicating minimal inter-aggregate exchange at reduced temperatures. However, upon warming and recooling the solution, the aggregate distribution shifted dramatically to favor the heteroaggregated A2B2 tetramer (Figure 1, Spectrum 2). This finding indicates that an energy barrier exists for rearrangement to the more thermodynamically stable enolate heteroaggregates.
Figure 1.
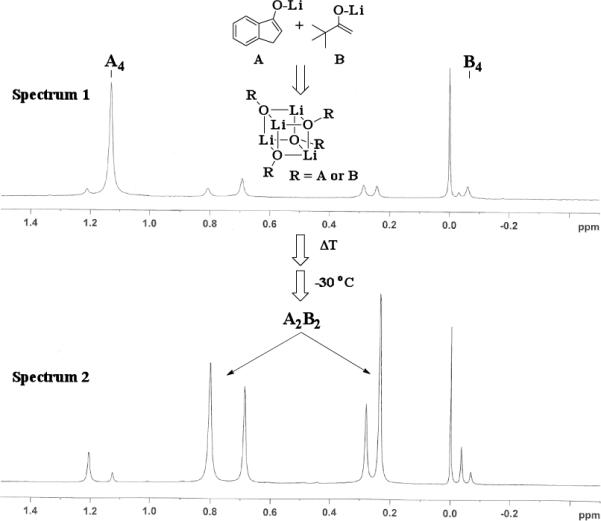
7Li NMR at −30 °C of 1 : 1 mixture of 7 and 9 with LiHMDS. Enolized separately and combined at −78 °C (Spectrum 1). Warming and recooling to −30 °C (Spectrum 2).
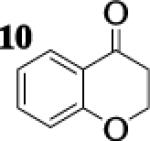
To assess the impact of substrate structure on the heteroaggregate distribution of equimolar mixtures of lithium enolates, the ratio between the most abundant heteroaggregate (A2B2) was compared to the individual homotetramers (A4 and B4) for every ketone-ketone mixture. As shown in Table 1, a unique ratio was obtained for each mixture of lithium enolates. Interestingly, even the ratio for the lithium enolates derived from 7 and 11, which was the lowest of the mixtures examined, was still above the statistically predicted distribution for an ensemble of tetramers.15
While placing substituents on the aromatic ring of ketone A (substrates 6 and 8) did not significantly impact the lithium aggregation, increasing the size of the adjacent ring (substrates 10 and 11) greatly reduced the amount of heteroaggregated tetramers. These observations are consistent with Collum's work on lithium heterodimers which showed that statistically predicted aggregate distributions were obtained when both enolates were sterically bulky.12
With the 7Li NMR data in hand, the question remains: are these non-statistical distributions of lithium aggregates involved in the selective oxidative heterocoupling of lithium enolates? To investigate the role of heteroaggregation, optimal reaction conditions were determined for the coupling of substrates 7 and 9. By screening several different oxidants, we found that CTAN and I2 provided the best yields and reproducibility. Iodine was employed as the oxidant in subsequent reactions (Table 2) since it is an attractive oxidant in terms of atom economy in that one equivalent of I2 carries out two single electron oxidations.16 Furthermore, oxidations using I2 benefited from improved synthetic work up procedures since the tetra-n-butylammonium counterions of CTAN are lipophilic and act as phase-transfer reagents complicating reaction workup.
Table 2.
Product distributions from the oxidative coupling of lithium enolatesa
| Ketone A | Ketone B | Heterocoupled Product | Product Ratiob | Yield (%)c,d |
|---|---|---|---|---|
| 6 | 7 |
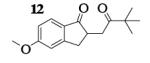
|
13.8 : 1 | 62 |
| 8 | 7 |
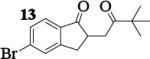
|
12.8 : 1 | 58 |
| 9 | 7 |
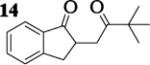
|
12.4 : 1 | 62 |
| 10 | 7 |
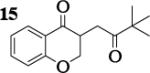
|
7.0 : 1 | 46 |
| 11 | 7 |
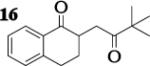
|
3.0 : 1 | 47 |
[A] = [B] = 0.12 M in THF, [LiHMDS] = 0.26 M in THF, [I2] = 0.12 M in THF.
Ratios (heterocoupled product:homodimer of 7) were determined by 1H NMR.
Determined by 1H NMR with ± 3 % error.
15–25% of ketone A was recovered in these reactions.
The oxidative coupling of equimolar mixtures of two different enolates preferentially generated the heterocoupled products (Table 2). More importantly, in all cases the product ratio of heterocoupled product to homodimer of 7 was better than statistically predicted (2 : 1). It is interesting to note that the homodimers of ketone A were never observed and instead the starting ketones were recovered in all cases. While experimental observations indicate that enolates derived from these ketones are oxidized, hydrogen atom abstraction from THF coordinated to the lithium centers of the aggregates becomes a competitive pathway.16, 17
With the synthetic studies completed, the degree of lithium enolate heteroaggregation was compared to the product ratios obtained after oxidation. As shown in Figure 2, there is a direct, linear correlation between the amount of lithium enolate heteroaggregation and the formation of heterocoupled product. Furthermore, the high degree of correlation between the heteroaggregate content and the degree of heterodimer product suggests that aggregation is the major driving force for the selective heterocoupling of two different lithium enolates. In the predominant A2B2 heteroaggregate, two different enolates are tethered to one another in solution. Having these enolates in proximity transforms a bimolecular oxidative carbon-carbon bond-forming event into a unimolecular process and provides a mechanism for non-statistical heterocoupling. As a consequence, equimolar mixtures of lithium enolates that exist predominantly as heteroaggregated enolates (A2B2) generate the most heterocoupled product when oxidized.
Figure 2.
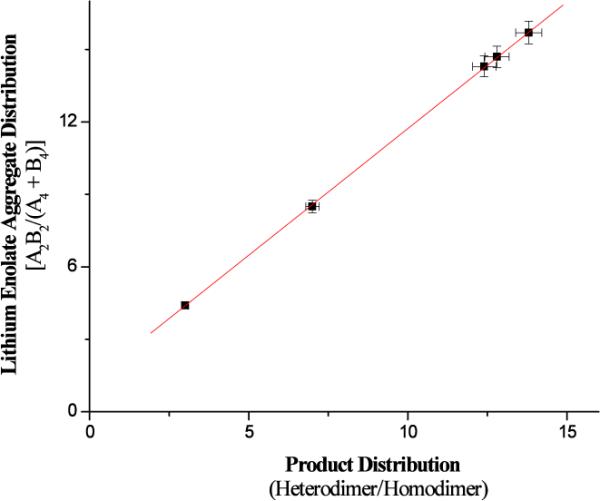
Impact of heteroaggregation on the oxidative heterocoupling of lithium enolates (R2 = 0.999).
Previous coupling reactions performed by both Saegusa and Baran have shown that synthetically useful yields of heterocoupled products can be obtained by employing an excess of one enolate relative to another.5, 7 To further demonstrate the importance of lithium aggregation in the oxidative coupling of lithium enolates, the 7Li NMR spectra for a 1 : 1 and a 2 : 1 mixture of enolates from substrates 10 and 7 were obtained (Figure 3). Spectrum 1 containing equimolar amounts of enolates derived from 7 and 10 exhibits a symmetric distribution of tetrameric aggregates. When oxidized, the heterocoupled product 15 to homodimer of 7 ratio was 7 : 1 (Table 2). Spectrum 2 shows the 7Li NMR spectrum of a 2 : 1 ratio of enolates derived from 10 and 7. Interestingly, the lithium enolate aggregate distribution dramatically shifts for the 2 : 1 mixture to favor A2B2 over the homotetramer of 7 (B4). When the 2 : 1 mixture was oxidized with I2, the selective formation of 15 improved to 26 : 1, well above the ratio expected from employing a one equivalent excess of 10 relative to 7.18 The enolate derived from 10 does not tend to homocouple upon oxidation (vide supra), and the homotetramer of 7 is drastically reduced in the 2 : 1 mixture. As a consequence, the likelihood of 7 being in proximity to 10 is significantly increased and the presence of excess A4 is not detrimental since 10 does not homocouple. This combination of factors leads to the increase in selectivity and yield, reaffirming the integral role that lithium aggregation plays in the oxidative coupling of enolates.
Figure 3.
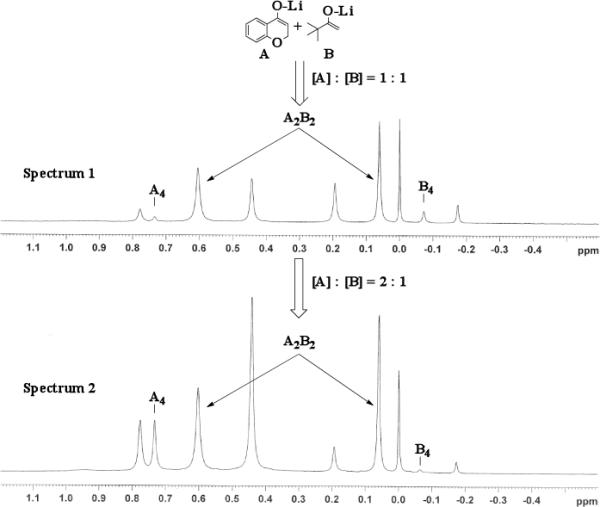
7Li NMR at −30 °C for 1 : 1 enolate mixture of 7 and 10 (Spectrum 1) and 2 : 1 mixture (Spectrum 2).
Taken together, the mechanistic experiments described herein show: (1) Equimolar mixtures of two different lithium enolates are ensembles of tetramers in THF. (2) The distribution of homoand heteroaggregates is dependent on substrate structure. (3) The major component of the mixture is heteroaggregate A2B2 when one enolate is sterically encumbered. (4) Single electron oxidation of solutions containing predominantly heteroaggregate A2B2 furnish the heterocoupled product selectively. (5) A direct correlation exists between the amount of heteroaggregate A2B2 and the ratio of heterocoupled to homocoupled products.
From a practical point of view, these data suggest that lithium aggregation may be responsible for the success (or failure) of previously reported reactions that proceed through the oxidation of enolates. In classic studies on oxidative cyclizations, Snider found that lithium enolates containing a pendant olefin dimerized and did not cyclize to produce a 5-membered ring as expected.19 In light of the present work, it is likely that lithium enolates tethered through an aggregate drive dimerization even over relatively fast intramolecular cyclizations. In another example, Alvarez-Ibarra and coworkers showed that lithium bases provided significantly improved yields and diastereoselectivities over potassium bases in the oxidative homocoupling of enolates derived from glycine esters.20
Overall, the results described herein highlight yet another example of lithium aggregation driving selectivity in organic reactions. The rational design of efficient syntheses is best facilitated by identifying and understanding the important mechanistic factors involved in the reaction system. Simple empirical models that discount aggregation are often insufficient to explain their role in bond-forming reactions. Given the large body of work on lithium aggregation, it is surprising the impact of lithium coordination chemistry in the design and mechanism of reactions is often over-looked. We are currently examining the role of lithium aggregates in more complex systems involving the oxidation of enolates derived from different carbonyl precursors (i.e. esters and amides). The results of these studies will be reported in due course.
Supplementary Material
ACKNOWLEDGEMENT
R.A.F. is grateful to the National Institutes of Health (1R15GM075960-01) for support of this work.
Footnotes
SUPPORTING INFORMATION AVAILABLE Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Ramig K, Kuzemko MA, McNamara K, Cohen T. J. Org. Chem. 1992;57:1968–1969. [Google Scholar]; (b) Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. J. Am. Chem. Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s. [DOI] [PubMed] [Google Scholar]; (c) Clift MD, Thomson RJ. J. Am. Chem. Soc. 2009;131:14579–14583. doi: 10.1021/ja906122g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martin CL, Overman LE, Rohde JM. J. Am. Chem. Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Babler JH, Sarussi SJ. J. Org. Chem. 1987;52:3462–3464. [Google Scholar]
- (3).Kobayashi Y, Taguchi T, Morikawa T, Tokuno E, Sekiguchi S. Chem. Pharm. Bull. 1980;28:262–267. [Google Scholar]
- (4).(a) Frazier RH, Harlow RL. J. Org. Chem. 1980;45:5408–5411. [Google Scholar]; (b) Langer T, Illich M, Helmchen G. Tetrahedron Lett. 1995;36:4409–4412. [Google Scholar]
- (5).(a) Ito Y, Konoike T, Saegusa T. J. Am. Chem. Soc. 1975;97:2912–2914. [Google Scholar]; (b) Ito Y, Konoike T, Harada T, Saegusa T. J. Am. Chem. Soc. 1977;99:1487–1493. [Google Scholar]
- (6).Avetta CT, Jr., Konkol LC, Taylor CN, Dugan KC, Stern CL, Thomson RJ. Org. Lett. 2008;10:5621–5624. doi: 10.1021/ol802516z. [DOI] [PubMed] [Google Scholar]
- (7).(a) Baran PS, DeMartino MP. Angew. Chem. Int. Ed. 2006;45:7083–7086. doi: 10.1002/anie.200603024. [DOI] [PubMed] [Google Scholar]; (b) DeMartino MP, Chen K, Baran PS. J. Am. Chem. Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y. [DOI] [PubMed] [Google Scholar]
- (8).(a) Prasad E, Flowers RA., II J. Am. Chem. Soc. 2002;124:6895–6899. doi: 10.1021/ja026074n. [DOI] [PubMed] [Google Scholar]; (b) Hansen AM, Lindsay KB, Antharjanam PKS, Karaffa J, Daasbjerg K, Flowers RA, II, Skrydstrup T. J. Am. Chem. Soc. 2006;128:9616–9617. doi: 10.1021/ja060553v. [DOI] [PubMed] [Google Scholar]; (c) Jiao J, Nguyen LX, Patterson DR, Flowers RA., II Org. Lett. 2007;9:1323–1326. doi: 10.1021/ol070159h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Casey BM, Eakin CA, Flowers RA., II Tetrahedron Lett. 2009;50:1264–1266. doi: 10.1016/j.tetlet.2008.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Choquette KA, Sadasivam DV, Flowers RA., II J. Am. Chem. Soc. 2010;132:17396–17398. doi: 10.1021/ja1088925. [DOI] [PubMed] [Google Scholar]
- (9).(a) Jones AC, Sanders AW, Bevan MJ, Reich HJ. J. Am. Chem. Soc. 2007;129:3492–3493. doi: 10.1021/ja0689334. [DOI] [PubMed] [Google Scholar]; (b) Jones AC, Sanders AW, Sikorski WH, Jansen KL, Reich HJ. J. Am. Chem. Soc. 2008;130:6060–6061. doi: 10.1021/ja8003528. [DOI] [PubMed] [Google Scholar]
- (10).(a) Seebach D. Angew. Chem. 1988;100:1685–1715. [Google Scholar]; (b) Seebach D. Angew. Chem. Int. Ed. 1988;27:1624–1654. [Google Scholar]
- (11).(a) Zuend SJ, Ramirez A, Lobkovsky E, Collum DB. J. Am. Chem. Soc. 2006;128:5939–5948. doi: 10.1021/ja060363k. [DOI] [PubMed] [Google Scholar]; (b) Singh KJ, Collum DB. J. Am. Chem. Soc. 2006;128:13753–13760. doi: 10.1021/ja064655x. [DOI] [PubMed] [Google Scholar]; (c) Collum DB, McNeil AJ, Ramirez A. Angew. Chem. Int. Ed. 2007;46:3002–3017. doi: 10.1002/anie.200603038. [DOI] [PubMed] [Google Scholar]; (d) Godenschwager PF, Collum DB. J. Am. Chem. Soc. 2007;129:12023–12031. doi: 10.1021/ja074018m. [DOI] [PubMed] [Google Scholar]; (e) Godenschwager PF, Collum DB. J. Am. Chem. Soc. 2008;130:8726–8732. doi: 10.1021/ja800250q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ma Y, Hoepker AC, Gupta L, Faggin MF, Collum DB. J. Am. Chem. Soc. 2010;132:15610–15623. doi: 10.1021/ja105855v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gruver JM, Liou LR, McNeil AJ, Ramirez A, Collum DB. J. Org. Chem. 2008;73:7743–7747. doi: 10.1021/jo801532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liou LR, McNeil AJ, Ramirez A, Toombes GES, Gruver JM, Collum DB. J. Am. Chem. Soc. 2008;130:4859–4868. doi: 10.1021/ja7100642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bordwell FG, Zhang XM, Filler R. J. Org. Chem. 1993;58:6067–6071. [Google Scholar]
- (15).Based on Pascal's triangle, the statistical distribution of an equimolar mixture of two enolates in an ensemble of tetramers should be 1 : 4 : 6 : 4 : 1. For this distribution, the ratio of heteroaggregate A2B2 to the homoaggregates A4 and B4 would be 3 : 1.
- (16).Renaud P, Fox MA. J. Org. Chem. 1988;53:3745–3752. [Google Scholar]
- (17).Kolonko KJ, Biddle MM, Guzei IA, Reich HJ. J. Am. Chem. Soc. 2009;131:11525–11534. doi: 10.1021/ja903479p. [DOI] [PubMed] [Google Scholar]
- (18).The yield of heterocoupled product 15 was also improved to 60 % (determined by 1H NMR).
- (19).Snider BB, Kwon T. J. Org. Chem. 1992;57:2399–2410. [Google Scholar]
- (20).Alvarez-Ibarra C, Csákÿ AG, Colmenero B, Quiroga ML. J. Org. Chem. 1997;62:2478–2482. doi: 10.1021/jo962116c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.