

Abstract
New dimensions in our understanding of immune cell trafficking in health and disease have been opened by the discovery of chemokines and their receptors. This family of chemo-attractant cytokines performs essential roles in the recruitment and subsequent positioning of leucocyte subsets within tissue microenvironments. Investigation of chemokine networks offers a novel approach to understand the mechanisms by which inflammatory cells persist in diseases such as rheumatoid arthritis (RA), where evidence is mounting that the inappropriate temporal and spatial expression of chemokines and/or their receptors may impair the resolution of leucocyte infiltrates. The recognition that stromal cells such as fibroblasts, as active components of tissue specific microenvironments, are able to determine the type and persistence of inflammatory infiltrates has opened new vistas in research. Stromal cells are active contributors to cytokine and inflammatory chemokine networks which result in immune cell recruitment and activation. However an intriguing role of stromal cells has been demonstrated in the inappropriate expression of constitutive, housekeeping chemokines, which contribute to the persistence of inflammation by actively blocking its resolution.
Keywords: Chemokine, chemokine receptor, Stromal, Fibroblast, Rheumatoid arthritis, Inflammation, Microenvironment, Review
2. INTRODUCTION
Chemokines are small, chemoattractant cytokines which enable the body to control the movement of cells of the immune system, and play key roles in the accumulation and activation of leukocytes at sites of inflammation. Chemokines and their receptors are therefore currently considered to be attractive therapeutic targets in several chronic inflammatory disorders, of which RA may be considered a prototype. In RA, hypertrophy of the synovial membrane, accompanied by persistent leukocyte infiltrates, is a characteristic histological feature. In health, the synovial membrane is composed of a thin layer of vascular connective tissue bounded at the synovial fluid compartment by a single cell layer composed of type A (macrophage-like) and type B (fibroblast-like) synoviocytes. In rheumatoid disease this structure undergoes marked hyperplasia, populated by numerous macrophages and proliferative synovial fibroblasts, the latter forming aggressive hyperplastic pannus lesions, which erode into adjacent cartilage and bone (70). The tissue becomes highly vascular, with areas of endothelium resembling high endothelial venules (20). The synovium also becomes infiltrated by numerous leukocytes, including B and T lymphocytes, which may be scattered diffusely, or form more organized focal aggregates (82). Recent research has implicated the expanded stromal cell populations of the inflamed rheumatoid synovium in maintaining these persistent inflammatory infiltrates via aberrant expression of chemokines and their receptors, resulting in recruitment and retention of inflammatory leukocytes, and the persistence of cytokine networks. In this review we will examine the evidence that stromal cells help maintain the persistence of the inflammatory infiltrate in the RA synovium, through their ectopic production of chemokines. We will also review the evidence from studies aimed at blocking the effects of chemokines which provide support for their role in inflammation and validate chemokines as potential therapeutic targets
3. CHEMOKINES
The chemokine family consists of some fifty chemoattractant cytokines (Table 1) which bind to members of the classical G-protein-coupled receptor (GPCR) family (72). Downstream signaling activates phospholipase C to mobilise intracellular calcium and utilises the Rho GTPase and phosphatidylinositol-3-OH kinase (PI3K) pathways (16). Such diverse signaling results in a variety of outcomes, including activation of leukocyte integrins and shape changes leading to chemotaxis (6;50). Chemokines share a common three dimensional structure despite remarkably low sequence homology of less than 20% (72). Structural similarity is maintained in part by the presence of a disulphide bond between two characteristic cysteine residues. Chemokines are classified according to the position these cysteine residues, which may be adjacent, as in CCL2 (MCP-1, monocyte chemoattractant protein 1), or separated by a single amino acid, as in CXCL12 (stromal cell-derived factor (SDF)-1). One important structural exception to this rule is CX3CL1 (fractalkine) in which the first two cysteines are separated by 3 amino acids; This chemokine is unique in that it may be secreted or expressed bound to cells by a mucin-rich, transmembrane stalk (3;62). It was Alisa Koch’s group who initially discovered that IL-8, in addition to having chemoattractant effects, also demonstrated angiogenic properties (48). Subsequently it became clear that among chemokines of the CXC family, the presence of an ELR motif defined angiogenic properties. Chemokines lacking the motif are largely angiostatic (72), a notable exception being CXCL12, which lacks an ELR motif but displays angiogenic properties (80). Fractalkine has also been shown to be angiogenic both in membrane bound and free forms (88). A universal property of chemokines is the presence of basic residues, particularly within the C terminal alpha helix region. Most chemokines are secreted, and in order to exert chemotactic effects or achieve activation of leucocytes at the endothelial surface, they must be immobilized on the surface of cells or within the extracellular matrix. This is achieved by binding to negatively charged glycosaminoglycans molecules (GAGs), for instance allowing presentation of chemokines to rolling leucocytes at the endothelial surface. This mechanism allows posting of secreted chemokines on the surface of local endothelium, where they can be sampled by rolling leukocytes (reviewed in (57)). Binding of chemokines such as CXCL12, CCL21 (SLC, secondary lymphoid tissue chemokine) and CCL19 (MIP3beta, macrophage inflammatory protein 3beta) activates integrins on the rolling cell surface, triggering firm lymphocyte adhesion, while CCL2 and CXCL8 trigger firm adhesion of monocytes and neutrophils respectively (13). Firm adhesion out of flowing blood is a prelude to migration across the endothelium, and through the extracellular matrix along immobilized haptotactic chemokine gradients, which may be fine-tuned by the oligomerisation of chemokine molecules on extracellular matrix GAGs (reviewed in (57)).
Table 1.
Human chemokines, receptor binding and function
| Human chemokine | Receptor(s) | Expression | Notes | |
|---|---|---|---|---|
| Nomenclature | ||||
| new | old | |||
| CXCL1 | GROα | CXCR2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL2 | GROβ | CXCR2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL3 | GROγ | CXCR2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL4 | PF4 | CXCR3 splice variant | ELR− | |
| CXCL5 | ENA-78 | CXCR2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL6 | GCP-2 | CXCR1,2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL7 | NAP-2 | CXCR2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL8 | IL-8 | CXCR1,2 | Inflammatory | ELR+ neutrophil chemotaxis |
| CXCL9 | Mig | CXCR3 | Inflammatory | ELR− Th1 inflammation |
| CXCL10 | IP-10 | CXCR3 | Inflammatory | ELR− Th1 inflammation |
| CXCL11 | I-TAC | CXCR3 | Inflammatory | ELR− Th1 inflammation |
| CXCL12 | SDF-1 | CXCR4 | Constitutive | ELR−, angiogenic, bone marrow, lymphoid organs |
| CXCL13 | BLC/BCA-1 | CXCR5 | Constitutive | Lymphoid follicles |
| CXCL14 | BRAK | Unknown | Unknown | ELR− |
| CXCL16 | SR-PSOX | CXCR6 | Heterogeneous | ELR− Th1 inflammation |
| XCL1 | lymphotactin | XCR1 | ||
| XCL2 | SCM-1β | XCR1 | ||
| CX3CL1 | fractalkine | CX3CR1 | Heterogeneous | |
| CCL1 | I-309 | CCR8 | Inflammatory | Th2 inflammation |
| CCL2 | MCP-1 | CCR2 | Inflammatory | Th1 inflammation |
| CCL3 | MIP-1α | CCR1,5 | Inflammatory | Th1 inflammation |
| CCL4 | MIP-1β | CCR5,8 | Inflammatory | Th1 inflammation |
| CCL5 | RANTES | CCR1,3,5 | Inflammatory | Th1, Th2 inflammation |
| CCL7 | MCP-3 | CCR1,2,3 | Inflammatory | Th1, Th2 inflammation |
| CCL8 | MCP-2 | CCR3,5 | Inflammatory | Th1, Th2 inflammation |
| CCL11 | eotaxin | CCR3 | Inflammatory | Th2 inflammation, allergy |
| CCL13 | MCP-4 | CCR2,3 | Inflammatory | Th1, Th2 inflammation, allergy |
| CCL14 | HCC-1 | CCR1 | Heterogeneous | |
| CCL15 | HCC-2/Lkn-1 | CCR1,3 | ||
| CCL16 | HCC-4/LEC | CCR1,2 | ||
| CCL17 | TARC | CCR4,8 | Heterogeneous | Th2 skin inflammation |
| CCL18 | DC-CK1 | Unknown | Constitutive | Lymphoid T cell zones |
| CCL19 | ELC/MIP-3β | CCR7 | Constitutive | Lymphoid T cell zones |
| CCL20 | LARC/MIP-3α | CCR6 | Inflammatory | Intestinal villi, skin |
| CCL21 | SLC/6Ckine | CCR7 | Constitutive | Lymphoid organs, high endothelial venules |
| CCL22 | MDC | CCR4,8 | Heterogeneous | Th2 inflammation, allergy |
| CCL23 | MPIF-1 | CCR1 | Heterogeneous | |
| CCL24 | Eotaxin-2 | CCR3 | Inflammatory | Th2 inflammation |
| CCL25 | TECK | CCR9 | Constitutive | Small intestine, thymus |
| CCL26 | Eotaxin-3 | CCR3 | Inflammatory | Th2 inflammation |
| CCL27 | CTACK | CCR10 | Constitutive | Skin |
| CCL28 | MEC | CCR3,10 | Heterogeneous | |
Functionally, chemokines and their receptors fall into two broad functional groups: constitutive (homeostatic) and inflammatory (inducible) chemokines (Table 1). Constitutive chemokines are continuously expressed and govern physiologically essential processes such as hematopoiesis and lymphocyte recirculation. They generally bind exclusively and monogamously as a single chemokine-receptor pair. For instance, CXCL12 is expressed by stromal cells of the bone marrow, where it is responsible for the recruitment and retention of CXCR4 expressing haematopoietic stem cells (18). Inflammatory chemokines such as CXCL8 are usually expressed only under conditions of inflammation and frequently bind promiscuously, with receptors being activated by more than one ligand and most chemokines activating more than one receptor. This highlights a fundamental difference in the regulation of chemokines and receptors which fall into either of the two groups. Tissues involved in homeostatic cell trafficking express constitutive chemokines persistently. Different cell types must therefore vary either the expression or sensitivity of chemokine receptors in order for selectivity in response to occur. For instance, pro- and pre-B cells are highly responsive to CXCL12, whilst dependent upon marrow stromal cells for mitogenic support, but at later stages of development lose this responsiveness (18). By contrast, receptors for inflammatory chemokines (such as CXCR1 and CXCR2, the receptors for CXCL8) must be persistently expressed by inflammatory cells such as neutrophils in order for them to be activated and chemo-attracted by infrequently expressed inflammatory chemokines. Regulation may therefore be achieved either at the level of ligand expression, receptor expression, or both.
4. THE STROMAL MICROENVIRONMENT AND PERSISTENCE OF DISEASE IN RA
Chemokines are intimately involved in site specific trafficking of leukocyte subsets, and this has provided the rational for antagonizing their function. However site specific recruitment of cells at the level of the endothelium is only one possible explanation for why certain subsets of leucocytes accumulate at sites of inflammation. Site specific retention of leucocytes by stromal cells may also play an important role. There is ample precedent for this. Stromal cells such as fibroblasts play well-documented roles in the selective retention of lymphocytes in organs such as the bone marrow and thymus. In the last ten years a paradigm shift has occurred in inflammation research. Haemopoietic cells such as leukocytes are no longer seen and analyzed in isolation, but need to be considered in the context of organ specific stromal microenvironments, made up of tissue specific cells such as fibroblasts, endothelial cells and resident macrophages, along with their highly specialised extracellular matrix (ECM) components.
4.1. The stromal microenvironment and resolution of inflammation
Physiological inflammation is not a stable state. In the absence of extrinsic stimuli such inflammation resolves and the tissue reverts to normal. In contrast, chronic persistent inflammation such as that present in the established rheumatoid synovium is highly stable. As an inflammatory process reaches its conclusion, the resolution of inflammatory leukocyte infiltrates within a microenvironment is governed by a number of dynamic factors: firstly the balance between cell recruitment and emigration, and secondly the balance between cell death and proliferation. Recent evidence suggests that tissue stromal cells are able to determine the type and duration of leukocyte infiltrates in an inflammatory response (10). At the resolution of such responses, stromal cells contribute to the withdrawal of survival signals and normalization of chemokine gradients, allowing infiltrating cells to undergo apoptosis or leave via draining lymphatics. Subversion of these pathways results in a switch to persistent inflammation which remains remarkably stable over time (63).
Ignoring the contribution of dynamic leucocyte-stromal interactions may account for the failure of conventional therapies to effect a permanent cure in RA since such therapies potentially miss many points where leukocyte-stromal interactions occur. Many therapeutic interventions that lead to elimination of inflammatory leukocytes from the synovium have been used with transient success, from intra-articular steroid injections to thoracic-duct drainage (66). However, these therapies do not affect the expanded fibroblast network, and within six to twelve weeks the inflammatory leukocytes return and with them, recurrence of disease symptoms. T lymphocytes have been shown to play an important role in TNF-alpha production by synovial monocytes, but crucially the T cells responsible appear to have the phenotype of cytokine-activated, rather than antigen-specific T cells (7). The role of a specific subtype of T helper lymphocyte, the Th17 cell, has recently become prominent. Studies suggest that the T lymphocyte phenotype observed in many chronic inflammatory diseases may be accounted for by this sub-population, the differentiation of which is dependent upon cytokines found plentifully in the RA synovial microenvironment (79). Recent evidence suggests that Th17 lymphocytes could also account for some specific pathological features of RA (76). Many studies throughout the 1990’s sought evidence for antigen-specific T cell clones in the synovium of patients with established RA. The overall outcome suggested that the synovial T cell population merely reflects recent immune events in the periphery rather than a synovial antigen-specific reaction (74). The perspective that the hyperplastic tissue microenvironment, rather than an antigen-specific immune process, maintains persistence of the inflammatory infiltrate has now received widespread support (64).
The most abundant cells of the stroma are fibroblasts which are responsible for the synthesis and remodeling of extracellular matrix components. In addition, their ability to produce and respond to growth factors allows reciprocal interactions with other stromal cell types and with adjacent epithelial and endothelial structures. As a consequence fibroblasts play a critical role during tissue development and homeostasis and are often described as having a “sentinel” or “landscaping” function. Moreover these functions contribute to the pathology of many diseases either directly, for example by overproduction of matrix components during fibrosis, and/or indirectly by influencing the behavior of neighboring cell types.
4.2. Rheumatoid synovial fibroblasts
Rheumatoid synovial fibroblasts provide a convincing example of how stromal cells contribute to the persistence of inflammation. RA synovial fibroblasts have been shown to regulate tissue injury and remodeling, and display an imprinted phenotype which is stable under in vitro culture conditions, and which extends to functionally important outcomes such as cartilage invasion, as demonstrated in SCID mouse models (58). Synovial fibroblast mediated erosion of cartilage and bone determine disease outcome for the majority of rheumatoid arthritis patients (24). Type I interferons are produced by the expanded stromal population of synovial fibroblasts and macrophages, resulting in a lack of proliferation, but also a block of the apoptotic signals which normally result in a coordinated wave of T lymphocyte death at the conclusion of an inflammatory response (67;75). The unique, imprinted phenotype of RA synovial fibroblasts bears remarkable phenotypic similarities to stromal cells of the bone marrow which are involved in the accumulation and support of haemopoietic cells (22). Recent studies have suggested that the phenotype of RA synovial fibroblasts is accounted for by the accumulation of blood borne stromal progenitor cells (Mesenchymal progenitor Cells) (22). Other possible sources of stromal cells in inflammatory diseases include epithelial to mesenchymal transition; a phenomenon observed in inflammatory diseases of the kidney at sites of epithelial injury (35). Targeting of such trans-differentiation processes may prove useful in retarding fibrotic diseases such as systemic sclerosis (69). Compelling evidence, discussed below, suggests that through their secretion of cytokines and chemokines, synovial fibroblasts play a role in the persistence of inflammation in the synovium (71).
5. CHEMOKINE AND CHEMOKINE RECEPTOR EXPRESSION IN THE INFLAMED SYNOVIUM
A considerable body of evidence has accumulated demonstrating sources of inflammatory chemokines which act to recruit inflammatory cells to the RA joint. Abundant monocytes and macrophages, and stromal elements such as synovial fibroblasts, are subject to a proinflammatory cytokine network and direct contact interactions with other infiltrating cells such as T lymphocytes (19;56), leading to high levels of expression of many inflammatory CK in the rheumatoid synovium (Figure 1). Neutrophil attracting chemokines are expressed at high levels by monocytes and stimulated fibroblasts and include CXCL8 (IL-8), CXCL5 (ENA-78, epithelial-cell-derived neutrophil attractant 78) and CXCL1 (GROalpha, growth related oncogene alpha) (44;46;47). Monocytes and T cells may be recruited by a range of CXC and CC chemokines found at high levels in the synovium; CXCL10 (IP-10) and CXCL9 (Mig) are highly expressed in synovial tissue and fluid (65). CXCL16 is also highly expressed in the RA synovium and acts as a potent chemoattractant for T cells .CCL2 (MCP-1) is found in synovial fluid and known to be produced by synovial fibroblasts; it is considered to be a pivotal chemokine for the recruitment of monocytes (45;87). CCL3 (Mip-1alpha), CCL4 (Mip-1beta) and CCL5 (RANTES) are chemotactic for monocytes and lymphocytes, expressed at high levels in inflamed rheumatoid synovium and known products of synovial fibroblasts (34;65). CCL20 (Mip-3alpha) is also over-expressed in the synovium, and has a similar chemoattractant profile via its specific receptor, CCR6 (14;54). CX3CL1 (Fractalkine) is also widely expressed in the rheumatoid synovium (73). A number of chemokine receptors have been shown to differ between peripheral blood and synovial leucocytes, suggesting that they are enriched in the synovium either though their selective recruitment by endothelial expressed chemokines, or following up-regulation by the microenvironment after recruitment. In RA patients, circulating monocytes express mainly CCR1, CCR2 and CCR4, whereas monocytes isolated from synovial fluid express higher levels of CXCR3 and CCR5 (38). Similarly, synovial CD4 T lymphocytes appear to express higher levels of CCR5, CXCR2, CXCR3 and CXCR6 than circulating cells while expressing low levels of CCR3, suggesting a Th1 selective recruitment bias (27). Clearly such exuberant expression of chemokines of an inflammatory type may be responsible for considerable recruitment of activated lymphocytes, monocytes and neutrophils, though once again, such expression does not constitute a disease-specific profile.
Figure 1.

Stromal codes regulating accumulation of leukocytes in the lymph node are aberrantly expressed during lymphoid neogenesis in rheumatoid arthritis. During physiological inflammation and in rheumatoid arthritis, inflammatory chemokines (CCL2-CCL5, CX3CL1 and CXCL1-CXCL11 and inflammatory mediators such as IFN-gamma, TNF-alpha and IL-1 are produced by stromal cells and lead to the recruitment of inflammatory cells (lymphocytes, neutrophils and monocytes). Homeostatic chemokines (CXCL12, CXCL13, CCL19, CCL21) are components of the stromal code that help define stromal niches such as the lymph node and bone marrow, governing leukocyte accumulation, differentiation and survival. Stromal cells express the appropriate chemokine that is recognized by cognate receptors on infiltrating leukocytes. In persistent, pathological inflammation such as occurs in RA, stromal cells begin to aberrantly express components of the physiological stromal code normally associated with lymphoid tissues, leading to lymphoid neogenesis.
Angiogenesis is a vital aspect of the proliferative RA synovium, and is characterised by the presence both of angiostatic chemokines such as CXCL9 (Mig) and CXCL10 (I-TAC) and angiogenic chemokines such as CXCL5 (ENA-78), CXCL8 (IL-8) and CXCL12 (4;49). Soluble fractalkine has also been shown by Alisa Koch’s group to contribute to the angiogenic effects of RA synovial fluid, suggesting an important role in disease (43). Given the degree of chemokine expression in the RA synovium, it appears that chemokines are important players in determining the balance between pro-angiogenic and anti-angiogenic factors in the RA synovium.
6. CONSTITUTIVE CHEMOKINES IN LEUKOCYTE-STROMAL CELL INTERACTIONS IN RA
The presence of high levels of inflammatory chemokines, particularly produced by stromal cells, is therefore a characteristic of RA. However recent data suggest that paradoxically there is also a role for constitutive chemokines, which previously had not been thought to be associated with inflammatory processes, but rather with the recruitment of lymphocytes to secondary lymphoid tissues. The constitutive chemokine CXCL12 (SDF-1) and its receptor CXCR4 have emerged as unexpected but crucial players in the accumulation of T lymphocytes within the rheumatoid synovial microenvironment (Figure 2). CXCR4 is expressed constitutively on naïve T cells, but not on highly differentiated CD45RO+ T cells in peripheral blood (9). Unexpectedly, CD45RO+ T lymphocytes were found to express CXCR4 receptors at high levels in the rheumatoid synovium. Its ligand CXCL12 was highly expressed on endothelial cells at the sites of T cell accumulation (9;59;61). In addition, stromal-cell derived TGF-beta is responsible for up-regulation of CXCR4 receptors on T cells in the synovium (9). Cross-talk between chemokine and cytokine networks may operate to reinforce the retention of T cells by CXCL12. For example, locally raised IL-1 or TNF-alpha levels cause synovial fibroblasts and macrophages to secrete IL-15. This cytokine also up-regulates CXCR4 on T cells, and may thus also contribute to the retention of T lymphocytes (59). Evidence also suggests that the stability of lymphocyte infiltrates is reinforced by a positive feedback loop, whereby tissue CXCL12 promotes CD40 ligand expression on T cells, which in turn stimulates further CXCL12 production by synovial fibroblasts (59). Interestingly, levels of CXCL12 secreted by synovial fibroblasts have recently been shown to be controlled in part by T cell derived IL-17 (40).
Figure 2.
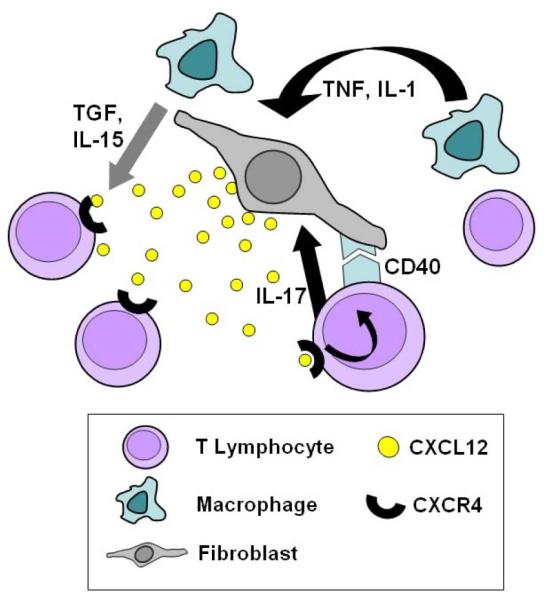
The constitutive chemokine CXCL12 and its receptor CXCR4 are crucial players in the accumulation of T lymphocytes within the rheumatoid synovial microenvironment. Activated T lymphocytes do not usually express high levels of CXCR4, the CXCL12 receptor, however high levels are expressed in activated T lymphocytes of the rheumatoid synovium. CXCL12 is highly expressed on endothelial cells and by synovial fibroblasts at sites of T cell accumulation. The mechanisms whereby both CXCR4 and CXCL12 are inappropriately upregulated are becoming clearer. Stromal-cell derived TGF-beta has been shown to be a major signal up-regulating CXCR4 receptors on T cells in the synovium. Furthermore, locally raised IL-1 or TNF-alpha levels cause synovial fibroblasts and macrophages to secrete IL-15. This cytokine also up-regulates CXCR4 expression by T cells. Additional evidence suggests that the stability of lymphocyte infiltrates is reinforced by a positive feedback loop, whereby tissue CXCL12 promotes CD40 ligand expression on T cells. Cell-contact mediated ligation of the fibroblast CD40 receptor and release of T cell derived IL-17 stimulates further CXCL12 production by synovial fibroblasts.
There is therefore clear evidence in support of the hypothesis that aberrant ectopic expression of constitutive CK by synovial fibroblasts contributes to the retention of T cells within the RA synovium. Other cell constituents of the rheumatoid inflammatory infiltrate may be affected by the CXCL12/CXCR4 axis. Blades and colleagues showed an increase expression of CXCL12/CXCR4 by monocyte/macrophage cells in RA compared with OA. In addition, using implanted human synovial tissue in SCID mice, they demonstrated that monocytes are recruited into transplanted synovial tissue by CXCL12 (5). Contact-mediated B cell survival induced by synovial fibroblasts has also been shown to depend upon CXCL12 and CD106 (VCAM-1)-dependent mechanisms which are independent of TNF-alpha (11). Over-expression of CXCL12 was also identified as a distinct feature of rheumatoid, as opposed to osteoarthritis synovia using cDNA arrays (85). Data validating these findings in vivo come from a CIA model in DBA/1 (interferon-γ receptor deficient) mice, where administration of the specific CXCR4 antagonist AMD3100 significantly ameliorated disease severity (55). In another murine CIA model the small molecule CXCR4 antagonist 4F-benzoyl-TN14003 ameliorated clinical severity and suppressed DTH (delayed type hypersensitivity) responses (83). The CXCL12/CXCR4 constitutive CK pair therefore seems to play an important role in cellular retention in RA.
The role of CXCL12 in angiogenesis in RA is also of interest; Pablos et al reported that rheumatoid synovial fibroblasts were responsible for secreting CXCL12, demonstrating by immunohistochemistry that it colocalised on endothelial cells with the angiogenesis marker αvβ3 integrin, consistent with a role for CXCL12 in both cell recruitment and angiogenesis (61). Synovial hypoxia may also play a role in determining chemokine expression, as both CXCL12 on synovial fibroblasts, and CXCR4 on monocytes have been shown to be upregulated under hypoxic conditions (32;77). It seems likely that CXCR4 antagonists will be of use in the therapy of rheumatoid arthritis, provided that toxicity issues due to stem cell mobilization from the bone marrow (in which CXCL12/CXCR4 interactions maintain the relationship between haemopoietic stem cells and bone marrow stromal cells) do not pose a major problem. Recent evidence suggests that the synovial microenvironment may in part be responsible for induction and continued expression of both inflammatory (CCL5 and CXCR3) and constitutive (CXCL12 and CCR7) CK receptors in synovial T cells. This provides an alternative explanation for the disproportionately high levels of some CK receptors seen on synovial T cells, and for the presence of constitutive CK receptors such as CCR7 and CXCR4 on retained lymphocytes (12). In summary, the inappropriate temporal and spatial expression of constitutive CK by stromal cells plays a crucial role in the accumulation of leukocytes within the synovium.
Until recently, the majority of research on leukocyte trafficking has concerned recruitment of cells via the vascular endothelium. However a recent breakthrough in the elucidation of trafficking pathways has been the development of specific markers for lymphatic endothelium. Of these, the most specific is LYVE-1, a hyaluronan receptor expressed exclusively on draining lymphatic vessels (36). Markers such as LYVE-1 now allow us to address some fundamental questions about differences between vascular and lymphatic vessels, and the part played by the latter in control of trafficking out of the joint. Recent work has identified gradients of expression of the constitutive chemokines CCL19 and CXCL12: In lymphoid tissue (tonsil), higher CK expression was seen on vascular than lymphoid vessels, consistent with the attraction and retention of lymphocytes. In RA synovium by contrast levels of chemokine expression were equally high in vascular and lymphoid vessels, suggesting that increased tissue expression of constitutive CK plays a role in lymphocyte retention by subverting the normal CK gradient which causes egress via lymphatics towards draining lymph nodes (12).
7. CHEMOKINES AND LYMPHOID NEOGENESIS IN RA
The lymphoid infiltrates in the rheumatoid synovium can be divided into at least 3 distinct histological groupings, varying from diffuse lymphocyte infiltrates to clear germinal center reactions (82). Moreover, there is evidence that such distinct histological types correlate with other serum indicators of disease activity (41;42). This long-described feature, termed lymphoid neogenesis, relies upon inappropriate, but highly organized temporal and spatial expression by stromal cells of the same constitutive CK, particularly CXCL13 and CCL21, which are associated with true lymphoid organogenesis (Figure 1). The elegant choreography of lymphocyte:stromal interactions within lymph nodes is organized by expression of adhesive and chemotactic cues in overlapping and combinatorial fashions. Once they have encountered new antigen, dendritic cells specialised in the presentation of antigen to lymphocytes undergo a process of maturation under the local influence of inflammatory cytokines and bacterial and viral products. As a result inflammatory CK receptors are down-regulated, and up-regulation of the constitutive receptors CCR4, CCR7 and CXCR4 occurs, causing DCs to migrate into local draining lymphatics (which express the CCR7 ligand CCL21 (secondary lymphoid tissue chemokine, SLC)) and thereby into peripheral lymph nodes (51). Trafficking of B and T cells is regulated by CXCL13 (BCA-1, B cell-attracting chemokine 1), its receptor CXCR5, and CCL21 and CCL19 (EBL-1-ligand chemokine, ELC), which are both CCR7 agonists. Within the lymph node CXCR5 bearing B cells are attracted to follicular areas, while T cells and DCs are maintained within parafollicular zones by local expression of CCL21 and CCL19 (17;21). Some T cells which have been successfully presented with their cognate antigen by DCs then up-regulate CXCR5, allowing them to migrate towards and interact with B cells.
Genesis of lymphoid follicular structures in diseases such as diabetes and RA appears to rely upon expression of such constitutive chemokines, in association with the lymphotoxins alpha and beta (LT-alpha and LT-beta) and TNF-alpha (33;82). In this context it is important to note that transgenic animals over-expressing the TNF-alpha gene display increased formation of focal lymphoid aggregates and develop a chronic arthritis similar to RA (39). Clearly one of the many mechanisms of action of anti-TNF therapy may involve the dissolution of such aggregates. In transgenic mouse models, expression of CXCL13 in the pancreatic islets was sufficient for the development of T and B cell clusters, but as they lacked follicular dendritic cells, was not sufficient for true germinal centre formation (52). CCL21 does appear to be sufficient in some cases for lymph node formation; murine pancreatic islet models have demonstrated formation of lymph node like structures in the presence of CCL21 (15;23), and lymphoid infiltrates in response to CCL19 expression, a possible common pathway being the induction of lymphotoxins on infiltrating lymphocytes (51). Lack of CCL21 signaling impairs T cell traffic into lymph node structures via high endothelial venules, and results in disorganization of T cell zones (25;53). Weyand and colleagues used the histological heterogeneity seen in RA to identify those factors critical to formation of lymphoid microstructures, showing that transcription levels of CXCL13 and CCL21 were increased 10- to 20-times in tissues with germinal centres compared to tissues with other histological patterns. Multivariate analysis showed that LT-beta and CXCL13 were necessary, but not sufficient for lymphoid neogenesis (82). It has also been shown that CXCR5 is overexpressed in the rheumatoid synovium, consistent with a role in recruitment and positioning of B and T lymphocytes within lymphoid aggregates of the RA synovium (78). It therefore seems likely that expression of lymphoid constitutive CK contributes significantly to the entry, local organization and exit of lymphocytes in the RA synovium. It also seems that the ectopic expression of chemokines is a general characteristic of a number of chronic rheumatic conditions, since another B cell attracting chemokine CXCL13 (BCA-1) is inappropriately expressed by stromal cells in the salivary glands of patients with Sjogren’s syndrome (1).
8. VALIDATION OF CHEMOKINES AS TARGETS IN RA
There is good evidence that blocking chemokine-receptor interactions in models of rheumatoid arthritis can have beneficial effects, thereby providing validation of chemokines as important mediators in inflammatory disease. Indirect evidence supporting the modulation of chemokine expression comes from the use of anti-TNF biological therapy, in which synovial expression of CXCL8 and CCL2 is reduced, accompanied by decreased cellularity of the synovium (84).
8.1. Evidence from animal models
An important study illustrating the principle that timing of therapeutic interventions is critical was that by Halloran et al, who showed that CXCL5 (ENA-78) expression correlated with disease progression at the synovial level, and demonstrated that administration of a polyclonal antibody against ENA-78 could attenuate adjuvant-induced arthritis in rats if given after the adjuvant but prior to the onset of disease. Antibody given after disease had become established was not effective (29). CCL3 (Mip-1α) is also expressed early in rodent disease models; treatment with an antibody to CCL3 delayed onset and reduced severity of collagen-induced arthritis (CIA) in mice (37). These studies have implications for the usefulness of chemokine antagonists in clinical disease, and for our need for information about when and how different chemokines become important players in disease pathogenesis. A number of other studies have shown the benefit of inhibiting CC chemokines in rodent models. A 67 amino acid sequence of CCL2, acting as an antagonist, has been shown to modify the severity of joint disease in MRL-lpr mice (28). However in when used in CIA in mice, anti-CCL2 antibodies ameliorated disease during its initiation, but aggravated disease once established (8). Inhibition of CCL5 (RANTES) binding appears to have beneficial effects: Administration of the partial agonist metRANTES, blocking the effects of CCL5 and CCL3, reduced the severity of CIA in mice (68), while an anti-CCL5 antibody had equivalent effects on CIA in rats (2). Yang et al used a small molecule antagonist of CCR5 and CXCR3 (TAK-779) to treat collagen-induced arthritis in DBA/1 mice (26;89). They found that antagonism of CCR5 decreased both the incidence and the severity of joint disease by inhibiting migration, with no effect on the development of T cell responses to collagen. Similarly positive results were seen when a CCR5 antagonist was used in CIA in rhesus monkeys (86). These results suggest that preventing the migration of activated T cells to the joint may be a viable approach for disease control (89). The CXCL16/CXCR6 chemokine receptor pair also shows promise as a means of decreasing T cell recruitment; anti-CXCL16 antibodies inhibited disease signs severity and bone destruction in murine CIA (60). Blockade of the CXCL12/CXCR4 axis has shown promise in animal models, as discussed above.
8.2. Chemokine blockade in RA patients
The most convincing validation of chemokine receptor antagonism as a therapeutic target in humans thus far comes from a phase I trial by Haringman et al, in which a selective antagonist of CCR1, the common receptor for CCL3 and CCL5, was administered to patients with active rheumatoid arthritis. Patients given the antagonist showed both clinical and immunohistochemical improvement after 14 days. Cells which might be expected to express CCR1 such as macrophages and T cells were reduced in number, whereas those not expressing CCR1 were unaffected; no significant adverse events were noted (31). This study forms the vanguard of a number of impending clinical trials using antagonists of receptors such as CCR1, CCR2, CCR5 and CXCR3. Despite excellent data demonstrating its production by stromal cells of the RA synovium, existing data for CCL2 (MCP1) blockade is however equivocal. A monoclonal antibody against CCL2 used in RA patients failed to show benefit, which may have resulted from a rise in serum CCL2 levels observed amongst treated patients (30). CXCL8 blockade has also been unsuccessful thus far in clinical trials, with a similar compensatory mechanism observed (81).
The next and more challenging family of targets is that of constitutive chemokines inappropriately expressed by stromal elements within the synovial tissue which directly contribute to maintaining the persistence of inflammatory leukocyte infiltrates.
9. PERSPECTIVE
It is now established that stromal cells are active participants in the development and resolution of physiological inflammatory responses within tissue microenvironments. It is also increasingly evident that the persistence of pathological inflammatory responses as seen in RA owes more to fundamental changes within the stromal microenvironment than it does to changes in leukocytes themselves. Understanding the roles played by stromal cells in maintaining leukocyte infiltrates and blocking their resolution offers the key to new therapeutic advances, and possibly the tantalizing prospect of curative therapies. Chemokines and their receptors appear to be central players in these pathological mechanisms and are thus tempting targets for intervention. It is evident that the rheumatoid synovium belongs to a select group of persistent inflammatory diseases characterised by subversion of the normal constitutive pathways which regulate the formation of lymphoid tissues, and trafficking of cells through them. The tissue phenotype resulting from these events is the formation of ectopic lymphoid deposits. Identifying further the mechanisms driving the aberrant expression of constitutive chemokines may offer new therapeutic opportunities to target inflammation in RA.
10. ACKNOWLEDGMENTS
The University of Birmingham Rheumatology Research group is supported by the Arthritis Research Campaign, The Medical Research Council, and is a member of the European Union Autocure consortium.
Abbreviations
- BCA-1
B cell-attracting chemokine 1
- BLC
B lymphocyte chemoattractant
- BRAK
breast and kidney-expressed chemokine
- CIA
collagen induced arthritis
- CTACK
cutaneous T cell-attracting chemokine
- DARC
Duffy antigen/receptor for chemokines
- DC-CK1
dendritic cell-derived CC chemokine 1
- ELC
EBL-1-ligand chemokine
- ENA-78
epithelial-cell-derived neutrophil attractant 78
- GAG
glycosaminoglycans
- GCP
granulocyte chemotactic protein
- GRO
growth related oncogene
- HCC
haemofiltrate CC chemokine
- IL
interleukin
- IP10
interferon-inducible protein 10
- I-TAC
interferon-inducible T cell alpha chemoattractant
- LARC
liver- and activation-regulated chemokine
- LEC
liver-expressed chemokine
- LCC-1
liver-specific CC chemokine 1
- Lkn
leukotactin
- LT
lymphotoxin
- MCP
monocyte chemoattractant protein
- MDC
macrophage-derived chemokine
- MEC
mammary-enriched chemokine
- Mig
monokine induced by interferon gamma
- MIP
macrophage inflammatory protein
- MPIF
myeloid progenitor inhibitory factor
- PF4
platelet factor 4
- RANTES
regulated on activation normal T cell expressed and secreted
- SCID
severe combined immunodeficient
- SDF
stromal cell-derived factor
- SLC
secondary lymphoid tissue chemokine
- TARC
thymus- and activation-regulated chemokine
- TECK
thymus-expressed chemokine
- TGF
transforming growth factor
- TNF
tumour necrosis factor
10. REFERENCES
- 1.Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, Hamburger J, Ainsworth J, Mathews J, Salmon M, Bowman SJ, Buckley CD. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren’s syndrome. Arthritis Rheum. 2001;44:2633–2641. doi: 10.1002/1529-0131(200111)44:11<2633::aid-art443>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 2.Barnes DA, Tse J, Kaufhold M, Owen M, Hesselgesser J, Strieter R, Horuk R, Perez HD. Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J.Clin.Invest. 1998;101:2910–2919. doi: 10.1172/JCI2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 4.Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, Strieter RM. CXC chemokines in angiogenesis. J.Leukoc.Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- 5.Blades MC, Ingegnoli F, Wheller SK, Manzo A, Wahid S, Panayi GS, Perretti M, Pitzalis C. Stromal cell-derived factor 1 (CXCL12) induces monocyte migration into human synovium transplanted onto SCID Mice. Arthritis Rheum. 2002;46:824–836. doi: 10.1002/art.10102. [DOI] [PubMed] [Google Scholar]
- 6.Bokoch GM. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–1660. [PubMed] [Google Scholar]
- 7.Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BM, Feldmann M. Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 2002;46:31–41. doi: 10.1002/1529-0131(200201)46:1<31::AID-ART10029>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlondorff D, Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J.Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- 9.Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, Amara A, Curnow SJ, Lord JM, Scheel-Toellner D, Salmon M. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J.Immunol. 2000;165:3423–3429. doi: 10.4049/jimmunol.165.6.3423. [DOI] [PubMed] [Google Scholar]
- 10.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 11.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J.Clin.Invest. 2001;107:305–315. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, Salmon M, Buckley CD. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J.Immunol. 2005;174:1693–1700. doi: 10.4049/jimmunol.174.3.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cascieri MA, Springer MS. The chemokine/chemokine-receptor family: potential and progress for therapeutic intervention. Curr.Opin.Chem.Biol. 2000;4:420–427. doi: 10.1016/s1367-5931(00)00113-7. [DOI] [PubMed] [Google Scholar]
- 14.Chabaud M, Page G, Miossec P. Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J.Immunol. 2001;167:6015–6020. doi: 10.4049/jimmunol.167.10.6015. [DOI] [PubMed] [Google Scholar]
- 15.Chen SC, Vassileva G, Kinsley D, Holzmann S, Manfra D, Wiekowski MT, Romani N, Lira SA. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J.Immunol. 2002;168:1001–1008. doi: 10.4049/jimmunol.168.3.1001. [DOI] [PubMed] [Google Scholar]
- 16.Curnock AP, Logan MK, Ward SG. Chemokine signalling: pivoting around multiple phosphoinositide 3-kinases. Immunology. 2002;105:125–136. doi: 10.1046/j.1365-2567.2002.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- 18.D’Apuzzo M, Rolink A, Loetscher M, Hoxie JA, Clark-Lewis I, Melchers F, Baggiolini M, Moser B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur.J.Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- 19.Dayer JM, Burger D. Cytokines and direct cell contact in synovitis: relevance to therapeutic intervention. Arthritis Res. 1999;1:17–20. doi: 10.1186/ar5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinther-Janssen AC, Pals ST, Scheper R, Breedveld F, Meijer CJ. Dendritic cells and high endothelial venules in the rheumatoid synovial membrane. J.Rheumatol. 1990;17:11–17. [PubMed] [Google Scholar]
- 21.Ebisuno Y, Tanaka T, Kanemitsu N, Kanda H, Yamaguchi K, Kaisho T, Akira S, Miyasaka M. Cutting edge: The B cell chemokine CXC chemokine ligand 13/B lymphocyte chemoattractant is expressed in the high endothelial venules of lymph nodes and Peyer’s patches and affects B cell trafficking across high endothelial venules. J.Immunol. 2003;171:1642–1646. doi: 10.4049/jimmunol.171.4.1642. [DOI] [PubMed] [Google Scholar]
- 22.Edwards JC. Fibroblast biology. Development and differentiation of synovial fibroblasts in arthritis. Arthritis Res. 2000;2:344–347. doi: 10.1186/ar110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan L, Reilly CR, Luo Y, Dorf ME, Lo D. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J.Immunol. 2000;164:3955–3959. doi: 10.4049/jimmunol.164.8.3955. [DOI] [PubMed] [Google Scholar]
- 24.Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 25.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 26.Gao P, Zhou XY, Yashiro-Ohtani Y, Yang YF, Sugimoto N, Ono S, Nakanishi T, Obika S, Imanishi T, Egawa T, Nagasawa T, Fujiwara H, Hamaoka T. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J.Leukoc.Biol. 2003;73:273–280. doi: 10.1189/jlb.0602269. [DOI] [PubMed] [Google Scholar]
- 27.Godessart N, Kunkel SL. Chemokines in autoimmune disease. Curr.Opin.Immunol. 2001;13:670–675. doi: 10.1016/s0952-7915(01)00277-1. [DOI] [PubMed] [Google Scholar]
- 28.Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J.Exp.Med. 1997;186:131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halloran MM, Woods JM, Strieter RM, Szekanecz Z, Volin MV, Hosaka S, Haines GK, III, Kunkel SL, Burdick MD, Walz A, Koch AE. The role of an epithelial neutrophil-activating peptide-78-like protein in rat adjuvant-induced arthritis. J.Immunol. 1999;162:7492–7500. [PubMed] [Google Scholar]
- 30.Haringman JJ, Gerlag DM, Smeets TJ, Baeten D, van den BF, Bresnihan B, Breedveld FC, Dinant HJ, Legay F, Gram H, Loetscher P, Schmouder R, Woodworth T, Tak PP. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:2387–2392. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 31.Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann.Rheum.Dis. 2003;62:715–721. doi: 10.1136/ard.62.8.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46:2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 33.Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am.J.Pathol. 2000;156:1133–1138. doi: 10.1016/S0002-9440(10)64981-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosaka S, Akahoshi T, Wada C, Kondo H. Expression of the chemokine superfamily in rheumatoid arthritis. Clin.Exp.Immunol. 1994;97:451–457. doi: 10.1111/j.1365-2249.1994.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J.Clin.Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson DG. The lymphatics revisited: new perspectives from the hyaluronan receptor LYVE-1. Trends Cardiovasc.Med. 2003;13:1–7. doi: 10.1016/s1050-1738(02)00189-5. [DOI] [PubMed] [Google Scholar]
- 37.Kasama T, Strieter RM, Lukacs NW, Lincoln PM, Burdick MD, Kunkel SL. Interleukin-10 expression and chemokine regulation during the evolution of murine type II collagen-induced arthritis. J.Clin.Invest. 1995;95:2868–2876. doi: 10.1172/JCI117993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katschke KJ, Jr., Rottman JB, Ruth JH, Qin S, Wu L, LaRosa G, Ponath P, Park CC, Pope r.m., Koch AE. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001;44:1022–1032. doi: 10.1002/1529-0131(200105)44:5<1022::AID-ANR181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 39.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim KW, Cho ML, Kim HR, Ju JH, Park MK, Oh HJ, Kim JS, Park SH, Lee SH, Kim HY. Up-regulation of stromal cell-derived factor 1 (CXCL12) production in rheumatoid synovial fibroblasts through interactions with T lymphocytes: role of interleukin-17 and CD40L-CD40 interaction. Arthritis Rheum. 2007;56:1076–1086. doi: 10.1002/art.22439. [DOI] [PubMed] [Google Scholar]
- 41.Klimiuk PA, Sierakowski S, Latosiewicz R, Cylwik JP, Cylwik B, Skowronski J, Chwiecko J. Soluble adhesion molecules (ICAM-1, VCAM-1, and E-selectin) and vascular endothelial growth factor (VEGF) in patients with distinct variants of rheumatoid synovitis. Ann.Rheum.Dis. 2002;61:804–809. doi: 10.1136/ard.61.9.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klimiuk PA, Sierakowski S, Latosiewicz R, Cylwik JP, Cylwik B, Skowronski J, Chwiecko J. Circulating tumour necrosis factor alpha and soluble tumour necrosis factor receptors in patients with different patterns of rheumatoid synovitis. Ann.Rheum.Dis. 2003;62:472–475. doi: 10.1136/ard.62.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann.Rheum.Dis. 2003;62(Suppl 2):ii60–ii67. doi: 10.1136/ard.62.suppl_2.ii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch AE, Kunkel SL, Burrows JC, Evanoff HL, Haines GK, Pope r.m., Strieter RM. Synovial tissue macrophage as a source of the chemotactic cytokine IL-8. J.Immunol. 1991;147:2187–2195. [PubMed] [Google Scholar]
- 45.Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, Burdick MD, Pope r.m., Strieter RM. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J.Clin.Invest. 1992;90:772–779. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, Pope r.m., Walz A, Strieter RM. Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J.Clin.Invest. 1994;94:1012–1018. doi: 10.1172/JCI117414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koch AE, Kunkel SL, Shah MR, Hosaka S, Halloran MM, Haines GK, Burdick MD, Pope r.m., Strieter RM. Growth-related gene product alpha. A chemotactic cytokine for neutrophils in rheumatoid arthritis. J.Immunol. 1995;155:3660–3666. [PubMed] [Google Scholar]
- 48.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 49.Koch AE, Volin MV, Woods JM, Kunkel SL, Connors MA, Harlow LA, Woodruff DC, Burdick MD, Strieter RM. Regulation of angiogenesis by the C-X-C chemokines interleukin-8 and epithelial neutrophil activating peptide 78 in the rheumatoid joint. Arthritis Rheum. 2001;44:31–40. doi: 10.1002/1529-0131(200101)44:1<31::AID-ANR5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 50.Kunkel EJ, Butcher EC. Chemokines and the tissue- specific migration of lymphocytes. Immunity. 2002;16:1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 51.Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, Matloubian M, Cyster JG. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J.Immunol. 2002;169:424–433. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 52.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 53.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc.Natl.Acad.Sci.U.S.A. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsui T, Akahoshi T, Namai R, Hashimoto A, Kurihara Y, Rana M, Nishimura A, Endo H, Kitasato H, Kawai S, Takagishi K, Kondo H. Selective recruitment of CCR6-expressing cells by increased production of MIP-3 alpha in rheumatoid arthritis. Clin.Exp.Immunol. 2001;125:155–161. doi: 10.1046/j.1365-2249.2001.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, De Clercq E, Billiau A, Schols D. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J.Immunol. 2001;167:4686–4692. doi: 10.4049/jimmunol.167.8.4686. [DOI] [PubMed] [Google Scholar]
- 56.McInnes IB, Leung BP, Liew FY. Cell-cell interactions in synovitis. Interactions between T lymphocytes and synovial cells. Arthritis Res. 2000;2:374–378. doi: 10.1186/ar115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- 58.Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, Gay S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am.J.Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 59.Nanki T, Hayashida K, El Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J.Immunol. 2000;165:6590–6598. doi: 10.4049/jimmunol.165.11.6590. [DOI] [PubMed] [Google Scholar]
- 60.Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 2005;52:3004–3014. doi: 10.1002/art.21301. [DOI] [PubMed] [Google Scholar]
- 61.Pablos JL, Santiago B, Galindo M, Torres C, Brehmer MT, Blanco FJ, Garcia-Lazaro FJ. Synoviocyte-derived CXCL12 is displayed on endothelium and induces angiogenesis in rheumatoid arthritis. J.Immunol. 2003;170:2147–2152. doi: 10.4049/jimmunol.170.4.2147. [DOI] [PubMed] [Google Scholar]
- 62.Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, Vath J, Gosselin M, Ma J, Dussault B, Woolf E, Alperin G, Culpepper J, Gutierrez-Ramos JC, Gearing D. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387:611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- 63.Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, Buckley CD. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26:150–156. doi: 10.1016/j.it.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, Buckley CD. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26:150–156. doi: 10.1016/j.it.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin.Immunol. 2001;98:39–45. doi: 10.1006/clim.2000.4957. [DOI] [PubMed] [Google Scholar]
- 66.Paulus HE, Machleder HI, Levine S, Yu DT, MacDonald NS. Lymphocyte involvement in rheumatoid arthritis. Studies during thoracic duct drainage. Arthritis Rheum. 1977;20:1249–1262. doi: 10.1002/art.1780200614. [DOI] [PubMed] [Google Scholar]
- 67.Pilling D, Akbar AN, Girdlestone J, Orteu CH, Borthwick NJ, Amft N, Scheel-Toellner D, Buckley CD, Salmon M. Interferon-beta mediates stromal cell rescue of T cells from apoptosis. Eur.J.Immunol. 1999;29:1041–1050. doi: 10.1002/(SICI)1521-4141(199903)29:03<1041::AID-IMMU1041>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 68.Plater-Zyberk C, Hoogewerf AJ, Proudfoot AE, Power CA, Wells TN. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol.Lett. 1997;57:117–120. doi: 10.1016/s0165-2478(97)00075-8. [DOI] [PubMed] [Google Scholar]
- 69.Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr.Opin.Rheumatol. 2004;16:733–738. doi: 10.1097/01.bor.0000139310.77347.9c. [DOI] [PubMed] [Google Scholar]
- 70.Qu Z, Garcia CH, O’Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining. Arthritis Rheum. 1994;37:212–220. doi: 10.1002/art.1780370210. [DOI] [PubMed] [Google Scholar]
- 71.Ritchlin C. Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000;2:356–360. doi: 10.1186/ar112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu.Rev.Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 73.Ruth JH, Volin MV, Haines GK, III, Woodruff DC, Katschke KJ, Jr., Woods JM, Park CC, Morel JC, Koch AE. Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis Rheum. 2001;44:1568–1581. doi: 10.1002/1529-0131(200107)44:7<1568::AID-ART280>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 74.Salmon M, Gaston JS. The role of T-lymphocytes in rheumatoid arthritis. Br.Med.Bull. 1995;51:332–345. doi: 10.1093/oxfordjournals.bmb.a072964. [DOI] [PubMed] [Google Scholar]
- 75.Salmon M, Scheel-Toellner D, Huissoon AP, Pilling D, Shamsadeen N, Hyde H, D’Angeac AD, Bacon PA, Emery P, Akbar AN. Inhibition of T cell apoptosis in the rheumatoid synovium. J.Clin.Invest. 1997;99:439–446. doi: 10.1172/JCI119178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J.Exp.Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J.Exp.Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmutz C, Hulme A, Burman A, Salmon M, Ashton B, Buckley C, Middleton J. Chemokine receptors in the rheumatoid synovium: upregulation of CXCR5. Arthritis Res.Ther. 2005;7:R217–R229. doi: 10.1186/ar1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat.Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 80.Szekanecz Z, Koch AE. Chemokines and angiogenesis. Curr.Opin.Rheumatol. 2001;13:202–208. doi: 10.1097/00002281-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 81.Tak PP. Chemokine inhibition in inflammatory arthritis. Best.Pract.Res.Clin.Rheumatol. 2006;20:929–939. doi: 10.1016/j.berh.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J.Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 83.Tamamura H, Fujisawa M, Hiramatsu K, Mizumoto M, Nakashima H, Yamamoto N, Otaka A, Fujii N. Identification of a CXCR4 antagonist, a T140 analog, as an anti-rheumatoid arthritis agent. FEBS Lett. 2004;569:99–104. doi: 10.1016/j.febslet.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 84.Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 85.van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, Fero M, Tak PP, Huizinga TW, Pieterman E, Breedveld FC, Alizadeh AA, Verweij CL. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–2145. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- 86.Vierboom MP, Zavodny PJ, Chou CC, Tagat JR, Pugliese-Sivo C, Strizki J, Steensma RW, McCombie SW, Celebi-Paul L, Remarque E, Jonker M, Narula SK, Hart B. Inhibition of the development of collagen-induced arthritis in rhesus monkeys by a small molecular weight antagonist of CCR5. Arthritis Rheum. 2005;52:627–636. doi: 10.1002/art.20850. [DOI] [PubMed] [Google Scholar]
- 87.Villiger PM, Terkeltaub R, Lotz M. Production of monocyte chemoattractant protein-1 by inflamed synovial tissue and cultured synoviocytes. J.Immunol. 1992;149:722–727. [PubMed] [Google Scholar]
- 88.Volin MV, Woods JM, Amin MA, Connors MA, Harlow LA, Koch AE. Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am.J.Pathol. 2001;159:1521–1530. doi: 10.1016/S0002-9440(10)62537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, Obika S, Imanishi T, Tsujimura T, Hamaoka T, Fujiwara H. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur.J.Immunol. 2002;32:2124–2132. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]