

Abstract
Originally identified as an angiogenic factor, vascular endothelial growth factor (VEGF-A) is now known to play multiple roles in the CNS, including the direct regulation of neuronal and astrocytic functions. Here, we ask whether VEGF-A can also have a novel role in white matter by modulating oligodendrocyte precursor cells (OPCs). OPCs were cultured from rat neonatal cortex. Expression of VEGF-receptor2/KDR/Flk-1 was confirmed with Western blot and immunostaining. VEGF-A did not affect proliferation or differentiation in OPC cultures, but VEGF-A promoted OPC migration in a concentration-dependent manner. Consistent with this migration phenotype, VEGF-A-treated OPCs showed reorganization of actin cytoskeleton in leading-edge processes. VEGF-A-induced migration and actin reorganization were inhibited by an anti-Flk-1 receptor-blocking antibody. Mechanistically, VEGF-A induced binding of focal adhesion kinase (FAK) with paxillin. The FAK inhibitor PF573228 reduced VEGF-A-induced OPC migration. VEGF-A signaling also evoked a transient rise in reactive oxygen species (ROS), and OPC migration was increased when antioxidants were removed from the culture media. Our findings demonstrate that VEGF-A can induce OPC migration via an ROS- and FAK-dependent mechanism, and suggest a novel role for VEGF-A in white-matter maintenance and homeostasis.
Introduction
Vascular endothelial growth factor (VEGF-A) is a primary regulator of angiogenesis by stimulating endothelial cell proliferation, migration, and tube formation (Greenberg and Jin, 2005). But it is now well recognized that VEGF-A is not solely an endothelial mediator. Indeed, VEGF-A may represent one of the best examples of common signaling mechanisms in the neurovascular unit (Rosenstein and Krum, 2004; Lambrechts and Carmeliet, 2006), a concept that emphasizes crosstalk between multiple cell types in the brain comprising neuronal, glial, and vascular compartments (Iadecola and Nedergaard, 2007; Zacchigna et al., 2008; Zlokovic, 2008; Moskowitz et al., 2010). VEGF-A not only underlies vascular homeostasis, but is also expressed in astrocytes (Chow et al., 2001), and VEGF-A signaling plays a key role in neuronal migration and CNS development (Carmeliet and Storkebaum, 2002).
The role of VEGF-A is well established in terms of common neuronal, glial, and vascular functions in gray matter. Given that so much overlap exists in cell–cell signaling in the neurovascular unit, is it possible that VEGF-A might also affect white matter in unknown ways? In this proof-of-concept study, we decided to ask whether VEGF-A affects oligodendrocyte precursor cells (OPCs), the primary cell type responsible for sustaining white-matter development and maintenance (Nishiyama et al., 2009).
Materials and Methods
Immunohistochemistry.
Rat brains (male and female Sprague Dawley rat, postnatal day 2) were taken after perfusion with PBS, pH 7.4, and quickly frozen in liquid nitrogen. Coronal sections of 12 μm thickness were cut on cryostat at −20°C and collected on glass slides. Sections were fixed by 4% PFA and rinsed three times in PBS, pH 7.4. After blocking with 3% bovine serum albumin (BSA), sections were then incubated at 4°C overnight in a solution containing the primary antibodies in PBS, 0.1% Tween 20, 0.3% BSA. Staining was performed for the OPC marker NG2 (1:50; Millipore) or VEGF-receptor2/KDR/Flk-1 (1:100; Santa Cruz Biotechnology). The sections were washed and incubated for 1 h with secondary antibodies with fluorescence conjugations. Subsequently, the slides were covered with Vectashield mounting medium with 4′, 6′-diamidino-2-phenylindole (DAPI; H-1200; Vector Laboratories). Immunostaining was analyzed with a fluorescence microscope (Olympus BX51) interfaced with a digital charge-coupled device camera and an image analysis system.
Cell culture.
OPCs were prepared following an institutionally approved protocol, as previously described (Arai and Lo, 2009). Briefly, cerebral cortices from 1- to 2-d-old Sprague Dawley rats were dissected, minced, and digested. Dissociated cells were plated in poly-d-lysine-coated 75 cm2 flasks and maintained in DMEM containing 20% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. After the cells were confluent (∼10 d), the flasks were shaken for 1 h on an orbital shaker (220 rpm) at 37°C. They were then changed to new medium and shaken overnight (∼20 h). The medium was collected and plated on noncoated tissue culture dishes for 1 h at 37°C. The nonadherent cells were collected and replated in Neurobasal medium containing glutamine, 1% penicillin/streptomycin, 10 ng/ml platelet-derived growth factor (PDGF), 10 ng/ml FGF, and 2% B27 supplement onto poly-dl-ornithine-coated plates. Four to five days after plating, the OPCs were used for the experiments. The purity of our OPCs is >98% as assessed with A2B5 staining. To differentiate OPCs from myelin-basic-protein-positive oligodendrocytes, the culture medium was switched to DMEM containing 1% penicillin/streptomycin, 10 ng/ml CNTF, 50 ng/ml T3, and 2% B27 supplement.
Immunocytochemistry.
After cells reached 70–80% confluence, they were rinsed with ice-cold PBS, pH 7.4, followed by 4% paraformaldehyde for 10 min. After being further washed three times in PBS containing 0.1% Triton X-100, they were incubated with 1% BSA in PBS for 1 h. Then cells were incubated with primary antibodies against the OPC markers NG2 (1:50; Millipore) or A2B5 (1:50; Abcam), or VEGF-receptor2/KDR/Flk-1 (1:100; Santa Cruz Biotechnology) at 4°C overnight. After washing with PBS, they were incubated with secondary antibodies conjugated with fluorescein isothiocyanate for 1 h at room temperature. Finally, nuclei were counterstained with DAPI, cells were rinsed, and cover slides were placed. Immunostaining was analyzed with a fluorescence microscope (Olympus BX51) interfaced with a digital charge-coupled device camera and an image analysis system.
Determination of cell proliferation/survival.
Cell proliferation/survival was assessed by water-soluble tetrazolium salt (WST) assay (Dojindo). This assay is based on the detection of dehydrogenase activity of viable cells. Twenty-four hours after treatment with VEGF-A (catalog number: V7259; Sigma) or VEGF-C (catalog number: SRP4633; Sigma), cells were incubated with 10% WST solution for 1 h at 37°C. Then the absorbance of the culture medium was measured with a microplate reader at a test wavelength of 450 nm and a reference wavelength of 630 nm.
OPC migration.
Once OPCs were 70–80% confluent, a wound was made across the cells with a cell scraper (MidSci). Then the cultures were washed once with Neurobasal medium. After VEGF-A, VEGF-C, or vehicle (PBS) was added into Neurobasal medium containing glutamine, 1% penicillin/streptomycin, and 2% B27, cells were allowed to migrate for 24 h. Next, the number of OPCs that moved across the injury line was counted. Because VEGF signaling is known to require low levels of reactive oxygen species (ROS), we further asked whether ROS were involved in our OPC migration system. To do this, we compared VEGF-A-induced OPC migration under conditions with or without ROS generation. We used Neurobasal medium containing glutamine and 1% penicillin/streptomycin and compared this with 2% B27AO−. B27AO− is a medium containing the same nutrients as B27, but missing five antioxidants (vitamin E, vitamin E acetate, superoxide dismutase, catalase, and glutathione).
Immunoprecipitation/Western blotting.
Immunoprecipitation with anti-FAK antibody was conducted using protein A/G plus-agarose (both from Santa Cruz Biotechnology) according to the manufacture's instruction, followed by Western blot analysis. For FLK-1/β-actin Western blotting, cultures were rinsed twice with ice-cold PBS and cells were collected into cell lysis buffer (Cell Signaling Technology) containing 2-mercaptoethanol. After cell lysates were heated at 95°C for 5 min, each sample was loaded onto 4–20% Tris-glycine gels following electrophoresis and transferring to polyvinylidene difluoride membranes (Novex). The membranes were then blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix) for 90 min at room temperature. Membranes were incubated overnight at 4°C with anti-Flk-1 antibody (1:200; Santa Cruz Biotechnology), anti-paxillin antibody (1:200; Santa Cruz Biotechnology), anti-MBP antibody (1:1000; Abcam), or anti-β-actin antibody (1:10,000; Santa Cruz Biotechnology). The next day, the membranes were incubated with peroxidase-conjugated secondary antibodies and visualized by enhanced chemiluminescence (GE Healthcare).
F-actin staining.
Twenty-four hours after cells were stimulated with 100 ng/ml VEGF-A, polymerized actin fibers were visualized by Alexa Fluor 350 phalloidin (Invitrogen) according to the manufacture's instruction. Briefly, cells were washed twice with prewarmed PBS, pH 7.4, followed by 4% paraformaldehyde for 10 min. After being further washed three times in PBS containing 0.1% Triton X-100, they were incubated with 165 nm Alexa Fluor 350 phalloidin in PBS containing 1% BSA for 20 min. After washing with PBS again, they were analyzed with the Olympus BX51 fluorescence microscope system.
ROS assay.
CM-H2DCFDA (Invitrogen) was used to detect ROS production. This compound is rapidly oxidized to the fluorescent 2′,7′-dichlorofluorescein in a reaction with oxidizing species such as H2O2. When OPCs were 70–80% confluent, the culture media were switched to Neurobasal medium containing glutamine, 1% penicillin/streptomycin, and B27 supplement minus antioxidant (Invitrogen) with or without 100 ng/ml VEGF-A. Thirty minutes later, cells were washed twice with prewarmed PBS, pH 7.4, followed by 4% paraformaldehyde for 10 min. After being further washed three times in PBS containing 0.1% Triton X-100, they were incubated with 10 μm CM-H2DCFDA in PBS for 30 min. After washing with PBS again, they were analyzed with the Olympus BX51 fluorescence microscope system.
Statistical analysis.
All of the experiments were blinded, performed in duplicate, and repeated three to five times independently. Quantitative data were analyzed using ANOVA followed by Tukey's honestly significant difference tests. Data are expressed as mean ± SD. Differences at p < 0.05 was considered significant.
Results
The endothelial receptor that is primarily responsible for VEGF-A-induced angiogenesis is VEGF-receptor2/KDR/Flk-1. In vivo, we first confirmed that OPCs in neonatal rat brain express Flk-1 (Fig. 1a). Next, to assess the potential role of VEGF-A signaling in OPCs, we asked whether this prototypical receptor is expressed in our cell system as well. OPCs were prepared from neonatal rat brains. As expected, immunostaining (Fig. 1b) and Western blot analysis (Fig. 1c) showed that our OPCs expressed Flk-1.
Figure 1.

a, Immunostaining showed that NG2-positive OPCs (green) in cerebral cortex of postnatal day 2 rat expressed VEGF-receptor 2/KDR/Flk-1 (red). Nuclei were stained with DAPI (blue). b, Immunostaining demonstrated that our OPC cultures expressed VEGF-receptor 2/KDR/Flk-1 (red) as well as OPC markers NG2 (green) and A2B5 (green). Nuclei were stained with DAPI (blue). c, Western blotting confirmed that OPCs expressed Flk-1. The rat brain endothelial cell line RBE.4 (endothelium) was used as a positive control. d, VEGF-A did not affect OPC proliferation in culture. Twenty-four hours after VEGF-A treatment, OPC proliferation was measured by a WST assay. e, VEGF-A did not differentiate OPCs. Twenty-four hours after VEGF-A treatment, cell lysates were subjected to Western blot with anti-myelin-basic protein (MBP; a marker for OPC maturation) and anti-β actin (internal control). Samples from cells differentiated with 10 ng/ml CNTF and 50 ng/ml T3 (C/T) were used as a positive control. f, VEGF-A induced the OPC migration in culture. Twenty-four hours after VEGF-A treatment, migrated OPCs was counted in a blind manner. Representative pictures demonstrated the increase in OPC migration upon VEGF-A treatment (100 ng/ml for 24 h). The dotted line represents the cut and the arrow indicates the direction of OPC movement. Quantitative data showed that VEGF-A migrated OPCs in a concentration-dependent manner. g, VEGF-C promoted the OPC proliferation in culture. Twenty-four hours after VEGF-A (100 ng/ml) or VEGF-C treatment, OPC proliferation was measured by a WST assay. In contrast, VEGF-C showed less efficacy than VEGF-A in the OPC migration in culture. Twenty-four hours after VEGF-A (100 ng/ml) or VEGF-C (100 ng/ml) treatment, migrated OPCs was counted in a blind manner. *p < 0.05 versus VEGF-A.
Since the receptor was present, we next asked whether VEGF-A signaling was active in OPCs. Cellular maturation, proliferation, and migration assays were used as surrogate measures of OPC function. OPCs were treated with exogenous VEGF-A and 24 h later, the cells were examined. VEGF-A treatment did not alter rates of OPC proliferation (Fig. 1d). VEGF-A did not appear to affect OPC maturation, as evidenced by the lack of myelin-basic-protein bands on Western blots (Fig. 1e). However, VEGF-A clearly amplified rates of OPC migration, as measured with a standard scratch migration assay (Fig. 1f). Because Le Bras et al. (2006) demonstrated that VEGF-C promoted OPC proliferation through VEGF-receptor-3, we tested the efficacy of VEGF-C in our cell culture system. As reported, VEGF-C also enhanced OPC proliferation but did not appear to have detectable effects on OPC migration (Fig. 1g).
But how does VEGF-A mediate OPC movement? Dynamic changes in actin cytoskeleton are needed for cells to move. Therefore, we next examined whether VEGF-A treatment changed the organization of actin cytoskeleton in OPC cultures. Twenty-four hours after VEGF-A treatments, OPCs were fixed and stained with phalloidin to visualize polymerized F-actin fibers. Migrated OPCs in the VEGF-A-treated group showed reorganization of actin cytoskeleton, especially in leading processes (Fig. 2a,b). A neutralizing antibody against Flk-1 inhibited both VEGF-A-induced actin reorganization and migration (Fig. 2a,c), confirming that this receptor mediated VEGF-A signaling for OPC migration.
Figure 2.
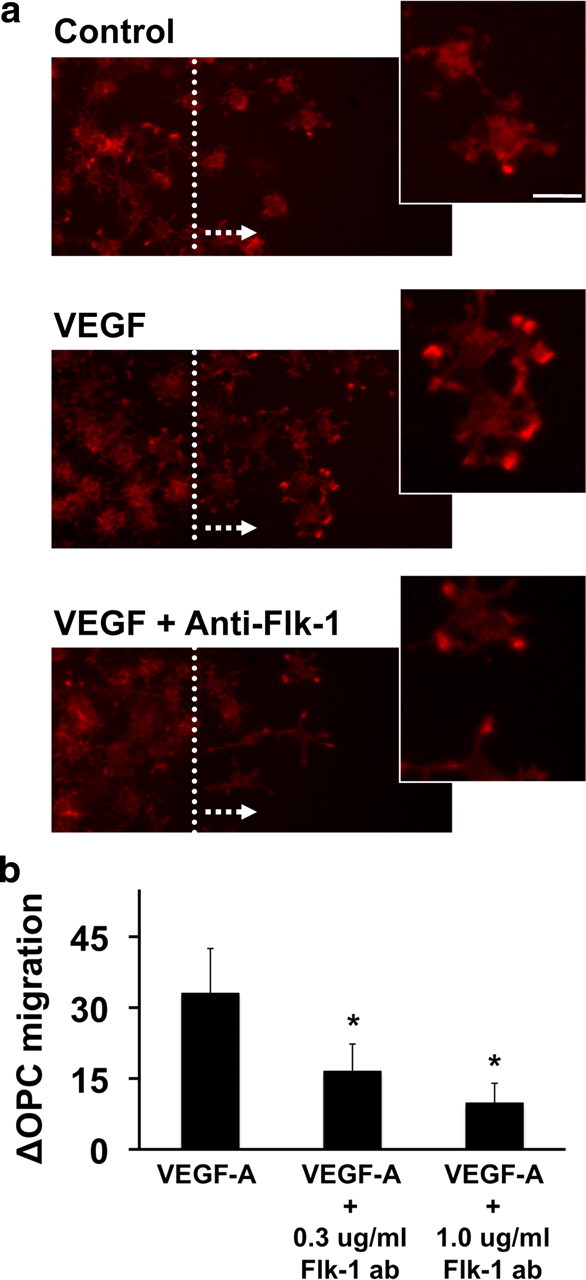
a, VEGF-A reorganized actin cytoskeleton in OPC cultures. Twenty-four hours after VEGF-A (100 ng/ml) treatment, cells were fixed and stained with phalloidin to visualize F-actin. Compared with control OPC group, VEGF-A-treated cells showed increased migration and polymerized F-actin fibers. Cotreatment with neutralizing anti-Flk-1 antibody reduced VEGF-A-induced OPC migration and actin reorganization. The dotted line represents the cut and the arrow indicates the direction of OPC movement. Phalloidin staining (high magnification panels on the right) showed that VEGF-A-induced actin reorganization occurred in OPC processes. Scale bar, 10 μm. b, Cotreatment with anti-Flk-1 antibody reduced VEGF-A-induced OPC migration. Twenty-four hours after treatments, migrated OPCs were counted in a blind manner. *p < 0.05 versus VEGF-A-treated group.
Focal adhesion kinase (FAK) and paxillin are both adhesion-associated proteins that transmit external signals into changes in cell motility. Paxillin works as a platform for protein tyrosine kinases such as FAK, and also binds to proteins that reorganize actin cytoskeleton during VEGF-induced cell migration. Therefore, we asked whether a similar signaling pathway was involved in our model system. OPCs were treated with VEGF-A for 24 h, then cell lysates were immunoprecipitated with anti-FAK antibody and subjected to Western blot analysis with anti-paxillin or anti-FAK antibodies. VEGF-A-treated OPCs showed augmented FAK-paxillin binding, and the binding was inhibited by cotreatment with anti-Flk-1 neutralizing antibody (Fig. 3a). Furthermore, the FAK inhibitor PF573228 reduced the number of VEGF-induced migrated OPCs (Fig. 3b) without affecting OPC cell numbers (Fig. 3c).
Figure 3.

a, VEGF-A increased the binding of FAK with paxillin. Twenty-four hours after VEGF-A (100 ng/ml) treatment, cell lysates were immunoprecipitated (IP) with anti-FAK antibody, followed by Western blotting with anti-paxillin or anti-FAK antibodies. b, c, A FAK inhibitor PF573228 decreased the number of migrated OPCs by VEGF-A treatment (100 ng/ml, 24 h). A WST assay confirmed that the compound did not induce cell damage in OPC cultures. *p < 0.05 versus VEGF-A-treated group.
Finally, we asked whether ROS were involved in VEGF-A-mediated OPC migration. Excess ROS generated in neurodegeneration are cytotoxic. But low levels of ROS may be essential for cellular homeostasis. Recently, it has been determined that VEGF signaling requires ROS (Ushio-Fukai, 2006). Therefore, we asked whether VEGF-induced OPC migration in our model system may also require ROS. A standard dichlorofluorescein assay demonstrated that ROS were rapidly generated after VEGF-A was added to OPCs (Fig. 4a). Removing anti-oxidants (vitamin E, vitamin E acetate, superoxide dismutase, catalase, and glutathione) from the experimental culture media significantly increased VEGF-A-induced OPC migration (Fig. 4b).
Figure 4.
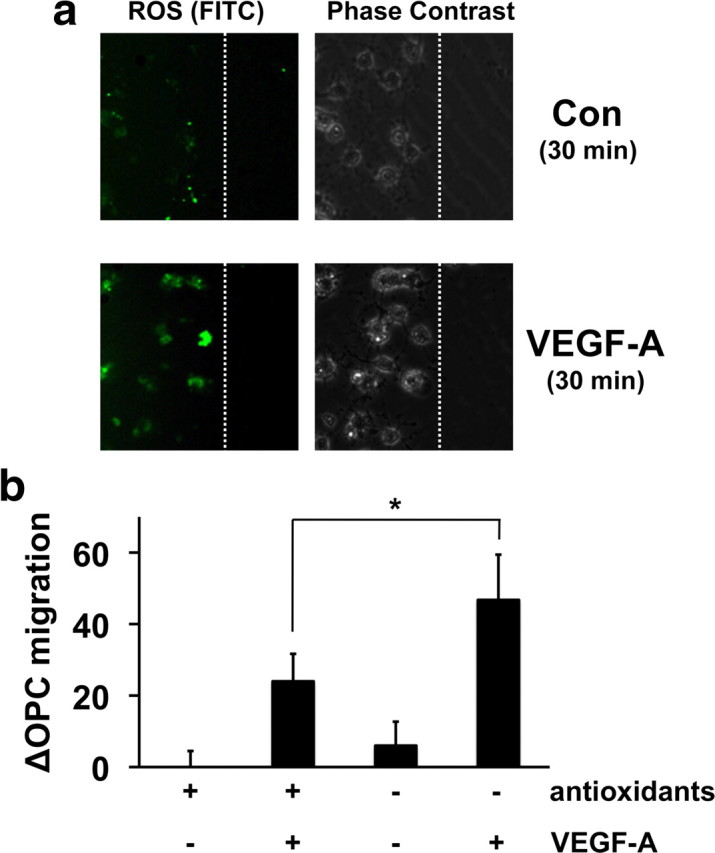
a, VEGF-A induced ROS production in OPC cultures. Thirty minutes after VEGF-A treatment (100 ng/ml), cells were fixed and stained with CM-H2DCFDA to visualize ROS generation. Representative images showed that positive DCF signals were observed in VEGF-A-treated OPCs. The dotted line represents the cut. Con, Control. b, VEGF-A-induced OPC migration was accelerated under antioxidant-free conditions. Antioxidants (vitamin E, vitamin E acetate, superoxide dismutase, catalase, and glutathione) were deprived from culture medium, and VEGF-A-stimulated (100 ng/ml, 24 h) migration was counted in a blinded manner. More cells moved in response to VEGF-A stimulation under antioxidant-free conditions. *p < 0.05.
Discussion
This study showed that the prototypical VEGF receptor Flk-1 is expressed in OPCs. VEGF-A did not affect proliferation or cause OPCs to differentiate into mature oligodendrocytes, but VEGF-A clearly stimulated OPC migration via FAK and ROS-dependent mechanisms. Together, our findings support a novel role for VEGF-A in OPC homeostasis and maintenance.
In recent years, the concept of the neurovascular unit has allowed a rethinking of how CNS function and dysfunction require crosstalk between multiple cell types in the brain (Iadecola and Nedergaard, 2007; Zacchigna et al., 2008; Zlokovic, 2008; Moskowitz et al., 2010). In part, many of these phenomena are mediated by the exchange of common signals and factors between different cell types. For example, cerebral endothelial cells produce BDNF and other growth factors that are neuroprotective and support neuronal function (Dugas et al., 2008; Guo et al., 2008). In the so-called neurovascular niche, angiogenic factors such as VEGF enhance both neurogenesis and angiogenesis (Storkebaum and Carmeliet, 2004; Ohab and Carmichael, 2008; Madri, 2009). Astrocytes release TSP-1, which promotes synaptic development in neurons (Eroglu et al., 2009). And of course, it is well known that astrocytes and pericytes release multiple factors that help maintain the blood–brain barrier phenotypes in brain endothelium (Iadecola and Nedergaard, 2007; Bell et al., 2010). Our present findings provide yet another example of how VEGF-A can play multiple roles in different cell types, including OPCs.
For the most part, the concept of the neurovascular unit is used to guide investigations in gray matter. Recently, however, we proposed that an oligo-vascular niche might also exist, wherein trophic support from brain endothelium help sustain OPCs and defend them against cellular stress (Arai and Lo, 2009). Furthermore, astrocyte-derived soluble factors were also reported to nourish oligodendrocyte lineage cells (Corley et al., 2001; Arai and Lo, 2010). Hence, cell–cell trophic interactions are likely to be important in white matter as well.
White matter primarily consists of axonal bundles ensheathed with myelin by mature oligodendrocytes, and plays an important role in passing signals between different areas of gray matter. During development, OPCs migrate from the ventricular zone to their destination and then differentiate to form myelin sheaths (Lee et al., 2000; Bhat, 2003). OPCs are also widely distributed in adult brain (Nishiyama et al., 2009), and after brain injury, they are guided to the site for contributing to myelin repair (Nishiyama et al., 2009). The maturation of OPCs into oligodendrocytes needs multiple steps, and several growth factors such as basic fibroblast growth factor (bFGF) and hepatocyte growth factor (HGF) are known to play key roles (Milner et al., 1997; Yan and Rivkees, 2002; Magy et al., 2003). In terms of OPC migration, PDGF is the most well examined growth factor involved. The mechanisms of PDGF-induced OPC migration are now thought to require phosphorylation of cyclin-dependent kinase 5 (Miyamoto et al., 2008) and tyrosine kinase modification of voltage-operated Ca2+ channels (Paez et al., 2010). Our current study identified VEGF as a new factor for this phenomenon. But one significant difference is that PDGF, bFGF, and HGF all promote OPC proliferation as well. In contrast, our data show that VEGF had no effects of OPC proliferation. Hence, VEGF might contribute to white matter homeostasis in different ways from other growth factors.
Although this proof-of-concept study supports a novel role for VEGF-A in OPCs, there are several caveats worth keeping in mind. First, we focused on only VEGF-A and Flk-1, but other VEGF families and VEGF receptors may also be involved. Le Bras et al. (2006) demonstrated that VEGF-C promoted OPC proliferation through VEGF-receptor-3. In our cell culture system, VEGF-C also enhanced OPC proliferation but did not appear to have detectable effects on OPC migration. Hence, OPC proliferation/migration might be differentially regulated by different VEGFs/VEGF receptors. Second, our VEGF effects seem to require ROS. Although excess ROS generated in neurodegeneration are obviously cytotoxic, it is now recognized that baseline levels of reactive species might also participate in normal cell function. For example, proangiogenic VEGF actions in endothelial cells require the functional production of radicals from the NADPH oxidase pathway (Ushio-Fukai, 2006). More work is warranted to investigate how VEGF-evoked ROS signals may contribute to the balance between deleterious (OPC death) versus potentially beneficial (OPC migration) effects in white-matter maintenance. Finally, our experiments are limited to functional assays. We do not have any data on whether the ability of VEGF to stimulate OPC migration may actually contribute to white-matter recovery after brain injury. When axons are damaged, OPCs are guided to the injury site to generate new oligodendrocytes. Indeed, OPCs from human embryonic stem cells are now proposed as a cell therapy for spinal-cord injury (Zhang et al., 2006). Hence, the ability of VEGF treatments to improve neurological deficits in rodent stroke models may involve not only angiogenesis (Zhang et al., 2000; Sun et al., 2003), but also oligodendrogenesis and plasticity. Future studies to dissect the roles of VEGF and OPCs in white-matter remodeling are warranted.
In conclusion, our findings demonstrate that VEGF-A promotes OPC migration via FAK- and ROS-dependent mechanisms. VEGF-A is not only an endothelial, neuronal, and astrocytic mediator; it also likely plays a role in OPC homeostasis. These findings provide yet another example of the commonality and overlap of signals in the neurovascular unit. How these pathways participate in white-matter maintenance and repair should be an interesting direction for future studies. White-matter dysfunction is a critically important part of stroke, brain injury, and neurodegeneration. Therefore, dissecting the mechanisms of how VEGF–OPC signaling works in white matter under normal and pathologic conditions may help us explore new therapeutic avenues for CNS disease.
Footnotes
This work was supported by the Deane Foundation, American Heart Association, and NIH.
References
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res. 2010;88:758–763. doi: 10.1002/jnr.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA. Molecular organization of axo-glial junctions. Curr Opin Neurobiol. 2003;13:552–559. doi: 10.1016/j.conb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Storkebaum E. Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin Cell Dev Biol. 2002;13:39–53. doi: 10.1006/scdb.2001.0290. [DOI] [PubMed] [Google Scholar]
- Chow J, Ogunshola O, Fan SY, Li Y, Ment LR, Madri JA. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Brain Res Dev Brain Res. 2001;130:123–132. doi: 10.1016/s0165-3806(01)00220-6. [DOI] [PubMed] [Google Scholar]
- Corley SM, Ladiwala U, Besson A, Yong VW. Astrocytes attenuate oligodendrocyte death in vitro through an alpha(6) integrin-laminin-dependent mechanism. Glia. 2001;36:281–294. doi: 10.1002/glia.1116. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Mandemakers W, Rogers M, Ibrahim A, Daneman R, Barres BA. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: therapeutic implications for motor neuron disease. Biochim Biophys Acta. 2006;1762:1109–1121. doi: 10.1016/j.bbadis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, Haiko P, Karkkainen MJ, Yuan L, Muriel MP, Chatzopoulou E, Bréant C, Zalc B, Carmeliet P, Alitalo K, Eichmann A, Thomas JL. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat Neurosci. 2006;9:340–348. doi: 10.1038/nn1646. [DOI] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS. Gliogenesis in the central nervous system. Glia. 2000;30:105–121. doi: 10.1002/(sici)1098-1136(200004)30:2<105::aid-glia1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Madri JA. Modeling the neurovascular niche: implications for recovery from CNS injury. J Physiol Pharmacol. 2009;60(Suppl 4):95–104. [PubMed] [Google Scholar]
- Magy L, Mertens C, Avellana-Adalid V, Keita M, Lachapelle F, Nait-Oumesmar B, Fontaine B, Baron-Van Evercooren A. Inducible expression of FGF2 by a rat oligodendrocyte precursor cell line promotes CNS myelination in vitro. Exp Neurol. 2003;184:912–922. doi: 10.1016/j.expneurol.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Milner R, Anderson HJ, Rippon RF, McKay JS, Franklin RJ, Marchionni MA, Reynolds R, ffrench-Constant C. Contrasting effects of mitogenic growth factors on oligodendrocyte precursor cell migration. Glia. 1997;19:85–90. doi: 10.1002/(sici)1098-1136(199701)19:1<85::aid-glia9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamauchi J, Tanoue A. Cdk5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase Fyn. J Neurosci. 2008;28:8326–8337. doi: 10.1523/JNEUROSCI.1482-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Ohab JJ, Carmichael ST. Poststroke neurogenesis: emerging principles of migration and localization of immature neurons. Neuroscientist. 2008;14:369–380. doi: 10.1177/1073858407309545. [DOI] [PubMed] [Google Scholar]
- Paez PM, Fulton DJ, Spreur V, Handley V, Campagnoni AT. Multiple kinase pathways regulate voltage-dependent Ca2+ influx and migration in oligodendrocyte precursor cells. J Neurosci. 2010;30:6422–6433. doi: 10.1523/JNEUROSCI.5086-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue: beyond blood vessels. Exp Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Carmeliet P. VEGF: a critical player in neurodegeneration. J Clin Invest. 2004;113:14–18. doi: 10.1172/JCI200420682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Yan H, Rivkees SA. Hepatocyte growth factor stimulates the proliferation and migration of oligodendrocyte precursor cells. J Neurosci Res. 2002;69:597–606. doi: 10.1002/jnr.10323. [DOI] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Denham J, Thies RS. Oligodendrocyte progenitor cells derived from human embryonic stem cells express neurotrophic factors. Stem Cells Dev. 2006;15:943–952. doi: 10.1089/scd.2006.15.943. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood–brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]