

Identifying disease-causing genetic variants in individual human genomes is a major challenge, even in protein-coding exons (the `exome'). Analysis of nucleotide-level sequence conservation may help address this challenge, on the assumption that purifying selection `constrains' evolutionary divergence at phenotypically important nucleotides. In contrast to functional classifiers (for example, non-synonymous mutations), constraint scores are quantitative and applicable to any genomic position1. However, it remains unclear if constraint scores can facilitate causal variant discovery, as statistical power is estimated to be marginal at the single-nucleotide level given current genome alignments1,2.
We therefore applied and assessed a nucleotide-level evolutionary metric to prioritize causal variants in genomes of 16 individuals. We analyzed exomes from four individuals with Freeman-Sheldon syndrome (FSS; Online Mendelian Inheritance in Man (OMIM) database identifier 193700), a dominant disease caused by mutations in MYH33; four individuals with Miller syndrome (OMIM identifier 263750), a recessive disease caused by mutations in DHODH4; and eight HapMap samples3. We generated constraint scores by Genomic Evolutionary Rate Profiling (GERP)1 on the mammalian subset of the 44-way MULTIZ/TBA alignments (http://genome.ucsc.edu; for details see2). For each aligned site, GERP defines a `rejected substitution' (RS) score by estimating the actual number of substitutions at that site and subtracting it from the number expected assuming neutrality (~5.82 substitutions per site). Selectively constrained sites tolerate fewer substitutions than neutral sites and have positive RS scores1,2.
We first defined the consensus nucleotide from the chimpanzee, gorilla, orangutan, and macaque genomes as ancestral and determined the derived allele frequency (DAF) for each variant in the eight HapMap exomes. We found a significant inverse correlation between DAF and RS score (Fig. 1; p < 0.0001; R2 = 1.4%, slope estimated as −1% DAF per RS). No correlation existed between DAF and the RS score for the nucleotide adjacent to the variant (Supplementary Fig. 1). While the DAF-RS correlation resulted partly from enrichment for singletons at sites with high RS scores, it was significant even within common variants (p < 0.0001; Supplementary Fig. 2). We also found that segregating sites, regardless of DAF, were enriched at sites with low RS scores and progressively depleted as the RS scores increased (Supplementary Fig. 3). Consistent with previous data2, these results suggest that RS scores enrich site-specifically for deleterious variants and non-variant positions at which new mutations would be deleterious.
Figure 1.
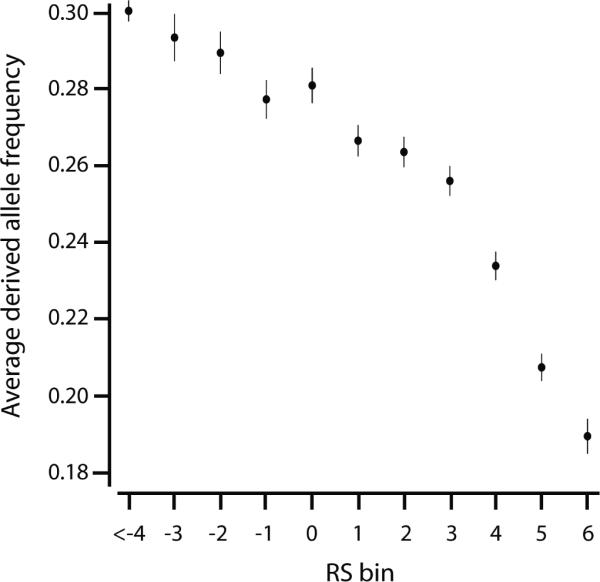
RS scores inversely correlate with DAF of single-nucleotide variants (n=48,750) in 8 HapMap exomes. The average DAF (Y-axis) is plotted for all variants at a site within a given RS bin (X-axis). Error bars show 1 standard error unit.
Next, we tested whether constraint scores could enrich for FSS or Miller syndrome causal variants. We identified candidate disease genes as those in which the affected individuals had variants not seen in the HapMap exomes that affected a nucleotide with a high constraint score. For a comparison, we used functional definitions of deleteriousness, namely non-synonymous, splice-site, or insertion-deletion (indel)3,4. We first used a threshold of RS > 0 (fewer substitutions than expected), and found that this narrowed candidate gene lists nearly as effectively as functional annotations. For example, there were 21 genes in which all FSS samples had a rare, functionally annotated variant3,4 versus 24 genes in which all FSS samples had a rare variant with RS > 0 (Fig. 2a). Increasing the RS threshold, which cannot readily be done with functional annotations, reduced candidate gene lists. At a threshold of RS > 4, for example, MYH3 was one of only five FSS candidate genes, while DHODH was the only Miller syndrome candidate (Fig. 2b).
Figure 2.

Constraint scores enrich for disease-causing genes. (a). Number of genes (Y-axis, log-scale) in which at least the given number of FSS individuals (X-axis) has a rare variant that is functionally defined (white), or with increasing RS scores (light gray to black). The total number of candidate genes defined at RS > 0 and the rank of MYH3 among those genes are indicated below the graph. (b). Similar to panel A, expect for Miller syndrome, caused by mutations in DHODH.
We note that protein-based approaches could similarly be used to reduce candidate gene lists. For example, there were only seven genes in which all FSS individuals harbored a rare variant annotated by PolyPhen5 as `possibly' or `probably' damaging. However, PolyPhen (and related approaches) is restricted to non-synonymous variants, does not facilitate ranking of candidates (see below), and excluded DHODH as a Miller syndrome candidate4.
Finally, we exploited the quantitative nature of constraint scores and ranked genes by the average score of all rare and deleterious (RS > 0) variants in the affected individuals. MYH3 and DHODH ranked highly for their associated diseases, even under models allowing for the possibility of multiple causal genes. For example, requiring only that at least two individuals shared the same causal gene, MYH3 ranked 9th among 666 genes. If we assumed FSS and Miller syndrome to be monogenic, MYH3 and DHODH ranked as the top candidates, respectively (Fig. 2).
RS scores for known or user-defined variants can be obtained from the Genome Variation Server (http://gvs.gs.washington.edu/GVS/) or SeattleSeq annotation pipeline (http://gvs.gs.washington.edu/SeattleSeqAnnotation/). Constraint scores facilitate threshold flexibility and candidate ranking and do not require functional annotations. Even in exomes, this allows for the possibility that synonymous variants contribute to disease6. More importantly, this independence offers exciting potential for the discovery of causal variation in arbitrary genomic segments (for example, linkage peaks) and ultimately resequenced genomes.
Supplementary Material
Acknowledgments
GMC is grateful for support from a Merck, Jane Coffin Childs Memorial Fund postdoctoral fellowship. DLG is supported by a Lucille P. Markey Biomedical Research Stanford Graduate Fellowship. This work was also supported by grants from the National Institutes of Health: U01 HL66682 and 5R01HL094976-02 (DAN), 5R01HD048895 (MJB), and 1R21HG004749-01 (JS).
References
- 1.Cooper GM, Stone EA, Asimenos G, et al. Genome Res. 2005;15(7):901. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goode DL, Cooper GM, Schmutz J, et al. Genome Res. 2010;20(3):301. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng SB, Turner EH, Robertson PD, et al. Nature. 2009;461(7261):272. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng SB, Buckingham KJ, Lee C, et al. Nat Genet. 2010;42(1):30. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sunyaev S, Ramensky V, Koch I, et al. Hum Mol Genet. 2001;10(6):591. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 6.Cartegni L, Chew SL, Krainer AR. Nat Rev Genet. 2002;3(4):285. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.