

Abstract
Kv2.1 channels exhibit a U-shaped voltage-dependence of inactivation that is thought to represent preferential inactivation from preopen closed states. However, the molecular mechanisms underlying so-called U-type inactivation are unknown. We have performed a cysteine scan of the S3-S4 and S5-P-loop linkers and found sites that are important for U-type inactivation. In the S5-P-loop linker, U-type inactivation was preserved in all mutant channels except E352C. This mutation, but not E352Q, abolished closed-state inactivation while preserving open-state inactivation, resulting in a loss of the U-shaped voltage profile. The reducing agent DTT, as well as the C232V mutation in S2, restored U-type inactivation to the E352C mutant, which suggests that residues 352C and C232 may interact to prevent U-type inactivation. The R289C mutation, in the S3-S4 linker, also reduced U-type inactivation. In this case, DTT had little effect but application of MTSET restored wild-type-like U-type inactivation behavior, suggestive of the importance of charge at this site. Kinetic modeling suggests that the E352C and R289C inactivation phenotypes largely resulted from reductions in the rate constants for transitions from closed to inactivated states. The data indicate that specific residues within the S3-S4 and S5-P-loop linkers may play important roles in Kv2.1 U-type inactivation.
Introduction
With prolonged depolarization, many voltage-gated potassium (Kv) channels inactivate and enter a nonconducting state that is distinct from the deactivated or closed state (1,2). Temporally and mechanistically, inactivation can be divided into two different processes: fast inactivation and slow inactivation. In Kv1 channels such as Shaker, slow inactivation typically occurs after channel opening. It is inhibited by extracellular tetraethylammonium (TEAo) (3–5) and elevated [K+]o (6–9), and is often called “P/C-type inactivation” (10,11). Based on these properties and studies on mutant channels (e.g., (12–14)), P/C-type inactivation is thought to involve a concerted conformational change in the selectivity filter and/or outer pore (1).
Slow inactivation of Kv channels is often described as “P/C-type” by default. However, inactivation is highly variable, even among members of the same Kv family, and various Kv channels exhibit inactivation properties that are inconsistent with those of classical P/C-type inactivation. For example, slow inactivation of Kv2.1 and Kv3.1 channels is enhanced by high [TEA]o or [K+]o (15–17). Also, although P/C-type inactivation tends to occur with maximal probability from the open state during strong depolarizations (e.g., 60 mV), the voltage-dependencies of Kv2.1 and Kv3.1 inactivation are U-shaped (15,16). That is, inactivation is more rapid and complete from preopen closed states occupied during moderate depolarizations (e.g., 0 mV). Thus, this U-type inactivation is enhanced by short, repetitive depolarizations associated with increased occupancy of partially activated closed states compared to a single prolonged depolarization (15,16). U-type inactivation has been observed in fast-inactivation removed Shaker (ShIR) channels (16,18) and in a naturally occurring Kv1.5 N-terminal deletion mutant (19–21).
U-type inactivation may be particularly important during repetitive stimulation, for example in nervous or cardiac tissue, during which it may dictate channel availability more so than P/C-type inactivation from the open state. Despite this, there is very little information on the structural mechanism(s) underlying U-type inactivation and it remains unclear whether U-type and P/C-type inactivation are variants of the same process.
This work examines the possibility that residues in the S3-S4 and S5-P-loop linkers are involved in U-type inactivation of the Kv2.1 channel. Previous studies with ShIR channels have suggested that P/C-type inactivation is dependent on the interaction of residues at the external end of S4 with those in the S5-P-loop linker (22–25). We hypothesized that similar interactions in Kv2.1 channels couple inactivation to voltage-sensor movement so that it exhibits a U-shaped voltage-dependence. A cysteine scan showed that the E352C mutation in the S5-P-loop linker removes U-type inactivation and that this may be due to a disulfide interaction with a native cysteine residue in S2. The R289C mutation in the S3-S4 linker also decreased inactivation from closed states, an effect that appears to be related to the loss of charge at this site. Both results can be explained using a previously described kinetic model for Kv2.1 U-type inactivation (15) by reducing the rate of inactivation from preopen closed states.
Materials and Methods
Molecular biology
Rat Kv2.1 (a gift from Dr. E. Bennett, University of South Florida, Tampa, FL) was expressed in Xenopus oocytes using a modified pBluescript SKII expression vector, pEXO. Mutant constructs were generated using QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA), using primers synthesized by Sigma Genosys (Oakville, Ontario). After sequencing (Eurofins MWG Operon, Huntsville, AL), constructs were linearized with SacII restriction endonuclease (New England Biolabs, Pickering, Ontario, Canada). cRNA was then synthesized using the mMessage mMachine T7 Ultra cRNA transcription kit (Ambion, Austin, TX). Xenopus oocytes were prepared as described previously (26).
Electrophysiology and solutions
Whole-cell current recordings from oocytes expressing wild-type (wt) and mutant Kv2.1 channels were made at 20–22°C using standard two-electrode voltage-clamp. Recordings were performed using an Axoclamp 900A amplifier and pClamp 10 software, via a Digidata 1440 interface (Axon Instruments, Foster City, CA). Microelectrodes were made from thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL) and had resistances of 0.2–2.0 MΩ (filled with 3 M KCl). Current signals were low-pass-filtered at 4 kHz (−3 dB, eight-pole Bessel filter) and digitized (16-bit) at a 10 kHz sampling frequency. A holding potential of −80 mV was standard. The standard bath solution for recording, ND96, contained (in mM): 96 NaCl, 3 KCl, 1 MgCl2, 0.5 CaCl2, 5 HEPES, titrated to pH 7.4 with NaOH. Solution with 30 mM K+ was prepared by isoosmotic replacement of NaCl. In some experiments, cells were pretreated for 30 min in ND96 supplemented with: dithiothreitol (DTT, 1 mM), 2-sulfonatoethyl methanethiosulfonate sodium salt (MTSES; 1 mM), or 2-trimethylammoniumethyl methanethiosulfonate bromide (MTSET; 1 mM). All reagents were obtained from Sigma-Aldrich (St. Louis, MO), except for MTSES and MTSET, which were from Toronto Research Chemicals (North York, Ontario, Canada).
Data analysis and modeling
Conductance-voltage (G-V) relationships were derived from peak tail current amplitudes (see the Supporting Material). The voltage-dependence of activation and inactivation were described by the Boltzmann functions: y = 1/(1 + exp[(V1/2–V)/k]) and y = (1–C)/(1 + exp[(V–V1/2)/k]) + C, respectively. In each function, V1/2 is the voltage at which 50% activation or inactivation has occurred, V is the test potential, and k is the slope factor. The value k is equal to RT/zF, where z is the apparent charge and R, T, and F have their usual meaning. The value C denotes the fraction of noninactivating channels at the potential where inactivation was maximal. The Gibbs free energy of activation at 0 mV(ΔG0) was given by: ΔG0 = 0.2389 zFV1/2 (27). The change in ΔG0 (ΔΔG0) caused by mutations could then be derived by: ΔΔG0 = ΔG0mut−ΔG0wt.
Curve fitting and statistical comparisons were performed in SigmaPlot 11.0 (Systat Software, San Jose, CA). Unless otherwise noted, data are shown as the mean ± SE; n represents the number of cells tested.
Macroscopic Kv2.1 currents were simulated in MATLAB v.7.8 (The MathWorks, Natick, MA) by calculating state occupancies as a function of time and voltage from the spectral expansion of the Q-matrix (28) generated from an allosteric model of Kv2.1 inactivation (15). Comparisons between the simulated and experimental data were performed by eye in Matlab. Rate constants for voltage-dependent transitions between horizontal states were described by equations of the form: kx = k0 · exp(zxFV/RT), where kx is the rate for transition x, k0 is the rate at 0 mV, zx is the apparent valence of the transition, and F, V, R, and T have their usual meaning.
Results
U-type inactivation of wt Kv2.1 channels
To examine the voltage-dependence of Kv2.1 inactivation, a three-pulse (3P) protocol (15) was used (Fig. 1 A). Current recorded during the first pulse (P1) to +60mV serves as a control for sweep-to-sweep variations in current, whereas the 15-s pulse (P2) induces inactivation at potentials between −100 and +60 mV. Current recorded during the final pulse (P3) to +60 mV, normalized to that from P1 (i.e., I3/I1), provides a measure of the inactivation occurring during P2. A plot of mean I3/I1 ratios (n = 7) against the P2 voltage (Fig. 1 B) exhibits a U-shaped profile, where inactivation is maximal at moderate potentials and decreases with further depolarization—suggestive of preferential inactivation from preopen closed states and consistent with previous reports (15,29). The descending portion of the inactivation-voltage relationship could be fit to a Boltzmann function (solid line, Fig. 1 B), with V1/2 and k values summarized in Table 1. The upturn has also been quantified as the percentage increase in the I3/I1 ratio at +60 mV, relative to the minimum I3/I1 value (Table 1).
Figure 1.
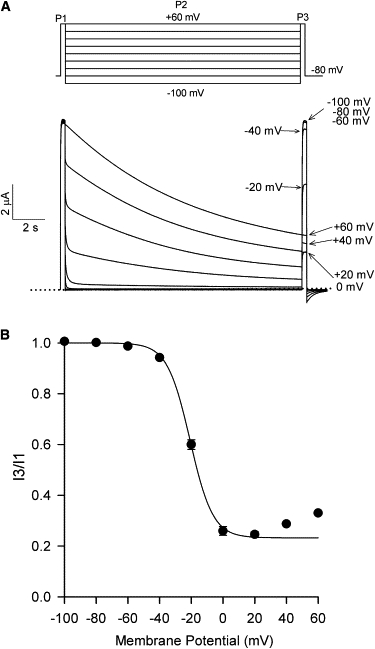
U-type inactivation of wt Kv2.1. (A, Top) Schematic of the three-pulse (3P) protocol used to study Kv2.1 inactivation. The voltage for the 15 s P2 was increased in 20-mV increments, whereas P1 and P3 were at +60 mV for 300 ms. (Bottom) Macroscopic currents recorded using the 3P protocol from an oocyte expressing wt Kv2.1 channels. Shown are nine superimposed traces obtained in ND96 solution. The sweep interval was 60 s. (B) The inactivation-voltage relationship for wt Kv2.1 was determined by taking the ratio of current (I3/I1) measured during P3 and P1 and plotting it against the P2 voltage. (Data points) Mean ± se; where not visible, error bars are hidden within symbols. n = 7. (Solid line) Fit of the descending portion of the inactivation-voltage relationship (i.e., between −100 and +20 mV) to a Boltzmann function. See Table 1 for a summary and the fit parameters.
Table 1.
Mutations in the S3-S4 and S5-P-loop linkers alter Kv2.1 inactivation
| Construct | V1/2 (mV) | k (mV) | Minimum I3/I1 | % Increase in I3/I1 at +60 mV∗ | n |
|---|---|---|---|---|---|
| wt | −21.0 ± 0.5 | 6.7 ± 0.3 | 0.24 ± 0.01 | 42.2 ± 6.3 | 7 |
| S3-S4 linker mutants | |||||
| N287C | −43.0 ± 4.3 | 16.6 ± 1.2† | 0.47 ± 0.02 | 36.6 ± 6.3 | 5 |
| V288C | −14.9 ± 2.7 | 11.4 ± 0.9† | 0.44 ± 0.02 | 26.6 ± 4.1 | 7 |
| R289C | −45.2 ± 2.6 | 17.8 ± 0.2† | 0.53 ± 0.02 | 27.0 ± 6.0 | 4 |
| R290C | −36.2 ± 2.3 | 14.1 ± 0.7† | 0.41 ± 0.03 | 69.8 ± 8.4 | 5 |
| V291C | −12.2 ± 1.8 | 6.1 ± 0.5 | 0.19 ± 0.03 | 52.6 ± 8.7 | 5 |
| V292C | −8.8 ± 3.7 | 15.0 ± 0.7† | 0.38 ± 0.04 | 32.2 ± 8.7 | 4 |
| S5-P-loop mutants | |||||
| K350C | −19.9 ± 2.3 | 11.2 ± 0.2† | 0.21 ± 0.02 | 33.6 ± 10.5 | 5 |
| D351C | −18.3 ± 0.6 | 9.4 ± 0.2 | 0.22 ± 0.02 | 28.5 ± 3.9 | 5 |
| E352C | −13.9 ± 0.9 | 9.9 ± 0.4 | 0.37 ± 0.03 | 6.6 ± 2.0† | 9 |
| D353C | −16.0 ± 2.1 | 8.1 ± 0.2 | 0.24 ± 0.03 | 43.2 ± 10.3 | 7 |
| D354C | −18.6 ± 0.9 | 6.7 ± 0.2 | 0.17 ± 0.01 | 66.6 ± 6.2 | 13 |
| T355C | −23.8 ± 1.6 | 7.9 ± 0.3 | 0.18 ± 0.03 | 58.8 ± 13.8 | 6 |
| K356C | −20.0 ± 1.3 | 6.4 ± 0.4 | 0.13 ± 0.01 | 75.3 ± 5.6 | 8 |
Relative to the minimum I3/I1.
Significant difference (P < 0.05) from wt values, based on the results of a Kruskal-Wallis ANOVA on ranks test.
Mutations in the S5-P-loop and S3-S4 linkers alter Kv2.1 inactivation
A cysteine scan of amino-acid residues in the C-terminal region of the S3-S4 linker (N287C–V292C) and in the S5-P-loop (K350C–K356C) (Fig. 2 A) was performed to investigate the molecular determinants of U-type inactivation. This provided the opportunity to assess both the effects of changing the residue at each site as well as chemical modification of the introduced cysteine.
Figure 2.
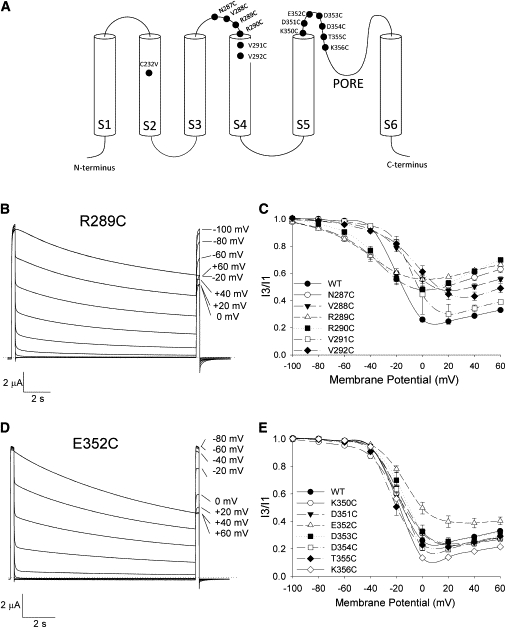
Cysteine substitutions in the S3-S4 and S5-P-loop linkers alter Kv2.1 inactivation. (A) Schematic of a Kv2.1 α-subunit showing the relative locations of the mutations studied. (B and D) Current recordings from oocytes expressing the R289C and E352C mutant channels, made using the 3P protocol. (C and E) The inactivation-voltage relationships for the S3-S4 and S5-P-loop linker mutants, respectively. The data for wt Kv2.1 is also shown for comparison. (Data points) Mean ± se. n > 4 for each mutant. (Lines) Spline connectors to guide the eye. For clarity, Boltzmann fits to the data are not shown; see Table 1 for a summary of the data and the fit parameters.
Changes in the voltage- and/or time-dependence with which channels move along the activation pathway may indirectly affect U-type inactivation by altering the occupancy of preopen closed states. Thus, before assessing the impact of the mutations on U-type inactivation, we first characterized their activation properties. Table 2 summarizes the G-V relationships for both wt and mutant Kv2.1 channels, as well as the change in the free energy of activation at 0 mV (ΔΔG0) caused by each mutation. Some mutations, particularly those in the S3-S4 linker (e.g., V292C), caused slight but statistically significant shifts in the V1/2 and k of activation. However, even for these mutations, the ΔΔG0 values were below 1 kcal mol−1, indicating that there were minimal perturbations in the relative stabilities of the open and closed states at 0 mV (27,30,31). We take these results to mean that the S3-S4 linker and S5-P-loop mutations resulted in only minor changes in voltage-dependent activation gating, especially during moderate depolarizations.
Table 2.
Voltage-dependent activation of the wt and mutant Kv2.1 channels
| Construct | V1/2 (mV) | k (mV) | ΔΔG (kcal mol−1) | n |
|---|---|---|---|---|
| wt | −1.6 ± 1.2 | 9.8 ± 0.33 | — | 9 |
| S3-S4 linker mutants | ||||
| N287C | −7.1 ± 1.3 | 12.4 ± 1.2∗ | −0.3 ± 0.08 | 5 |
| V288C | 3.5 ± 1.9 | 11.4 ± 0.2∗ | 0.3 ± 0.1 | 7 |
| R289C | 2.7 ± 2.0 | 14.3 ± 0.4∗ | 0.2 ± 0.1 | 5 |
| R290C | 4.0 ± 1.3 | 12.2 ± 0.3∗ | −0.1 ± 0.09 | 4 |
| V291C | 5.6 ± 1.9∗ | 11.2 ± 1.0 | 0.5 ± 0.1 | 5 |
| V292C | 14.2 ± 0.5∗ | 11.7 ± 0.4∗ | 0.8 ± 0.2 | 6 |
| S5-P-loop mutants | ||||
| K350C | 1.8 ± 3.6 | 12.1 ± 0.1 | 0.3 ± 0.2 | 5 |
| D351C | −9.4 ± 0.7∗ | 9.4 ± 0.3 | −0.5 ± 0.09 | 5 |
| E352C | −2.6 ± 1.1 | 10.4 ± 0.5 | −0.07 ± 0.09 | 5 |
| D353C | 0.3 ± 1.1 | 10.1 ± 0.3 | 0.1 ± 0.1 | 7 |
| D354C | −1.6 ± 1.0 | 9.1 ± 0.2 | −0.04 ± 0.1 | 10 |
| T355C | −8.2 ± 0.9∗ | 10.4 ± 0.1 | −0.4 ± 0.09 | 5 |
| K356C | 3.8 ± 1.9 | 10.1 ± 0.3 | 0.3 ± 0.1 | 8 |
Significant difference (P < 0.05) from wt values, based on the results of a Kruskal-Wallis ANOVA on ranks test.
Current recordings made with the 3P protocol from oocytes expressing an S3-S4 linker (R289C) and S5-P-loop (E352C) mutant are shown in Fig. 2, B and D, respectively. The mean inactivation-voltage relationships for each set of mutant channels are summarized in Fig. 2, C and E, and in Table 1. Relative to wt Kv2.1, mutations in the S5-P-loop caused little to no shift of the inactivation-voltage curves (Fig. 2 E), although K350C did show an increase in the slope factor k (Table 1). The minimum I3/I1 and the degree of upturn at +60 mV were also similar between wt and most S5-P-loop mutant channels (Table 1), indicative of preserved U-type inactivation. The striking exception to this was the E352C mutant, which showed reduced inactivation at moderate potentials and no upturn in the inactivation-voltage relationship, which could be fully described by a Boltzmann function (Fig. 2 E). That none of the S5-P-loop mutations, including E352C, caused major changes to Kv2.1 channel activation (see above; Table 2), suggests that the altered inactivation-voltage relationship of the E352C mutant is due to a specific effect on U-type inactivation.
Unlike the S5-P-loop mutants, the S3-S4 linker mutations caused variable but statistically insignificant shifts in the V1/2 of inactivation (Table 1). Additionally, with the exception of V291C, all of the S3-S4 linker mutations caused a significant (P < 0.05) increase in the slope factor k (Table 1). However, this may be related to the increased k values for the G-V curves of these mutants (Table 2). Of greater interest was the observation that, although the U-shaped inactivation-voltage relationship was preserved, all S3-S4 linker mutants, except for V291C, exhibited a decrease in the maximum extent of inactivation (Fig. 2 C, Table 1). In sum, U-type inactivation is reduced, but not eliminated by N287C, V287C, R289C, R290C, or V292C.
E352C selectively removes U-type inactivation from Kv2.1
A hallmark of U-type inactivation is that it is enhanced by increasing [K+]o (2,15,16,20,21). This is in contrast to the inhibitory effects of high [K+]o on P/C-type inactivation (7,32,33). To examine further the apparent loss of U-type inactivation in the E352C mutant, the effects of increasing [K+]o on the inactivation-voltage relationships of wt and E352C channels were studied (Fig. 3 A). As reported previously (15), changing [K+]o from 3 to 30 mM enhances the extent of U-type inactivation of wt Kv2.1 channels, particularly at moderate depolarizations (e.g., 0 mV), suggesting that external K+ ions promote and/or stabilize closed-inactivated states. In contrast, high [K+]o had no effect on the inactivation-voltage relationship of E352C channels. These results support the observation that the E352C mutation abolishes U-type inactivation of Kv2.1 channels.
Figure 3.
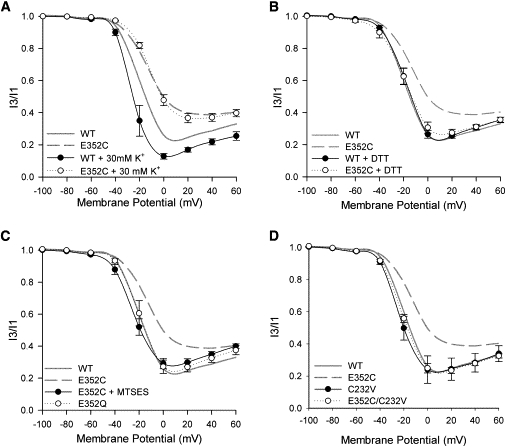
U-type inactivation can be restored to the E352C mutant by removing disulfide bonds. Each panel shows the inactivation-voltage relationship of wt and E352C channels under different test conditions. Unless otherwise stated, recordings were performed in ND96. Data are shown as mean ± SE, connected by simple spline curves; n > 3 for each data set. The inactivation-voltage relationships of wt and E352C channels under control conditions in ND96 are also shown for comparison (shaded and dashed spline curves, respectively). (A) Inactivation of the E352C mutant is not enhanced by 30 mM [K+]o. (B) Thirty-minute DTT pretreatment restores U-type inactivation to E352C channels. (C) Thirty-minute MTSES pretreatment restores U-type inactivation in E352C channels. Note that before treatment with MTSES, the oocyte was exposed to DTT for 30 min. U-type inactivation is unaffected by the E352Q mutation. (D) U-type inactivation is restored in the E352C/C232V double mutant. The inactivation-voltage relationship of the C232V mutation alone is not different from that of wt Kv2.1.
E352C may form a disulfide bond to remove U-type inactivation
Potential reasons for the loss of U-type inactivation in the E352C mutant are that the introduced cysteine residue forms a disulfide bond that inhibits inactivation from pre-open closed states or that the negatively charged glutamate residue is required for U-type inactivation. To test for the first possibility, oocytes expressing the E352C mutant were pretreated with the reducing agent, DTT (1 mM) before recording. Fig. 3 B shows that DTT had no effect on wt Kv2.1, but restored U-type inactivation to the E352C mutant, consistent with the hypothesis that E352C may interact with a native cysteine to remove U-type inactivation.
To ensure that the loss of charge caused by the E352C mutation does not contribute to the reduced U-type inactivation, the effect of restoring the charge by pretreating E352C expressing oocytes with the cysteine modifying agent, MTSES (1 mM), was examined. MTSES modification of all of the available introduced cysteines was ensured by first pretreating oocytes with DTT. We also examined the effect of the charge-neutralizing E352Q mutation. Fig. 3 C shows that U-type inactivation is maintained in both the MTSES-modified E352C channels and in E352Q channels. Together, these results show that the charge at position 352 is not required for U-type inactivation and instead suggest that the chemical nature of the introduced cysteine residue causes the loss of U-type inactivation of the E352C mutant.
Having shown that the E352C-induced loss of U-type inactivation is dependent on the oxidative state of the introduced cysteine residue and thus, likely due to disulfide bond formation, the next goal was to find the disulfide bond partner. Potential candidates are cysteine residues in S2 (C232) and in S6 (C393, C394). The possibility of an interaction between E352C and C232 was examined using an E352C/C232V double-mutant channel. In the background of the C232V mutation, E352C no longer prevented U-type inactivation and the double mutant had an inactivation-voltage relationship that is nearly identical to that of wt Kv2.1 (Fig. 3 D). C232V alone does not change the inactivation-voltage relationship of the channel (Fig. 3 D, see also (34)), indicating that the U-type inactivation in the double mutant is not due to enhanced inactivation brought about by the C232V mutation. To test the specificity of the interaction, cysteine substitutions were made in the background of the E352C/C232V mutant at positions proximal to C232 (L225, V228, or F236) on the same face of the S2 helix. Fig. S1 in the Supporting Material shows that U-type inactivation was preserved in each of these triple mutants and was not affected by DTT. Together, the results support the hypothesis that in the E352C mutant channel, E352C interacts specifically with C232 to remove U-type inactivation from preopen closed states. To test whether the disulfide bond occurs within a single subunit, or between adjacent subunits, Western blot analysis was performed (see Fig. S2). Previously, it had been shown that disulfide interactions between adjacent channel subunits produce dimers that can be disrupted into monomers after DTT pretreatment (35–37). The data in Fig. S2 show that both wt and E352C channel subunits form monomers rather than dimers, suggesting that the E352C-C232 disulfide bonds likely form within the same subunit. Similar to Zühlke et al. (34), we found that the C393V and C394V mutations cause dramatic changes to inactivation gating (not shown) that complicate the interpretation of any interaction with E352C and that require further investigation.
The reduced inactivation in the S3-S4 linker mutants may involve both disulfide bond and charge effects
The maximum extent of inactivation in N287C, V288C, R289C, R290C, and V292C mutant channels was decreased compared to wt (Fig. 2, Table 1). Compared to ShIR, Kv2.1 has two additional arginine residues (R289 and R290) in the C-terminal S3-S4 linker that may be important in the robust voltage- and state-dependent U-type inactivation observed in Kv2.1 but not ShIR channels. To assess this, the effect of restoring the charge with MTSET was examined (Fig. 4 C). DTT treatment of the R289C mutant (to ensure the availability of the introduced cysteines for MTSET modification) caused only a slight right-shift and increase in the extent of inactivation. However, subsequent treatment with MTSET resulted in an inactivation-voltage relationship very similar to that of wt, suggesting that the loss of positive charge at position 289 underlies the reduced inactivation in the R289C mutant. In contrast, the positive charge at R290 does not appear to be important for U-type inactivation, as there was no difference between the effects of pretreatment of the R290C mutant with DTT or MTSET (Fig. 4 D), both of which enhanced U-type inactivation to similar degrees.
Figure 4.
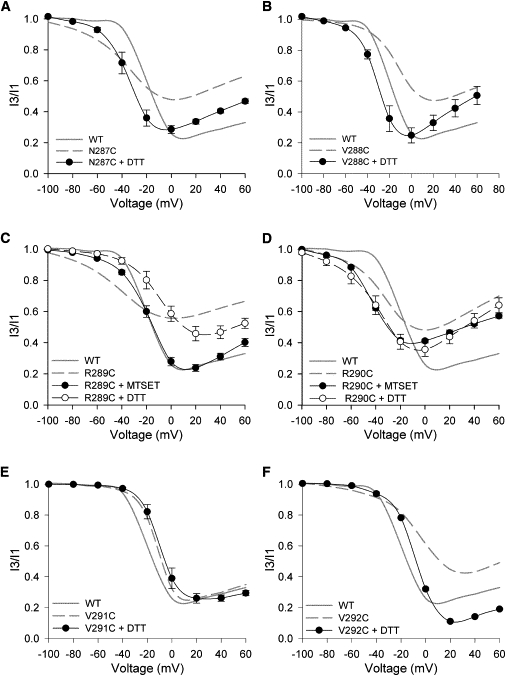
Modulation of U-type inactivation in the S3-S4 linker mutants. Each panel shows the inactivation-voltage relationship of a given mutant channel recorded after pretreatment with DTT and/or MTSET. Each panel also shows the inactivation-voltage relationship of wt Kv2.1 and the mutant channel (shaded and dashed lines, respectively) recorded under control conditions for comparison. (Data points) Mean ± SE, connected by spline curves; n ≥ 3 for each test condition.
Because the N287C, V288C, and V292C mutations are charge-neutral, the reduced inactivation is likely caused by other factors. As for E352C, the possibility of inhibitory disulfide-bond formation in these mutants was investigated. After DTT treatment, the N287C, V288C and V292C mutant channels exhibited similar maximum levels of inactivation to wt channels (Fig. 4, A, B, and F). Also, the slope of the descending portion of the inactivation-voltage relationship for each mutant after DTT treatment was similar to that of wt Kv2.1. As Fig. 4 E shows, DTT treatment had little to no effect on the inactivation-voltage relationship of the V291C mutant channel, which was already similar to that of wt. In summary, the decreased extent of U-type inactivation in the R289C mutation appears to be largely a result of the loss of positive charge at that position. In the case of the S3-S4 linker mutants N287C, V288C, R290C, and V292C, the decreased U-type inactivation appears to be due to disulfide interactions involving the introduced cysteine.
Modeling the effects of E352C and R289C
An allosteric model of U-type inactivation has previously been described for Kv2.1 (15) and, in modified form, for Kv3.1, ShIR channels (16), and N-terminally truncated Kv1.5 channels (19). The same 12-state model (Fig. 5 A) was used here to simulate current through wt, E352C, and R289C channels. Satisfactory simulations of the G-V (not shown) and inactivation-voltage relationships (Fig. 5 B) for wt Kv2.1, which were somewhat right-shifted compared to those described by Klemic et al. (15), required slight changes to some of the model's 11 free parameters. The voltage-dependent properties of wt Kv2.1 channel activation and deactivation have been described previously (38–40) and will not be addressed in detail here but are presented in Fig. S3 and Table S1. As in the original model, the forward rate constant (ko) from the final closed state (C4) to the open state (O) was voltage-independent and determined from the minimum time constant for current activation at strongly depolarized potentials (see Fig. S3). The reverse transition between these two states was exponentially dependent on voltage (see Materials and Methods), with a rate constant (k−o) and apparent valence (z−o) that were constrained by the voltage-dependence of the fast time constant for deactivation measured between −140 and −80 mV (see Fig. S3).
Figure 5.
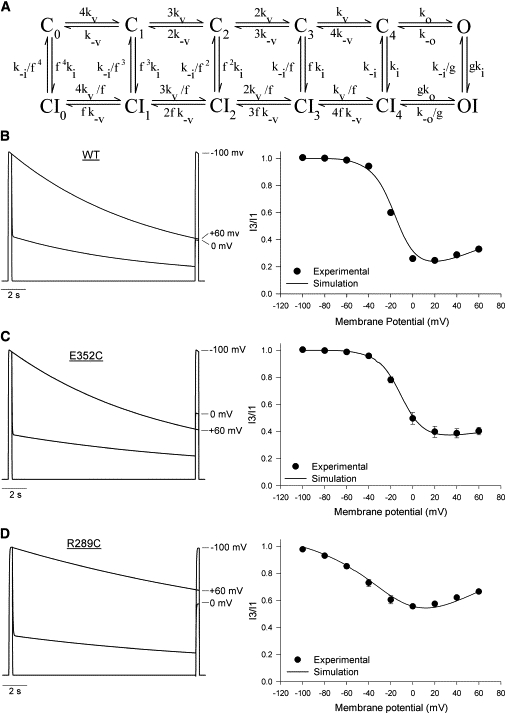
Kinetic modeling of wt and mutant Kv2.1 inactivation. (A) Allosteric model used to simulate channel gating. State transitions are governed by 11 free parameters, as described in the text and Table 3. (B–D, left side) Simulated macroscopic currents for wt, E352C, and R289C channels, respectively. These simulations were performed using the parameters shown in Table 3. Three superimposed traces are shown for each construct. (Right side) Comparison of the experimental and simulated inactivation-voltage relationships. (Data points) Mean ± SE experimental i3/i1 values. (Solid lines) Simulated I3/I1 values, plotted against voltage in 1-mV increments.
With iterative manipulation of the forward and backward rate constants for the remaining horizontal transitions, we were able to reproduce the experimentally observed kinetics and voltage dependence of activation (not shown). A final constraint was placed on the parameters k−i and f, an allosteric factor that governs how much inactivation is favored by activation of the voltage sensors. Time constants for recovery from inactivation, measured at very negative potentials (e.g., −120 mV; see Fig. S3), were used to fix the rate of recovery (k−i/f 4) from the earliest closed-inactivated state (CI0). As described by Kurata et al. (19), this was based on the assumption that the strongly hyperpolarized recovery potential would result in almost all of the channels occupying CI0 and a single exponential time-course for recovery (see Fig. S3). Thus, any changes in k−i would cause reciprocal changes in f. With these constraints, and with the allosteric factor g set to allow for some inactivation from the open state, a reasonable simulation of the macroscopic currents and inactivation-voltage relationship of wt Kv2.1 channels was achieved (Fig. 5 B) with the parameters shown in Table 3.
Table 3.
List of model parameters used in simulations
| wt | E352C | R289C | |
|---|---|---|---|
| kv | 110 | 110 | 90 |
| k−v | 50 | 50 | 45 |
| ko∗ | 77 | 77 | 41 |
| k−o∗ | 43 | 43 | 28 |
| ki | 0.33 | 0.14 | 0.13 |
| k−i∗ | 0.0058 | 0.0058 | 0.0058 |
| f∗ | 0.20 | 0.20 | 0.43 |
| g | 0.082 | 0.39 | 0.001 |
| zv | 1.25 | 1.25 | 0.32 |
| z−v | −0.1 | −0.1 | −0.39 |
| z−o∗ | −0.41 | −0.41 | −0.40 |
Values for kv, k−v, ko, k−o, ki, k−i are given in s−1.
Values that were constrained by experimental measurements (see text and Table S1).
Having established model parameters to describe the U-type inactivation of wt Kv2.1, the kinetic changes required to simulate the inactivation of the E352C and R289C mutants were determined. For E352C, which showed no statistical change in either the properties of the G-V relationship (Table 2) or in the kinetics of activation or recovery from inactivation (see Table S1), the constraints on the parameters were unchanged and the main challenge was to simulate the reduction in the maximum extent of inactivation and the loss of the upturn in the inactivation-voltage relationship. A satisfactory fit (Fig. 5 C) was obtained by decreasing the value for ki, and increasing the value of g (Table 3). The reduction in the rate of inactivation from the closed states and the ∼100% increase in the rate of inactivation from the open state is consistent with a selective loss of U-type inactivation in favor of inactivation from the open state.
In contrast to E352C, the R289C mutant causes an increase in the slope factor for the G-V relationship and slows channel gating (Table 2 and Table S1). R289C also caused a left-shift and increased slope factor of the inactivation-voltage relationship, a decreased maximum extent of inactivation, and a slowing of the time-course for recovery from inactivation (Table 1 and Fig. S1). Adjustment of the constrained and unconstrained parameters for horizontal transitions (see Table 3) allowed for simulation (not shown) of the altered G-V relationship and also largely accounted for the shift and altered slope of the inactivation-voltage relationship, suggesting that these latter effects largely result from modified activation gating transitions that consequently affect inactivation. Further adjustment of the value of k-i/f4 to match the constraint of the recovery rate, along with iterative decreases in the values of ki and g, were required to achieve a reasonable simulation (Fig. 5 D) of macroscopic currents and the inactivation-voltage relationship. The resulting reductions in the rates of inactivation from the preopen closed states (i.e., C3 and C4) and the open state, as well as the greatly accelerated rate of recovery from the open-inactivated state, are consistent with our experimental results and the conclusion that the R289C mutation inhibits inactivation from both the open and the closed states.
Discussion
Despite its frequent use in the study of Kv channel gating and structure/function relationships (e.g., (10,17,41,42)), there is relatively little information about how Kv2.1 channels inactivate. More specifically, although it is thought that Kv2.1 channels may undergo both P/C-type inactivation and U-type inactivation, little is known about the molecular mechanisms underlying U-type inactivation and how they are different from those of P/C-type inactivation. The experiments described here were performed with the aim of identifying residues in the external S3-S4 and S5-P-loop linkers potentially involved in the regulation of Kv2.1 U-type inactivation. We found that E352C in the S5-P-loop specifically inhibits inactivation from pre-open closed states and U-type inactivation. In contrast, the S3-S4 residues, with the exception of V291C, tended to reduce the extent of inactivation overall.
E352C selectively removes U-type inactivation
The E352C mutation selectively abolished the U-shaped inactivation-voltage profile without any other changes in the voltage- and/or time-dependence of activation, deactivation, or recovery from inactivation. That U-type inactivation is restored to the E352C mutant by DTT pretreatment (Fig. 3 B) or the C232V mutation (Fig. 3 D) strongly suggests that a disulfide bond between E352C and C232 underlies the loss of U-type inactivation. Although recent x-ray crystal structures for Kv1.2 and Kv1.2/2.1 chimeric channels (43–45) place C232 too far away from E352C to form either inter- or intrasubunit disulfide bonds, our data (including Western blot analysis, see Fig. S2) are consistent with recent evidence that suggests the S1-S2 linker and perhaps the S2 segment undergo dynamic movements that may include interactions with the pore domain (46,47). It is interesting to note that the intrasubunit E352C-C232 interaction we propose appears to be specific and localized, because substitution of cysteine residues at locations on the same face of the S2 helix as C232 does not remove U-type inactivation of E352C/C232V mutant channels (see Fig. S1). We envision, then, motions of S2 during gating that orient C232 to within close proximity of E352C in the S5-P-loop linker. Although we did not assess the possibility of disulfide interactions between E352C and the native cysteines in S6 (C393 and C394), given our experimental results it is difficult to come to an explanation for the loss of U-type inactivation in the E352C mutant other than that of disulfide bond formation with C232.
How does E352C inhibit inactivation from closed but not open states?
The finding that E352C and C232 interact to selectively inhibit U-type inactivation from closed states during moderate depolarizations while leaving inactivation from the open state at strong depolarizations mostly intact suggests that two mechanistically distinct processes underlie inactivation from the closed and open states of Kv2.1. This is in agreement with previous suggestions that both P/C-type inactivation and U-type inactivation can occur in Kv2.1 and ShIR channels (15,18,41). Why U-type inactivation is dominant in Kv2.1 channels whereas P/C-type inactivation takes precedence in ShIR and other channels of the Kv1 family, however, remains unclear. It has been reported that a chimeric channel created by inserting the S5-S6 regions of Kv1.3 into the Kv2.1 backbone exhibits characteristics of P/C-type inactivation only (32,41), which would imply that U-type inactivation depends on the pore domain formed by S5-S6. The importance of the pore domain is highlighted by the inhibition of Kv2.1 U-type inactivation by internal TEA (18,48), which is largely removed by the replacement of residues in this region with an analogous segment from Kv3.1 (49).
Further support for the involvement of the pore includes U-type inactivation-induced changes in ion selectivity, and the sensitivity of inactivation to [K+]o, TEAo (17,41), and to mutation of C393 in the S6 segment (34,50). Although the precise changes in the pore that cause U-type inactivation are as yet unclear, it is intriguing to consider that the E352C-C232 interaction may distort the conformation and/or restrict movement of the S5-P-loop linker in a manner that inhibits the U-type inactivation mechanism while leaving P/C-type inactivation intact. In other words, the observation that the E352C-C232 interaction abolishes inactivation from closed states suggests that protein motions involving E352 underlie U-type inactivation and that the engineered E352C-C232 interaction prevents these rearrangements. An alternative interpretation is that the E352C-C232 interaction may inhibit both the mechanism for allosteric coupling of the voltage sensor and the U-type inactivation gate that is thought to underlie preferential inactivation from preopen closed states (2). Further studies are required to distinguish between these possibilities.
The importance of positive charge at R289 for inactivation
Unlike E352C, R289C in the S3-S4 linker inhibits inactivation during both moderate and strong depolarizations (Figs. 4 C and 5 D). That inactivation was rescued in R289C channels by MTSET (Fig. 4 C), which suggests that the R289 charge is important for both closed and open state inactivation in Kv2.1 channels. A simple explanation may be that an electrostatic interaction between R289 and E352 is important for U-type inactivation, and that mutation of either of these residues inhibits that interaction and, thus, we have inactivation. However, this does not account for the specific effects of E352C on closed-state inactivation versus the reduction of both closed- and open-state inactivation induced by R289C. Furthermore, the effects of E352C were not the result of the loss of charge at this position (see Fig. 3 C), which argues against such an electrostatic interaction.
It is possible that the insensitivity of U-type inactivation to charge neutralization at position 352 is due to the surrounding negative charges (i.e., D351, D353, D354) that may buffer the effects of a loss of charge in this region. In contrast, R289 is relatively isolated from other charges within the α-helical portion of the S3-S4 linker (31) and this region may therefore be more sensitive to the loss of positive charge. An alternative explanation is that R289 is involved in voltage sensing and removal of the charge at this position decreases voltage sensitivity and interferes with the allosteric coupling of the voltage sensor and the inactivation machinery. That R289C impedes the movement of the S4 voltage sensor is suggested by the slowed activation and deactivation kinetics (see Table S1), and the shallower slopes of the G-V and inactivation-voltage relationships (Tables 1 and 2). As both U-type and P/C-type inactivation are coupled to voltage-sensor movement (1,15,16,18), this could account for the generalized inhibition of inactivation from both the closed and open states.
The C-terminal half of the S3-S4 linker may also modulate Kv2.1 inactivation
Cysteine substitutions from N287 to V292 showed variable effects on the inactivation-voltage relationship (Fig. 4). As discussed above, the effects of R289C appear to be largely due to the loss of charge at this position. V291C had little effect on inactivation, whereas the other constructs (N287C, V288C, R290C, and V292C) caused a decrease in the maximum steady-state extent of inactivation, a shallower slope of the inactivation-voltage relationship and, in some cases, a shift in the voltage-dependence of inactivation.
The effects of these four mutations, particularly the reduced slope and extent of inactivation, were significantly inhibited by DTT pretreatment (Fig. 4). This suggests that disulfide bond formation involving S3-S4 linker residues inhibits U-type inactivation, perhaps by restricting the movement of the S4 voltage-sensor itself, or by altering allosteric coupling to the pore. That mutation at multiple sites within the S3-S4 linker modulates the inactivation process implies an intrinsic role for this region. Indeed, voltage-clamp fluorimetry data suggest that the C-terminal portion of the S3-S4 linker is involved in the dynamics of inactivation (22,23). Here, we have identified sites important in Kv2.1 inactivation. Further studies should be directed at understanding their dynamic involvement.
In sum, we have found that substitution of a cysteine residue at position E352 in the S5-P-loop linker results in a profound and specific inhibition of U-type inactivation that is likely related to an inhibitory disulfide bond between E352C and C232 in the S2 segment. Substitutions in the S3-S4 linker also inhibited inactivation: in the case of R289, this resulted directly from loss of positive charge; in other cases, it was apparently due to the formation of disulfide bonds.
Acknowledgments
This work was supported by Natural Sciences and Engineering Research Council of Canada and Canada Foundation for Innovation Leader's Opportunity Fund infrastructure grants to T.W.C., who was also supported by a Heart and Stroke Foundation of Canada New Investigator Award and a Michael Smith Foundation for Health Research Career Scholar Award. C.M.N. was supported by a Natural Sciences and Engineering Research Council of Canada Undergraduate Student Research Award.
Supporting Material
References
- 1.Kurata H.T., Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Bähring R., Covarrubias M. Mechanisms of closed-state inactivation in voltage-gated ion channels. J. Physiol. 2011;589:461–479. doi: 10.1113/jphysiol.2010.191965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grissmer S., Cahalan M. TEA prevents inactivation while blocking open K+ channels in human T lymphocytes. Biophys. J. 1989;55:203–206. doi: 10.1016/S0006-3495(89)82793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi K.L., Aldrich R.W., Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molina A., Castellano A.G., López-Barneo J. Pore mutations in Shaker K+ channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J. Physiol. 1997;499:361–367. doi: 10.1113/jphysiol.1997.sp021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 7.López-Barneo J., Hoshi T., Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 8.Baukrowitz T., Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 9.Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 10.De Biasi M., Hartmann H.A., Kirsch G.E. Inactivation determined by a single site in K+ pores. Pflugers Arch. 1993;422:354–363. doi: 10.1007/BF00374291. [DOI] [PubMed] [Google Scholar]
- 11.Hoshi T., Zagotta W.N., Aldrich R.W. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 12.Ogielska E.M., Zagotta W.N., Aldrich R.W. Cooperative subunit interactions in C-type inactivation of K channels. Biophys. J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y., Jurman M.E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 14.Panyi G., Sheng Z., Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klemic K.G., Shieh C.C., Jones S.W. Inactivation of Kv2.1 potassium channels. Biophys. J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemic K.G., Kirsch G.E., Jones S.W. U-type inactivation of Kv3.1 and Shaker potassium channels. Biophys. J. 2001;81:814–826. doi: 10.1016/S0006-3495(01)75743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Immke D., Wood M., Korn S.J. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J. Gen. Physiol. 1999;113:819–836. doi: 10.1085/jgp.113.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González-Pérez V., Neely A., Naranjo D. Slow inactivation in Shaker K channels is delayed by intracellular tetraethylammonium. J. Gen. Physiol. 2008;132:633–650. doi: 10.1085/jgp.200810057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurata H.T., Soon G.S., Fedida D. Altered state dependence of c-type inactivation in the long and short forms of human Kv1.5. J. Gen. Physiol. 2001;118:315–332. doi: 10.1085/jgp.118.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurata H.T., Soon G.S., Fedida D. Amino-terminal determinants of U-type inactivation of voltage-gated K+ channels. J. Biol. Chem. 2002;277:29045–29053. doi: 10.1074/jbc.M111470200. [DOI] [PubMed] [Google Scholar]
- 21.Kurata H.T., Doerksen K.W., Fedida D. Separation of P/C- and U-type inactivation pathways in Kv1.5 potassium channels. J. Physiol. 2005;568:31–46. doi: 10.1113/jphysiol.2005.087148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loots E., Isacoff E.Y. Molecular coupling of S4 to a K+ channel's slow inactivation gate. J. Gen. Physiol. 2000;116:623–636. doi: 10.1085/jgp.116.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandhi C.S., Loots E., Isacoff E.Y. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 2000;27:585–595. doi: 10.1016/s0896-6273(00)00068-4. [DOI] [PubMed] [Google Scholar]
- 24.Ortega-Sáenz P., Pardal R., López-Barneo J. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K+ channels. J. Gen. Physiol. 2000;116:181–190. doi: 10.1085/jgp.116.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elinder F., Männikkö R., Larsson H.P. S4 charges move close to residues in the pore domain during activation in a K channel. J. Gen. Physiol. 2001;118:1–10. doi: 10.1085/jgp.118.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Slyke A.C., Rezazadeh S., Claydon T.W. Mutations within the S4-S5 linker alter voltage sensor constraints in hERG K+ channels. Biophys. J. 2010;99:2841–2852. doi: 10.1016/j.bpj.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li-Smerin Y., Hackos D.H., Swartz K.J. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 2000;25:411–423. doi: 10.1016/s0896-6273(00)80904-6. [DOI] [PubMed] [Google Scholar]
- 28.Colquhoun D., Hawkes A.G. A Q-matrix cookbook: how to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In: Sakmann B., Neher E., editors. Single-Channel Recording. Plenum Press; New York: 1995. pp. 589–633. [Google Scholar]
- 29.Lvov A., Chikvashvili D., Lotan I. VAMP2 interacts directly with the N terminus of Kv2.1 to enhance channel inactivation. Pflugers Arch. 2008;456:1121–1136. doi: 10.1007/s00424-008-0468-7. [DOI] [PubMed] [Google Scholar]
- 30.Yifrach O., MacKinnon R. Energetics of pore opening in a voltage-gated K+ channel. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- 31.Li-Smerin Y., Hackos D.H., Swartz K.J. Alpha-helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 2000;115:33–50. doi: 10.1085/jgp.115.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiss L., Korn S.J. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng Y.M., Fedida D., Kehl S.J. Kinetic analysis of the effects of H+ or Ni2+ on Kv1.5 current shows that both ions enhance slow inactivation and induce resting inactivation. J. Physiol. 2010;588:3011–3030. doi: 10.1113/jphysiol.2010.191544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zühlke R.D., Zhang H.J., Joho R.H. Role of an invariant cysteine in gating and ion permeation of the voltage-sensitive K+ channel Kv2.1. Receptors Channels. 1994;2:237–248. [PubMed] [Google Scholar]
- 35.Aziz Q.H., Partridge C.J., Sivaprasadarao A. Depolarization induces intersubunit cross-linking in a S4 cysteine mutant of the Shaker potassium channel. J. Biol. Chem. 2002;277:42719–42725. doi: 10.1074/jbc.M207258200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H.J., Liu Y., Joho R.H. Oxidation of an engineered pore cysteine locks a voltage-gated K+ channel in a nonconducting state. Biophys. J. 1996;71:3083–3090. doi: 10.1016/S0006-3495(96)79502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang M., Zhang M., Tseng G.N. Dynamic conformational changes of extracellular S5-P linkers in the hERG channel. J. Physiol. 2005;569:75–89. doi: 10.1113/jphysiol.2005.093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shieh C.C., Klemic K.G., Kirsch G.E. Role of transmembrane segment S5 on gating of voltage-dependent K+ channels. J. Gen. Physiol. 1997;109:767–778. doi: 10.1085/jgp.109.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi G., Kleinklaus A.K., Trimmer J.S. Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J. Biol. Chem. 1994;269:23204–23211. [PubMed] [Google Scholar]
- 40.Kerschensteiner D., Stocker M. Heteromeric assembly of Kv2.1 with Kv9.3: effect on the state dependence of inactivation. Biophys. J. 1999;77:248–257. doi: 10.1016/S0006-3495(99)76886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiss L., LoTurco J., Korn S.J. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao X., MacKinnon R. Functional analysis of Kv1.2 and paddle chimera Kv channels in planar lipid bilayers. J. Mol. Biol. 2008;382:24–33. doi: 10.1016/j.jmb.2008.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Wang Q., Ma J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based x-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA. 2010;107:11352–11357. doi: 10.1073/pnas.1000142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 46.Chakrapani S., Cuello L.G., Perozo E. Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer. Structure. 2008;16:398–409. doi: 10.1016/j.str.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horne A.J., Peters C.J., Fedida D. Fast and slow voltage sensor rearrangements during activation gating in Kv1.2 channels detected using tetramethylrhodamine fluorescence. J. Gen. Physiol. 2010;136:83–99. doi: 10.1085/jgp.201010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerschensteiner D., Monje F., Stocker M. Structural determinants of the regulation of the voltage-gated potassium channel Kv2.1 by the modulatory α-subunit Kv9.3. J. Biol. Chem. 2003;278:18154–18161. doi: 10.1074/jbc.M213117200. [DOI] [PubMed] [Google Scholar]
- 49.Hartmann H.A., Kirsch G.E., Brown A.M. Exchange of conduction pathways between two related K+ channels. Science. 1991;251:942–944. doi: 10.1126/science.2000495. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y., Joho R.H. A side chain in S6 influences both open-state stability and ion permeation in a voltage-gated K+ channel. Pflugers Arch. 1998;435:654–661. doi: 10.1007/s004240050566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.